
Fabricating polyacrylamide microbeads by inverse emulsification to mimic the size and elasticity of living cells†
Nicholas R.
Labriola
a,
Edith
Mathiowitz
b and
Eric M.
Darling
 *bc
*bc
aCenter for Biomedical Engineering, Brown University, Providence, RI 02912, USA
bDepartment of Molecular Pharmacology, Physiology, and Biotechnology, Brown University, Providence, RI 02912, USA
cSchool of Engineering and Department of Orthopaedics, Brown University, Providence, RI 02906, USA. E-mail: Eric_Darling@brown.edu; Fax: +1 401 863 1595; Tel: +1 401 863 6818
First published on 9th December 2016
Abstract
Inverse emulsification was used to fabricate polyacrylamide (PAAm) microbeads with size and elastic properties similar to typical, mammalian cells. These biomimicking microbeads could be fluorescently stained and functionalized with a collagen type-I coating, post-polymerization, for tracking bead locations and promoting cell recognition/binding, respectively. By occupying a previously unfilled range of sizes and mechanical properties, these microbeads may find unique use in both biomedical and materials applications.
Polymer microparticles have been used extensively throughout the biomedical sciences (e.g., drug delivery, tissue engineering, encapsulation, etc.). Such particles have been produced through a variety of methodologies, including microfluidics,1 layer-by-layer deposition,2 particle replication in non-wetting templates,3 hydroelectrodynamic jetting,4 dispersion polymerization,5 and emulsification,6 among others. The inherent differences associated with these methodologies result in varying levels of compatibility with specific polymers, as well as different particle elasticity and size distributions (Fig. 1). Importantly, there is currently a gap for the sizes (5–50 μm)7 and elasticities (0.1–5 kPa)8 that are similar to living cells. To the best of our knowledge, our study is the first to demonstrate the fabrication of microbeads with characteristics that mimic cell elasticity, size, and spherical shape.
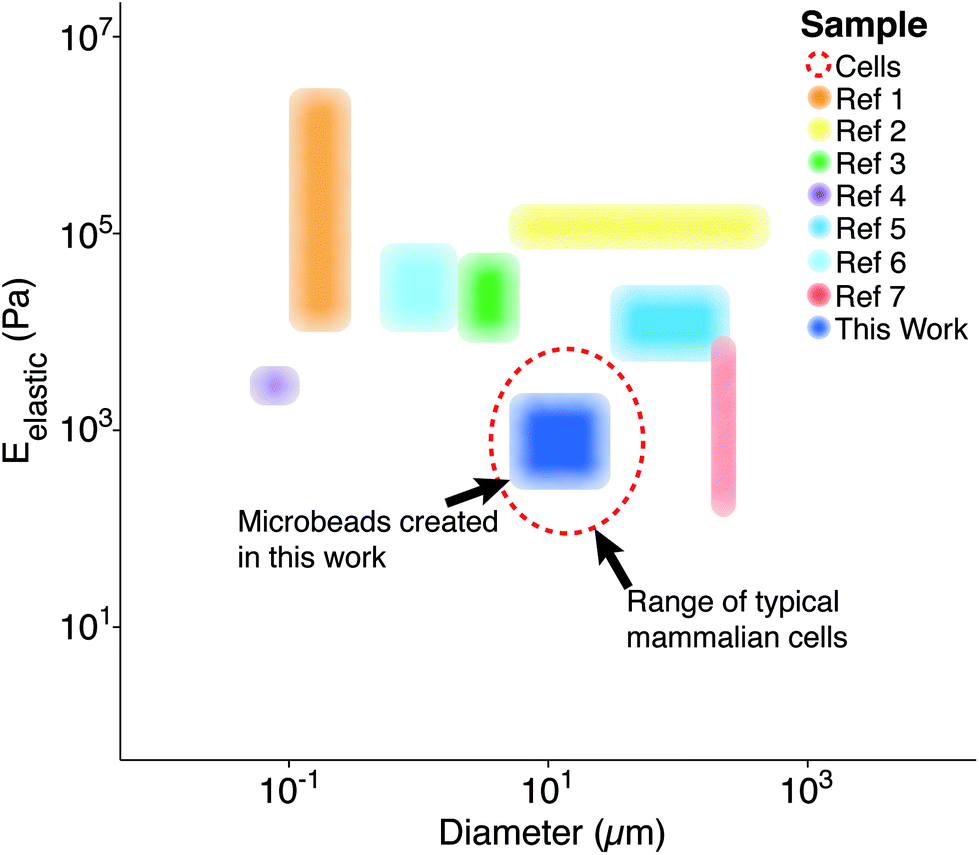 | ||
| Fig. 1 Previously reported particle/bead size and elasticity ranges. Each region represents the span of microbead diameters and elastic moduli documented in other studies alongside that developed for this work (dark blue).9 The dashed, red line represents the approximate diameter (∼10 μm) and elastic moduli (∼1 kPa) associated with mammalian cells in their spherical morphology.8 *Ref. 9 contains each of the references from the legend of this figure, in order. | ||
Cellular mechanical properties, or mechanophenotype, have increasingly been used as a novel biomarker for identifying specific cell types or disease states. A mechanophenotype can be used for both diagnostic and research purposes, with relevant examples including cancer, sickle cell, and diabetes.10 Microfluidic devices are one means by which cellular mechanophenotypes are being identified and explored, e.g., to isolate rare, circulating tumor cells (CTCs) from blood based on their unique size and deformability.11 However, standardization of this type of equipment can be complicated, especially since reference materials combining size and stiffness do not currently exist. The microbeads presented in this study could be used to test and calibrate devices or methodologies intended to manipulate, characterize, or sort cells.
Additional motivation for creating microbeads with high mechanical compliance is to mimic the stimulatory effect cells can receive when adhered to materials with biologically relevant mechanical properties.12 Through mechanosensing, cells recognize the stiffness of their substrates and undergo cytoskeletal remodelling that can alter cell fate. Because this effect does not require exogenous molecules, there has been increased interest in developing new cell culture systems that use material mechanical properties to direct cell behavior and physiological responses in general.13 As such, polymer microbeads that mimic the size and mechanical properties of cells will have a variety of applications in research involving three-dimensional (3D) culture systems used to study cell responses to physiologically relevant substrate mechanical properties, various surface coatings, and localized delivery of bioactive molecules.
Emulsification is a common and straightforward technique used to produce micro/nano-beads and is compatible with many polymers.14 The method described herein uses polyacrylamide (PAAm), a mechanically tunable polymer12 that relies on free radical initiation15 to form a hydrogel mesh structure, in conjunction with water-in-oil emulsion, or inverse emulsification, to produce “cell-like” microbeads. PAAm offers simple chemistry, rapid polymerization, long-term mechanical and morphological stability, functionalization, and compatibility with protein coatings through NHS ester-mediated cross-linking. Of paramount importance is the capability of PAAm to allow for reproducible formulation of cross-linked gels with Young's moduli below 1 kPa, which can be problematic for other materials.
The aim of this work was to fabricate PAAm microspheres with diameters and mechanical properties similar to cells exhibiting a spherical morphology. Here we describe for the first time a method that can create cell-sized (5–40 μm) PAAm microbeads with tunable mechanical properties (0.25–2 kPa) through inverse emulsification. The microbeads were additionally modified post-polymerization by fluorescent staining and collagen coating.
PAAm microbead fabrication was accomplished through inverse emulsion polymerization in a 250 mL Erlenmeyer flask using 200 mL of cyclohexane (HPLC grade, Thermo Fisher Scientific, Madison, WI, USA). The surfactant polysorbate 85 (Span 85, Sigma-Aldrich, Nantic, MA) was dispersed at a 1% (v/v) concentration in the cyclohexane solvent to stabilize microbeads in spherical morphologies and to minimize particle aggregation using a magnetic stirrer (375 Hotplate/Stirrer, VWR Scientific Products, Bridgeport, CT) with a cylindrical stir bar (39 × 10 mm, 2 × 11 mm pivot ring). Since free radicals are required to initiate the polymerization of PAAm, it was critical to remove oxygen, a free radical trap, from the system for synthesis to proceed uninhibited.16 The solubility of dissolved gases was decreased by reducing the local environmental pressure with a vacuum pump.17 While stirring at 700 RPM, a −25′′ Hg vacuum was applied to the surfactant/solvent mixture for 30 minutes by linking a vacuum pump to the flask's rubber stopper (Note: An additional large volume vessel should be connected in series between the pump and reaction vessel, and can be additionally cooled, to condense any evaporated cyclohexane). During the degassing period, a 10 mL PAAm solution was prepared using acrylamide (Bio-Rad, Hercules, CA), bis-acrylamide (Bio-Rad), ammonium persulfate (APS, Sigma-Aldrich), and phosphate buffered saline (PBS, Thermo Fisher Sci.). PBS was prepared with ultrapure, Milli-Q water (18 MΩ resistivity, Merck Millipore, Billerica, MA). For the three formulations used in this study, the final concentration of acrylamide and APS were kept constant at 4% and 0.1% (v/v), respectively. The final concentrations of bis-acrylamide used were 0.05%, 0.1%, or 0.2% (v/v) to create varying levels of crosslinking and elasticity. Immediately after degassing, N,N,N′,N′-tetramethylethylenediamine (TEMED, Thermo Fisher Sci.) was added to the PAAm solution to yield a final concentration of 0.1% (v/v). The mixture was vortexed for ten seconds and added drop-wise into the cyclohexane/Span 85 mixture. Vacuum was reapplied for one hour, and the stirring rate was increased to produce microbeads of the desired size. For this study, a stir rate of ∼1500 RPM yielded a range similar to typical mammalian cells (Fig. 2).
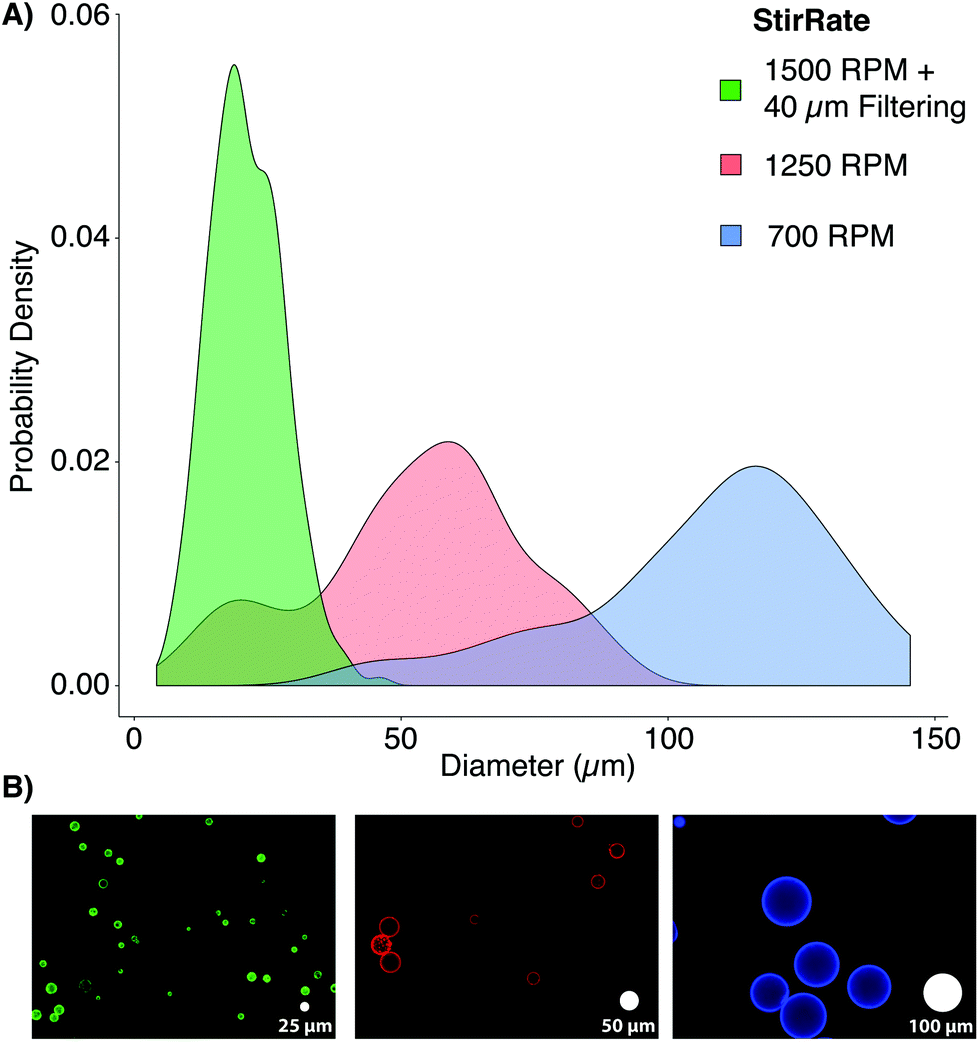 | ||
| Fig. 2 Microbead size distributions for various stir rates: (A) the population density histograms for microbeads generated using 0.1% bis-acrylamide at 1500 RPM with 40 μm filtering (green, 20 ± 7 μm), 1250 RPM (red, 48 ± 20 μm), and 700 RPM (blue, 104 ± 24 μm) show the expected inverse relationship between stir speed and particle size. The inclusion of 40 μm filtering yielded a much more monodisperse population within the target size range by removing large beads and aggregates. (B) Representative images of each bead population (false colored to match the distribution plots) show bead sizes relative to white “scale microbeads” with 25, 50, and 100 μm diameters (left to right). | ||
Once polymerization was completed (∼1 h), stirring was stopped, vacuum was released, the stir bar was removed, and microbeads were allowed to settle for 30 minutes. The solvent was removed, and the remaining solution containing microbeads (∼10 mL, viscous white fluid) was split between two, 50 mL conical tubes (Genesee, San Diego, CA). The microbeads were washed twice with 100% ethanol and pelleted by 5 minutes centrifugation at 400g. The microbeads were rehydrated, overnight, with 45 mL of PBS on a shaker. After rehydration, the microbeads were consecutively passed through 100, 70, and finally 40 μm cell strainers (Thermo Fisher Sci.), increasing the monodispersity of the bead populations to more closely mimic the distribution of cell populations through the removal of large beads and aggregates. Proper polymerization of PAAm microbeads was confirmed by FT-IR (ESI Fig. S1†).
The elastic properties of individual microbeads were characterized through indentation testing with an MFP-3D-Bio atomic force microscope (AFM, Asylum Research, Santa Barbara, CA) equipped with a spherically tipped cantilever, made by adhering a 5 μm diameter, polystyrene bead (Microbeads AS, Skedsmoorset, Norway) to a tip-less, silicon nitride cantilever (Bruker Corporation, MLCT10, k ∼ 0.03 N m−1), using an approach velocity of 10 μm s−1 and a trigger force of 5 nN (∼1–2 μm indentation). This testing set up has routinely been used to characterize live cells by our group.18 The elastic/Young's moduli (Eelastic) of the microbeads were determined from force vs. indentation curves using a modified Hertz model, as described previously.19 As has been demonstrated in two-dimensional gels,12 the elastic modulus of PAAm hydrogel microbeads was positively correlated to the concentration of the bis-acrylamide cross-linker, which connects linear chains of acrylamide together. Thus, we were able to generate mechanically distinct microbead populations by changing only the volume of bis-acrylamide in the polymer solution (Fig. 3). Interestingly, microbead moduli were approximately half that of 2D gels using the same PAAm formulation (Fig. S2†). This phenomenon is hypothesized to be due the surfactant adsorbing in place of acrylamide sub-units as well as interacting with looped regions of the polymer chains, altering the amorphous structure of the polymer and resulting in reduced mechanical stability.20 The elastic modulus for a given microbead batch typically had a standard deviation of less than 15% of the mean and consistent size distributions. While successive batches of microbeads created using the same formulation did not always yield the same average elastic modulus (ESI Table S1 and Fig. S2†), size distributions after 40 μm filtering remained in good agreement. This finding emphasizes the need to mechanically sample individual microbead batches to confirm properties align with those needed for a given application.
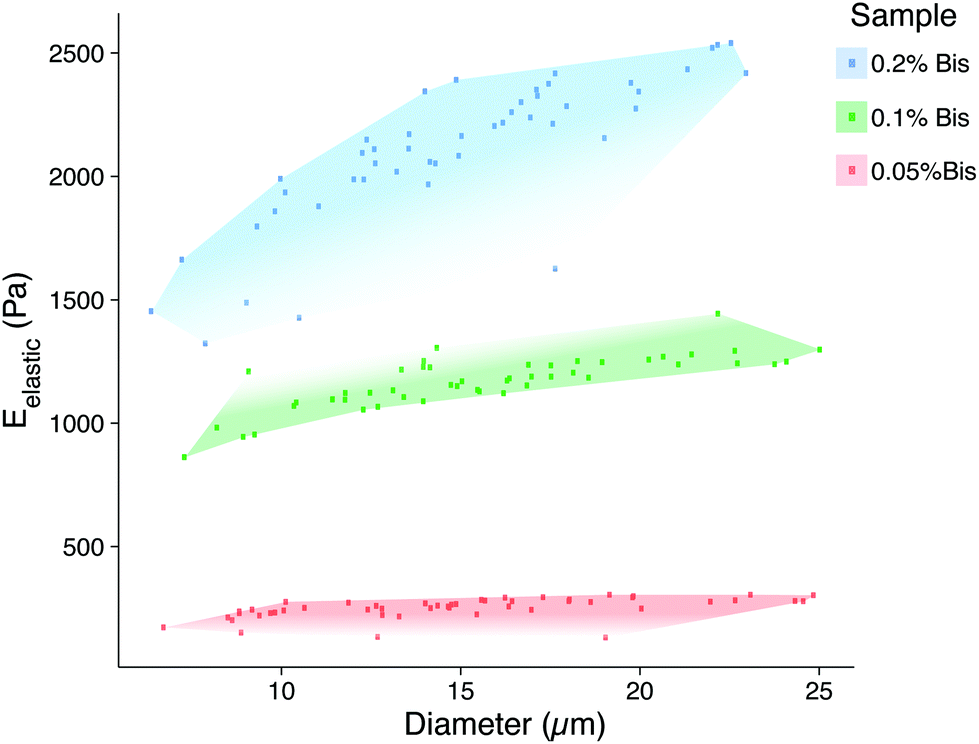 | ||
| Fig. 3 Microbead elasticity and size distributions measured by atomic force microscopy. This scatter plot illustrates the ranges of sizes and elastic moduli for each PAAm formulation. The red region represents the most compliant 0.05% bis-acrylamide formulation (250 ± 10 Pa, 14 ± 5 μm), green represents 0.1% bis-acrylamide (1200 ± 100 Pa, 15 ± 4 μm), and blue represents 0.2% bis-acrylamide (2100 ± 300 Pa, 14 ± 4 μm). | ||
Following polymerization, microbeads were stained fluorescently with pyrene, rhodamine, or triphenylmethane dye(s) (Sharpie, Oak Brook, IL, ESI Table S2 & Fig. S3†). A suspension of 10 million microbeads per mL in deionized water was spiked with 20 μL mL−1 of dye (dilution factor 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50), vortexed, and further diluted 1:1 with 100% ethanol. The suspension was then centrifuged for 5 minutes at 400g, supernatant removed, and pellet resuspended in 15 mL of deionized water. Following this wash, the suspension was centrifuged for 5 minutes at 1000g with the brake disabled to reduce resuspension of the microbead pellet during rapid deceleration. All subsequent centrifugations followed this procedure. The pellet was resuspended in PBS at the desired final concentration for the intended application. The dye is hypothesized to bind to the PAAm via hydrogen bonding between the carboxylic groups of the dye and the amide of the acrylamide sub-units. Although dye can likely be incorporated directly into the PAAm solution, the option to stain after fabrication allows a single microbead batch to be used easily with multiple dyes. The addition of fluorescence is also useful for determining bead sizes (see ESI†), tracking their movement in culture systems, or for distinguishing different microbead formulations.
:50), vortexed, and further diluted 1:1 with 100% ethanol. The suspension was then centrifuged for 5 minutes at 400g, supernatant removed, and pellet resuspended in 15 mL of deionized water. Following this wash, the suspension was centrifuged for 5 minutes at 1000g with the brake disabled to reduce resuspension of the microbead pellet during rapid deceleration. All subsequent centrifugations followed this procedure. The pellet was resuspended in PBS at the desired final concentration for the intended application. The dye is hypothesized to bind to the PAAm via hydrogen bonding between the carboxylic groups of the dye and the amide of the acrylamide sub-units. Although dye can likely be incorporated directly into the PAAm solution, the option to stain after fabrication allows a single microbead batch to be used easily with multiple dyes. The addition of fluorescence is also useful for determining bead sizes (see ESI†), tracking their movement in culture systems, or for distinguishing different microbead formulations.
To make the microbead surfaces recognizable for cell adhesion, Sulfo-SANPAH (CovaChem, LLC, Loves Park, IL) was used to conjugate rat tail collagen type-I (Millipore) to the PAAm bead surface. Briefly, this reaction proceeds by covalently linking the UV-sensitive nitrophenylazide group of the sulfo-SANPAH to the PAAm surface after exposure to a UV light source. The collagen then binds to the free N-hydroxysuccinimide ester to create a recognizable surface for cells to interact with.21 After the microbeads were washed and stained, they were centrifuged and resuspended in 500 μL of 1 mg mL−1 Sulfo-SANPAH solution. The tube was uncapped and exposed to ultraviolet light in a Rayonet UV reaction chamber (The Southern New England Ultraviolet Co., Branford, CT) for 15 minutes. Samples were then flooded with 14.5 mL of deionized water, centrifuged, and subjected to a second Sulfo-SANPAH treatment.
After re-pelleting the microbeads, they were resuspended in 5 mL of deionized water and transferred to a polyethylene terephthalate (PET) tube (Corning Inc., Corning, NY), which exhibited reduced microbead adhesion compared to other plastic alternatives. Collagen type-I was added to the suspension to yield a final concentration of 100 μg mL−1, greater than 1000-fold molar excess to accessible amide groups of the microbeads. The suspension was vortexed and placed on a shaker overnight at 4 °C. The next day, 50 μL of 1 M HCl was added to create a slightly acidic environment (pH ∼ 6.9), intended to prevent collagen gelation that can aggregate and entrap microbeads. After 5 minutes, 10 mL of deionized water was added to the tube followed by centrifugation and a second wash in 15 mL deionized water spiked with 50 μL of 1 M HCl. The microbead pellet was then resuspended in PBS to yield the desired final concentration. Significant loss of microbeads can occur during treatment and wash steps (30–80%, ESI Table S3†), due primarily to aggregation and adhesion to plastic after being coated with collagen. To investigate how cells interacted with the compliant PAAm microbeads, MG-63, osteosarcoma cells (ATCC, Manassas, VA) were seeded into 2% agarose 3D Petri Dishes® (#24-96-Small, Microtissues, Inc., Providence, RI) either alone, with only uncoated or collagen-coated microbeads, or with both uncoated and collagen-coated microbeads (Eelastic ∼ 1 kPa) at a 4:1 ratio (cells:microbeads, 100000 particles per well). Results showed that cells incorporated collagen-coated microbeads into self-assembled spheroids, confirming cell recognition (Fig. 4). Cells interacted differently with uncoated microbeads, which lacked a complementary ligand, either excluding them from the spheroid or randomly entrapping them in a dispersed manner. The demonstrated ability to functionalize the microbeads for cellular recognition makes them a promising component for 3D scaffolding technologies. While PAAm, as a material, is non-ideal for tissue engineering applications because it is non-biodegradable, there are biocompatible polymer alternatives that may be compatible with the presented methodology. For investigations purely into the effects of a passive mechanical signal on cell behavior, a stable polymer such as PAAm is ideal since a biodegradable polymer would likely undergo drastic changes in mechanical properties.
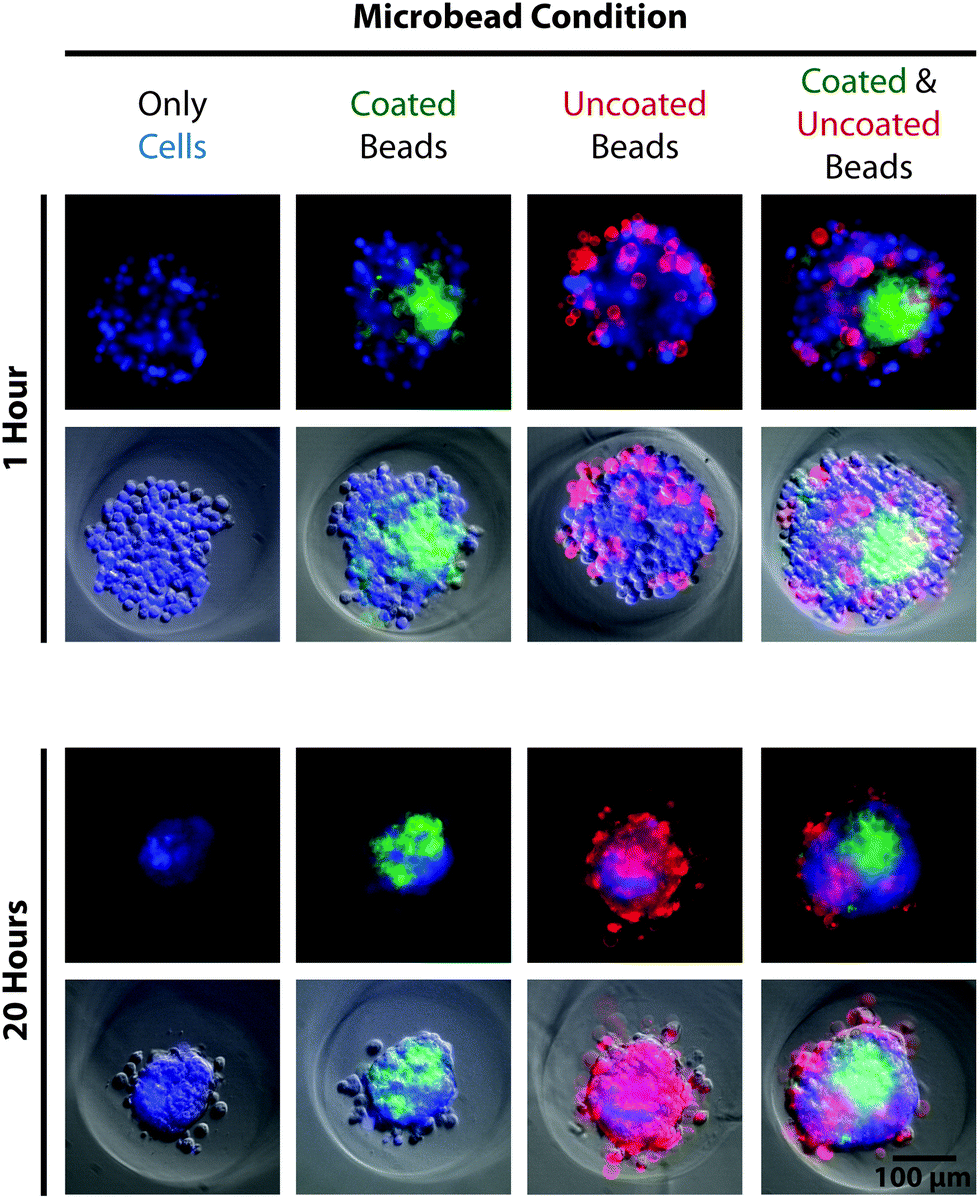 | ||
| Fig. 4 Cells recognize and attach to protein-coated microbeads. Fluorescence and phase contrast images illustrate the self-assembly of MG-63, osteoscarcoma cells stained with calcein AM blue (blue, 10 μg mL−1, AnaSpec Inc. Fremont, CA) in the presence of collagen-coated (green, pyrene-based dye, Sharpie) and uncoated (red, rhodamine-based dye, Sharpie) microbeads 1 hour (top) and 20 hours (bottom) after seeding. Spheroids containing cells alone (left column) assembled into compact structures over 20 hours. Spheroids containing cells and only coated microbeads (center-left column), only uncoated microbeads (center-right column), or both types of microbeads (right column) formed similar, compact spheroids but with different incorporation behavior of the microbeads. | ||
Conclusions
In this work, we have described a method for generating PAAm microbeads that mimic the size and elastic modulus distributions of typical cell populations using a vacuum-maintained inverse emulsification process. We successfully controlled microbead elasticity by altering cross-linker concentration and diameter by varying stir rate in conjunction with filtering. The fully polymerized microbeads were also compatible with fluorescent dyes that allow for easy particle visualization. Additionally, microbeads can be functionalized with a protein coating to promote cell recognition and binding. The relatively tight distributions of elastic moduli and diameters of microbeads within individual bead populations makes them ideal calibration particles for microfluidic devices designed to examine, quantify, or exploit the elastic moduli of cells. Combining mechanical tunability with the ability of cells to recognize and bind to the microbeads after protein coating will also make it possible to investigate the mechanosensitive responses seen in 2D culture in 3D microtissue culture/scaffold systems.Acknowledgements
The authors would like to thank Dr. Jeffrey Morgan for providing 3D Petri Dish molds for spheroid production and Dr. Ralph Milliken for performing FTIR scans. This work was supported by awards from the National Science Foundation (EMD, CAREER Award, CBET 1253189; EMD, EAGER Award, CBET 1547819), National Institute of Arthritis and Musculoskeletal and Skin Diseases (EMD, R01 AR063642), and National Institute of General Medical Sciences (EMD, P20 GM104937). The content of this article is solely the responsibility of the authors and does not necessarily represent the official views of the National Science Foundation or National Institutes of Health.References
- S. Xu, Z. Nie, M. Seo, P. Lewis, E. Kumacheva, H. A. Stone, P. Garstecki, D. B. Weibel, I. Gitlin and G. M. Whitesides, Angew. Chem., Int. Ed., 2005, 117, 734–738 CrossRef; G. F. Christopher and S. L. Anna, J. Phys. D: Appl. Phys., 2007, 40, 319–336 CrossRef.
- Y. Lvov, K. Ariga, M. Onda, I. Ichinose and T. Kunitake, Langmuir, 1997, 13, 6195–6203 CrossRef CAS.
- J. P. Rolland, B. W. Maynor, L. E. Euliss, A. E. Exner, G. M. Denison and J. M. DeSimone, J. Am. Chem. Soc., 2005, 127, 10096–10100 CrossRef CAS PubMed.
- N. Doshi, A. S. Zahr, S. Bhaskar, J. Lahann and S. Mitragotri, Proc. Natl. Acad. Sci. U. S. A., 2009, 106(51), 21495–21499 CrossRef CAS PubMed; K. H. Roh, D. C. Martin and J. Lahann, Nat. Mater., 2005, 4, 759–763 CrossRef PubMed.
- K. P. Lok and C. K. Ober, Can. J. Chem., 1985, 63, 209–216 CrossRef CAS; C. M. Tseng, Y. Y. Lu, M. S. El-Aasser and J. W. Vanderhoff, J. Polymer Sci. Pt., A, 1986, 24, 2995–3007 CrossRef.
- Y. S. Leong and F. Candau, J. Phys. Chem., 1982, 86(13), 2269–2271 CrossRef CAS; K. McAllister, P. Sazani, M. Adam, M. J. Cho, M. Rubinstein, R. J. Samulski and J. M. DeSimone, J. Am. Chem. Soc., 2002, 124, 15198–15207 CrossRef PubMed.
- J. Lo Surdo and S. R. Bauer, Tissue Eng., Part C, 2012, 18(11), 877–889 CrossRef CAS PubMed; A. H. Hielscher, J. R. Mourant and I. J. Bigio, Appl. Opt., 1997, 36(1), 125–135 CrossRef PubMed.
- E. M. Darling, M. Topel, S. Zauscher, T. P. Vail and F. Guilak, J. Biomech., 2008, 41, 454–464 CrossRef CAS PubMed; R. D. Gonzalez-Cruz, V. C. Fonseca and E. M. Darling, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, E1523–E1529 CrossRef PubMed; M. Kanthilal and E. M. Darling, Cell. Mol. Bioeng., 2014, 7(4), 585–597 CrossRef PubMed; E. M. Darling and D. Di Carlo, Annu. Rev. Biomed. Eng., 2015, 17, 35–62 CrossRef PubMed.
- A. C. Anselmo, M. Zhang, S. Kumar, D. R. Vogus, S. Menegatti, M. E. Helgeson and S. Mitragotri, ACS Nano, 2015, 9(3), 3169–3177 CrossRef CAS PubMed; M. Singh, B. Sandhu, A. Scurto, C. Berkland and M. S. Detamore, Acta Biomater., 2010, 6(1), 137–143 CrossRef PubMed; T. J. Merkel, S. W. Jones, K. P. Herlihy, F. R. Kersey, A. R. Shields, M. Napier, J. C. Luft, H. Wu, W. C. Zamboni, A. Z. Wang, J. E. Bear and J. M. DeSimone, Proc. Natl. Acad. Sci. U. S. A., 2011, 108(2), 586–591 CrossRef PubMed; L. J. Puig, J. C. Sánchez-Díaz, M. Villacampa, E. Mendizábal, J. E. Puig, A. Aguiar and I. Katime, J. Colloid Interface Sci., 2001, 235, 278–282 CrossRef PubMed; M. Platen, E. Mathieu, S. Lück, R. Schubel, R. Jordan and S. Pautot, Biomacromoledules, 2015, 16, 1516–1524 CrossRef PubMed; E. Marsich, M. Borgona, I. Donati, P. Mozetic, B. L. Strand, S. Gomez Salvador, F. Vittur and S. Paoletti, J. Biomed. Mater. Res., Part A, 2007, 84(2), 364–376 Search PubMed; B. P. Chan, C. H. Li, K. L. Au-Yeung, K. Y. Sze and A. H. W. Ngan, Ann. Biomed. Eng., 2008, 36(7), 1254–1267 CrossRef PubMed.
- E. M. Darling and D. Di Carlo, Annu. Rev. Biomed. Eng., 2015, 17, 35–62 CrossRef CAS PubMed.
- S. C. Hur, N. K. Henderson-MacLennan, E. R. B. McCabe and D. DiCarlo, Lab Chip, 2011, 11, 912 RSC.
- A. J. Engler, S. Sen, H. L. Sweeney and D. E. Discher, Cell, 2006, 126, 677–689 CrossRef CAS PubMed.
- D. R. Gossett, W. M. Weaver, A. J. Mach, S. C. Hur, H. T. K. Tse, W. Lee, H. Amini and D. DiCarlo, Anal. Bioanal. Chem., 2010, 397, 3249–3267 CrossRef CAS PubMed; D. R. Gossett, H. T. K. Tse, S. A. Lee, Y. Ying, A. G. Lindgren, O. O. Yang, J. Rao, A. T. Clark and D. DiCarlo, Proc. Natl. Acad. Sci. U. S. A., 2012, 109(20), 7630–7635 CrossRef PubMed.
- A. C. Anselmo and S. Mitragotri, Adv. Drug Delivery Rev., 2016 DOI:10.1016/j.addr.2016.01.007.
- P. Menter, “Acrylamide Polymerization—A Practical Approach”, Tech Note, Bio-Rad Laboratories, 2000, p. 1156 CrossRef; I. Capek, Des. Monomers Polym., 2012, 6(4), 399–409 CrossRef.
- A. Chrambach and D. Rodbard, Science, 1971, 171(3982), 440–451 Search PubMed.
- R. Battino and H. L. Clever, Chem. Rev., 1966, 66(4), 395–463 CrossRef CAS.
- E. M. Darling, S. Zauscher and F. Guilak, Osteoarthr. Cartilage, 2006, 14(6), 571–579 CrossRef CAS PubMed; N. R. Labriola and E. M. Darling, J. Biomech., 2015, 48, 1058–1066 CrossRef PubMed.
- E. K. Dimitriadis, F. Horkay, J. Maresca, B. Kachar and R. S. Chadwick, Biophys. J., 2002, 82, 2798–2810 CrossRef CAS PubMed.
- B. Kronberg, K. Holmberg and B. Lindman, Surface Chemistry of Surfactants and Polymers, John Wiley & Sons, Hoboken NJ, 2014 Search PubMed.
- J. R. Tse and A. J. Engler, Curr. Protoc. Cell Biol., 2010, 10(16), 1–16 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: FT-IR analysis, batch-to-batch variation, size characterization, and additional experimental details. See DOI: 10.1039/c6bm00692b |
| This journal is © The Royal Society of Chemistry 2017 |