
Effects of ovalbumin protein nanoparticle vaccine size and coating on dendritic cell processing†
Timothy Z.
Chang
,
Samantha S.
Stadmiller
,
Erika
Staskevicius
and
Julie A.
Champion
*
School of Chemical and Biomolecular Engineering, Georgia Institute of Technology, 950 Atlantic Drive NW, Atlanta, GA 30332, USA. E-mail: julie.champion@chbe.gatech.edu; Tel: +1 (404) 894 2874
First published on 5th December 2016
Abstract
Nanoparticle vaccine delivery platforms are a promising technology for enhancing vaccine immunogenicity. Protein nanoparticles (PNPs), made entirely from antigen, have been shown to induce protective immune responses against influenza. However, the fundamental mechanisms by which PNPs enhance component protein immunogenicity are not understood. Here, we investigate the role of size and coating of model ovalbumin (OVA) PNPs on particle uptake and trafficking, as well as on inflammation and maturation factor expression in dendritic cells (DCs) in vitro. OVA PNPs enhance antigen uptake in a size-independent manner, and experience attenuated endosomal acidification as compared to soluble OVA. OVA PNPs also trigger Fc receptor upregulation. Expression of cytokines IL-1β and TNF-α were PNP size- and coating-dependent, with small (∼270 nm) nanoparticles triggering greater inflammatory cytokine production than large (∼560 nm) particles. IL-1β expression by DCs in response to PNP stimulation implies activation of the inflammasome, a pathway known to be activated by certain types of nanoparticulate adjuvants. The attenuated acidification and pro-inflammatory profile generated by PNPs in DCs demonstrate that physical biomaterial properties can modulate dendritic cell-mediated antigen processing and adjuvancy. In addition to nanoparticles’ enhancement of DC antigen uptake, our work suggests that vaccine nanoparticle size and coating are uptake-independent modulators of immunogenicity.
1. Introduction
As vaccinology looks to move beyond the empirical “isolate-inactivate-inject” paradigm of traditional vaccination,1 nanoparticles have emerged as an attractive platform technology.2 Vaccine nanoparticles are designed to present specific antigens and epitopes in the context of enhanced immunogenicity conferred by particulate matter. Since the discovery of alum-mediated adjuvancy in influenza vaccines,3 numerous efforts have been made to understand and harness particulate-based vaccine adjuvancy.4–6 However, the immunomodulatory effects of nanoparticles have been shown to be a function of many factors, including nanoparticle size,7–9 shape,10,11 charge,12 and administration route.13 Given the diversity of vaccine nanoparticle types14 and immunological responses observed, universal design principles have been difficult to elucidate.Vaccine nanoparticle design generally follows two strategies: internal encapsulation of antigen and/or native antigen display on a particle surface. Biodegradable polymers such as chitosan and poly lactic-co-glycolic acid (PLGA) have been studied extensively for nanoparticle vaccine encapsulation.15 Incorporation of antigens in a nanoparticulate polymer matrix has shown that controlled release of antigen to the immune system gives an advantage over vaccination with soluble antigen.16–18 However, recently surface-based display of antigen on nanoparticles, such as on virus-like particles16 or protein particles19 has gained increasing attention. The shift towards surface antigen display has been driven by two insights: (1) an immunological understanding that surface receptor engagement on antigen presenting cells (APC) is essential for optimal interfacing with the innate and adaptive immune systems,14 and (2) the discovery that APC engagement with nanoparticles themselves triggers inflammatory responses.4,20,21
While multiple cell types interact with vaccine nanoparticles, dendritic cells (DCs) have been identified as the most potent antigen presenting cells.22 These cells are responsible for processing and presenting antigens to cells of the adaptive immune system, transforming an innate immune response into an adaptive one essential for protective immunity.23 The current model of antigen presentation requires three types of signaling to be initiated by dendritic cells: (1) presentation of antigen-derived peptide on the MHC complex, (2) costimulatory surface presentation of CD80 or CD86, and (3) soluble cytokine secretion. While the first two signals are essential for DC maturation and successful antigen presentation, it is hypothesized that the types of cytokines secreted vary in response to the type of antigen being presented.24
Different subpopulations of DCs, such as tissue-resident or lymph-node (LN)-resident DCs, can elicit different types of immune responses.25 Consequently, the effect of vaccine nanoparticle parameters, such as size, on immune responses is complex and model-system-dependent. On a system-wide level, vaccine targeting to the draining lymph nodes is a strategy for enhancing antigen delivery to dendritic cells,26 and 100–200 nm has been found to be the upper limit on nanoparticle size for passive drainage to the lymphatic system.26,27 Particles in the 500–2000 nm range are actively trafficked to the lymph nodes by antigen presenting cells. At the cellular level, size effects also tend to vary among different nanoparticle materials.7,8,28,29 However even within the extensively-studied category of polystyrene nanoparticles, nanoparticle sizes ranging from 40 nm (ref. 28) to 3 μm (ref. 30) have been suggested as optimal.
Protein nanoparticles (PNPs) are formed entirely from protein by solvent-directed assembly. Desolvation introduces an unfavorable solvent to a protein solution to increase protein–protein interactions, causing proteins in solution to coalesce into nanoparticles. These PNPs are in contrast to those formed via a self-assembly sequence in the protein, such as cages or vaults.31 Without an engineered self-assembly tag on the antigen to effect nanoparticle formation, the chances of an off-target immune response32 to the tag are also decreased. Desolvated PNPs were originally made from albumin.33 Recently, PNPs have been shown capable of intracellular delivery of folded, active enzymes.34 The delivery of properly-folded antigen protein is especially desirable in nanoparticle vaccine design.
In prior work, PNPs made with the conserved influenza peptide M2e were found to induce protective immune responses in mice against two subtypes of influenza.35 Mice immunized with M2e PNPs showed anti-M2e IgG and IgA responses in lung and nasal washes as well as M2e-inducible splenic IL-4 and IFN-γ responses, indicating both strong humoral and T-cell mediated immune responses. In contrast, immunization with soluble M2e triggered no protective immunity or immune correlates. We have also found nanoparticles made from trimerized, H7 hemagglutinin confer protection against an influenza challenge, and that coating the hemagglutinin nanoparticles with an additional layer of hemagglutinin protein increased the nanoparticles’ hemagglutinating capabilities.36 To understand how PNPs confer protective immunity and how particle properties, such as size and coating, affect the resulting immune response, an in vitro examination of nanoparticulate antigen processing, primarily dendritic cell interaction with PNPs, is needed.
In this work, we examine mechanisms of PNP and soluble ovalbumin (OVA) uptake and processing, and resulting markers of inflammation and maturation in dendritic cells. PNP size and coating were varied due to the immunomodulatory importance of particle size28 and properly-conformed surface antigen.37 PNP size and coating are the two fundamental parameters of protein nanoparticles made entirely from antigen, while other nanoparticle properties such as charge and hydrophobicity are intrinsically linked to the antigen protein of choice. We found both PNP size and coating to be important modifiers of DC inflammatory cytokine production. Other aspects of PNP antigen processing, such as uptake and acidification, were found to be significantly different from soluble antigen processing, but particle size- and coating-independent.
2. Methods
2.1 PNP synthesis
Nanoparticles were made by a modified desolvation method,35 and size was adjusted through the exact conditions as listed in Table S1.† To make the particles, 0.4 mL pure ethanol was added at a constant rate to 0.1 mL of 6.2 mg mL−1 OVA (Invivogen, San Diego, CA) in phosphate-buffered saline (PBS) under constant stirring at 600 rpm. The amine-reactive crosslinker 3,3′-dithiobis[sulfosuccinimidylpropionate] (DTSSP) (ThermoFisher Scientific, Waltham, MA) was used to stabilize the resulting nanoparticles. The nanoparticles were crosslinked while stirring at room temperature for one hour, followed by centrifugation to collect the particles. Nanoparticles were resuspended by sonication in either 1 mL of 6.2 mg mL−1 OVA in PBS or 1 mL of PBS for coating or non-coating, respectively. Coating was performed for 2 hours while stirring at 4 °C. Following collection by centrifugation, particles were resuspended in 0.5 mL of PBS, and additional DTSSP was added to a concentration of 6.18 mM to stabilize the outer coating. Coat crosslinking was performed for 2 hours while stirring at 4 °C. The coat crosslinking reaction was quenched with 50 mM Tris base, and particles were collected by centrifugation and resuspended in 0.5 mL PBS.2.2 PNP characterization
Nanoparticle size distribution and zeta potential were assessed by dynamic light scattering (DLS) and electrophoretic light scattering (ELS) respectively with a Malvern Zetasizer Nano ZS (Malvern Instruments, Westborough, MA). Protein concentration in the nanoparticle solution was assessed with a BCA assay according to the manufacturer's instructions (Thermo Scientific, Waltham, MA). Nanoparticles were resuspended in water, air-dried, and sputter-coated with carbon prior to visualization with a Zeiss Ultra60 FE (Carl Zeiss Microscopy, Cambridge, UK) scanning electron microscope at 5.0 kV.Fluorescent PNP coating was confirmed by flow cytometry. Coat antigenicity was assessed with ELISA using a horseradish peroxidase (HRP)-conjugated polyclonal anti-ovalbumin antibody (Thermo Scientific, Rockford, IL). 5.4 μg mL−1 OVA PNPs in PBS were coated on a 96-well plate overnight at room temperature. Each reagent incubation step was followed by three washes with 0.05% Tween-20 in PBS. Non-specific binding was blocked by a 1 hour incubation of the plate at 4 °C with 1% bovine serum albumin (BSA) in PBS. The HRP-conjugated anti-OVA antibody in PBS was incubated at 1 μg mL−1 on the plate for 2 hours at 4 °C. Chromogenic quantification was assessed by the oxidation of tetramethylbenzidine by hydrogen peroxide (R&D Systems, Minneapolis, MN) according to the manufacturer's instructions.
2.3 Cell culture
The JAWS II immature dendritic cell line was obtained from the American Type Culture Collection (Manassas, VA) and cultured in MEM-alpha (Corning, Manassas, VA) supplemented to 4 mM glutamine and 5 ng mL−1 GM-CSF (Peprotech, Rocky Hill, NJ), 20% fetal bovine serum (FBS), and 1% penicillin/streptomycin (Amresco, Solon, OH). Cells were grown in a humidified incubator at 37 °C in 5% CO2. For all experiments, cells were plated at a density of 105 cells per mL and incubated for 24 hours prior to stimulation unless indicated otherwise.2.4 Nanoparticle uptake
Nanoparticle uptake was assessed by flow cytometry. Fluorescent nanoparticles were made as described but with OVA containing 5 wt% AlexaFluor 488-conjugated OVA (Life Technologies, Grand Island, NY). JAWS II dendritic cells were plated in 24 well plates, and stimulated with 20 μg mL−1 fluorescent OVA PNPs or fluorescent soluble OVA. Cells were washed once with PBS, briefly trypsinized, collected by centrifugation, and resuspended in chilled trypan blue (Corning, Manassas, VA) to quench external green fluorescence. Cell fluorescence was measured at 6, 15 and 24 hours post-stimulation with a BD Accuri C6 flow cytometer (BD Biosciences, San Jose, CA). Nanoparticle internalization was confirmed by confocal microscopy.2.5 Endosomal pH
Endosomal pH was assessed by ratiometric fluorescence. FITC-(pH-sensitive fluorophore) and AlexaFluor 647-(pH insensitive fluorophore) conjugated OVA (Life Technologies, Grand Island, NY) were added to a final concentration of 15 wt% and 1 wt%, respectively, to non-fluorescent OVA to make green/red-fluorescent nanoparticles as described above. Standard curves were generated in pH-adjusted PBS to correlate fluorescent intensity ratios to pH (Fig. S1†). 100 μg mL−1 green/red PNPs were incubated with JAWS DCs in 96-well plates for 2 hours. The nanoparticle solution was removed, the cells were washed with PBS, and fresh media was added. Following an additional incubation for 0–6 hours, the media was replaced with PBS, and fluorescence measurements were taken on a plate reader (BioTek, Atlanta, GA) or a flow cytometer (BD Biosciences, San Jose, CA). Fluorescent intensities were calculated as averages of either a 5 × 5 fluorescent area scan of each well or the median fluorescence intensity, respectively.2.6 Nanoparticle uptake inhibition
Fluorescent nanoparticle uptake was assessed in the presence of 42 μM chlorpromazine or 49 μM cytochalasin D. Concentrations of inhibitors were chosen based on (1) maximizing positive control uptake inhibition of fluorescent transferrin or 3 μm polystyrene beads as assessed by flow cytometry or confocal microscopy, respectively, and (2) minimizing cytotoxicity as assessed by LDH. Percent inhibition of positive control and nanoparticle uptake was calculated by normalizing inhibited uptake by uninhibited uptake. Uptake inhibition studies were performed in 48-well plates. Cells were first pre-treated with uptake inhibitors for one hour. Afterwards, complete media containing fluorescent PNPs or soluble OVA at a concentration of 20 μg mL−1 and each appropriate uptake inhibitor was added. After 3 hours of incubation, the cells were washed once with PBS, fixed with paraformaldehyde, washed with PBS again, and resuspended in trypan blue to quench extracellular fluorescence. Cells were scraped from the plate and their fluorescence measured by flow cytometry. Degree of uptake inhibition was calculated by median fluorescence intensities of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells (eqn (1)).
000 cells (eqn (1)).
2.7 In vitro inflammatory and maturation markers
JAWS DCs were plated in either 24- or 48-well plates for measuring in vitro maturation and inflammation markers, respectively. Cells were stimulated with 20 μg mL−1 of soluble OVA or PNPs. CD86 was assessed by flow cytometry after 24 hours of stimulation. Cells were fixed with 3.7% paraformaldehyde and blocked with 1% BSA in PBS for 1 hour. Cells were then incubated with 2 μg mL−1 rat-anti-mouse CD86 (clone GL-1) or isotype control (clone aRTK2758) primary antibodies for 30 minutes, washed twice with 1% BSA in PBS, then stained with 1 μg per 106 cells polyclonal PE-conjugated goat-anti-rat Fc secondary antibody for 30 minutes. All antibodies were purchased from Abcam (Cambridge, MA). Cells were washed twice with PBS, scraped from the plate, and analyzed by flow cytometry. TNF-α was assessed in supernatants after 6 hours of stimulation by ELISA (R&D Systems, Minneapolis, MN) according to the manufacturer's instructions.2.8 Bone marrow derived dendritic cell (BMDC) generation and cell culture
Bone marrow from euthanized, ten-week old Balb/c mouse femurs and tibias was collected and cultured as previously described.38 All animal work was compliant with the NIH Guide for the Care and Use of Laboratory Animals, as well as with relevant laws and institutional guidelines as prescribed by Emory University's Institutional Animal Care and Use Committee. Bone marrow progenitor cells were cultured in RPMI 1640 (ATCC, Manassas, VA) supplemented with 10% heat-inactivated FBS, 50 μM β-mercaptoethanol, 1% penicillin–streptomycin, 2 mM sodium pyruvate, 1× non-essential amino acids (Thermo Scientific, Grand Island, NY), and 25 ng mL−1 each of IL-4 and GM-CSF (Peprotech, Rocky Hill, NJ). Cells were matured for one week, and the media was changed on days 2, 4 and 6. On day 7, cells were harvested by scraping and plated for experiments.2.9 IL-1β measurement
BMDCs were plated at a density of 5 × 105 cells per mL in 24-well plates. Cells were stimulated with 20 μg mL−1 of soluble OVA or PNPs. Supernatants were collected after 48 hours and stored at −80 °C. IL-1β was assessed by ELISA (R&D Systems, Minneapolis, MN).2.10 BSA coating of nanoparticles
100 μg of small coated and uncoated OVA PNPs were incubated stirring in 0.5 mL of 10 mg mL−1 bovine serum albumin (BSA) in PBS for 2 hours at 4 °C, collected by centrifugation, resuspended in PBS and crosslinked as described in Table S1.† Nanoparticles were then assessed for BSA presence with an HRP-conjugated anti-BSA antibody (Life Technologies, Grand Island, NY) using a standard ELISA protocol (R&D Systems, Minneapolis, MN).2.11 Statistics
Significance was assessed by a one-way ANOVA or Student's unpaired t-test at a significance level of p < 0.05. All data shown is representative of at least three independent measurements unless indicated otherwise.3. Results
3.1 OVA PNP synthesis and characterization
Ovalbumin PNPs were made by a modified desolvation procedure.34 Small- (∼270 nm diameter) and medium-sized (∼350 nm) PNPs were made by desolvation of ovalbumin at 1 mL min−1. Increasing the rate of ethanol addition created large (∼560 nm diameter) PNPs. Crosslinking nanoparticles in the desolvation media led to medium and large PNPs, while crosslinking in PBS created small PNPs (Fig. 1a, Table S2†). Incorporation of fluorophore-conjugated OVA into the PNPs had no effect on size (Fig. S2†). Zeta potential was unaffected by nanoparticle size (Fig. 1b).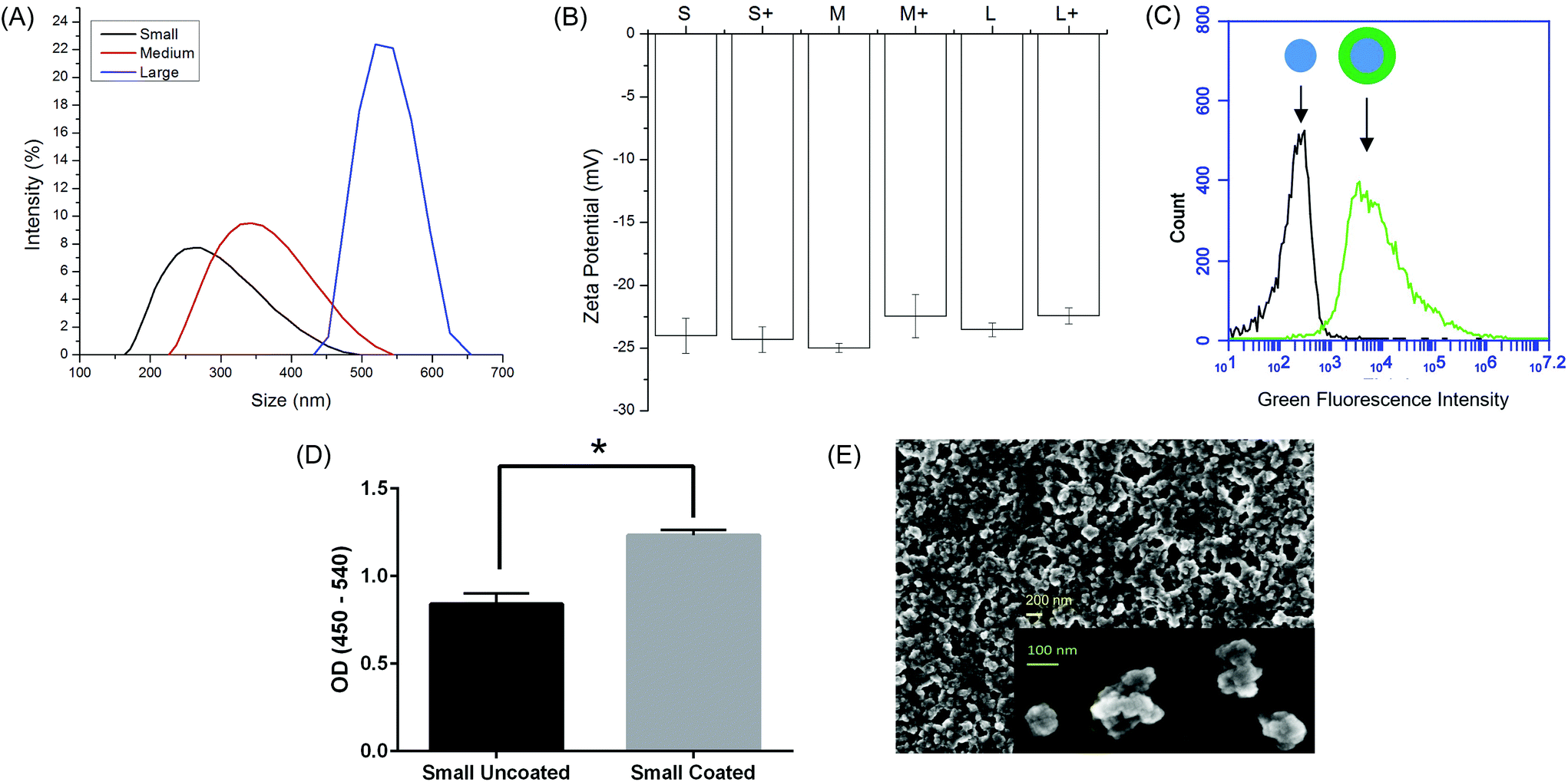 | ||
| Fig. 1 Physical characterization of OVA PNPs. (A) Representative size distributions of OVA PNPs of different sizes. (B) Zeta potential of all PNP types measured in PBS, average of three independent replicates. (C) Fluorescent OVA-coated PNPs (right) had greater fluorescence than uncoated PNPs (left) as measured by flow cytometry. (D) Anti-ovalbumin antibody binding was significantly enhanced (*, p < 0.05) upon PNP coating with OVA. (E) Scanning electron microscope images of air-dried PNPs. | ||
While addition of ethanol to soluble protein is necessary for the desolvation process, the resulting solvent environment can lead to denaturation of some proteins. Given the importance of antigen conformation on the particle surface to trigger pro-immunogenic surface receptor-mediated pathways,39 we added a coating step after particle synthesis. Nanoparticles were resuspended in a solution of soluble OVA in PBS to adsorb antigen to the nanoparticle surface in the absence of desolvent. Successful coating was confirmed by flow cytometry (Fig. 1c). ELISA with an anti-OVA antibody demonstrated increased antibody binding on the nanoparticle surface after coating (Fig. 1d). Coating the nanoparticles had no effect on particle size or zeta potential (Fig. 1b, Table S2†). Scanning electron micrographs of dried PNPs demonstrate a roughly spherical morphology and appear to have size distributions smaller than those obtained by DLS in the hydrated state (Fig. 1e).
3.2 PNP uptake in dendritic cells
Using fluorescent PNPs and soluble OVA, we tracked antigen uptake in the JAWS II dendritic cell line by flow cytometry. Given the same dose of antigen, cells took up approximately 5–8 times more antigen as PNPs than the soluble form (Fig. 2). Internalization of nanoparticles was confirmed by confocal microscopy (Fig. S3†). PNP enhancement of antigen uptake relative to soluble protein was greater than any uptake differences among PNPs of different sizes and coatings (Fig. 2).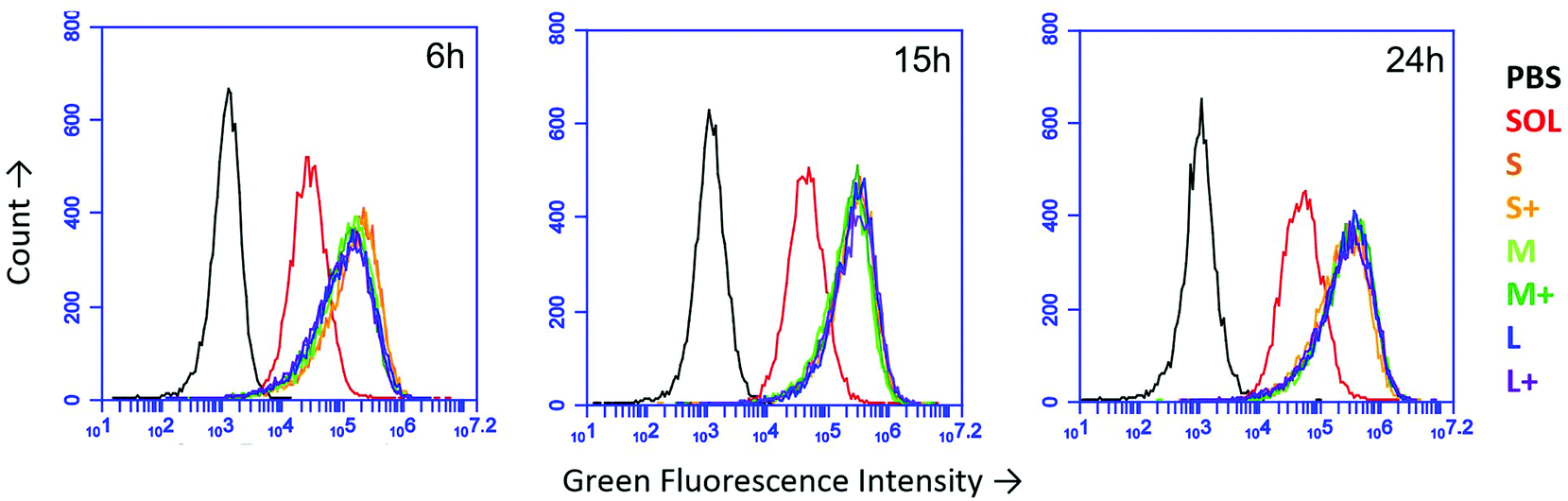 | ||
| Fig. 2 Uptake of OVA PNPs by JAWS II DCs as measured by flow cytometry after 6 hours, 15 hours, and 24 hours. PBS = PBS-treated cells, SOL = soluble OVA, S,M,L = small, medium, large, respectively, + = coated PNPs. Traces were consistent across two independent replicates. | ||
3.3 OVA nanoparticles experience attenuated acidification
Upon uptake, endosomal pH was assessed with ratiometric fluorescence.30 AlexaFluor 647- (a pH-insensitive red fluorophore) and FITC- (a pH-sensitive green fluorophore) conjugated OVA were incorporated into nanoparticles. A standard curve correlating pH to fluorescence ratio was generated by exposing the nanoparticles to pH-adjusted PBS in the presence and absence of cells (Fig. S1†). While soluble protein was exposed to acidic conditions, all PNPs experienced attenuated acidification in comparison (Fig. 3). No significant differences in pH were observed among PNPs of different sizes or coatings.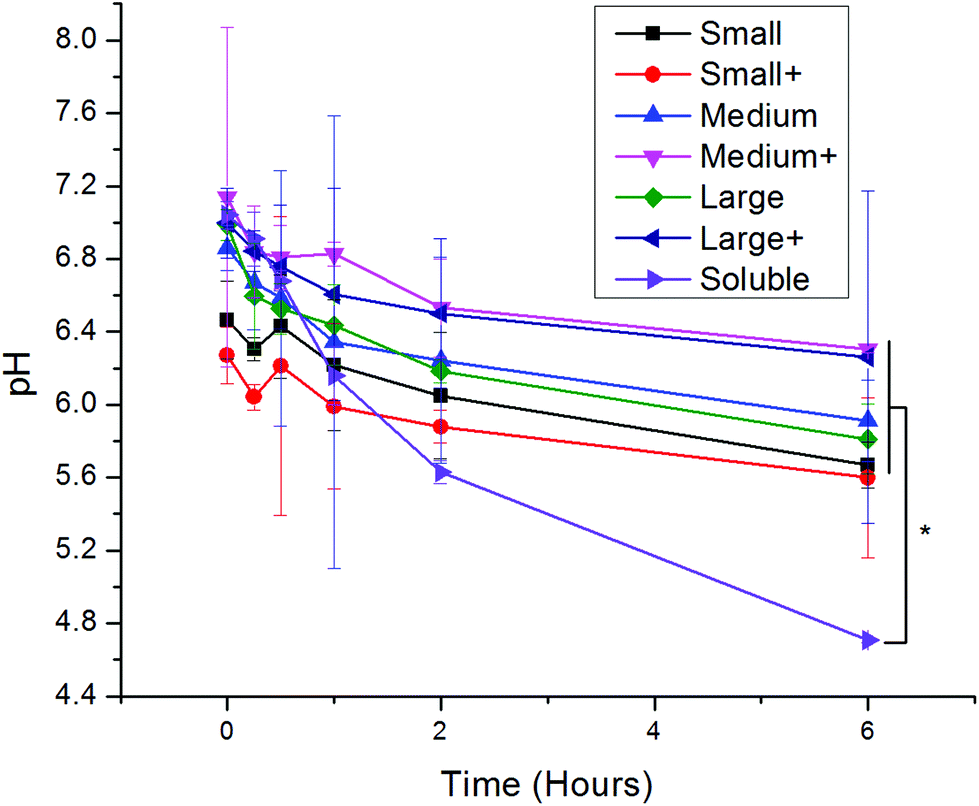 | ||
| Fig. 3 Log-mean pH of red/green PNPs upon incubation with DCs for various times following a two-hour pulse of PNPs. Differences in log-mean pH were significant (p < 0.05) between soluble OVA and all PNPs except small coated (red circles). Each point is shows an average and standard deviation of three independent replicates (n = 3). | ||
3.4 Differences in uptake pathways
Uptake inhibitors were used to assess PNP uptake routes in JAWS II DCs. Soluble ovalbumin is internalized via clathrin-mediated endocytosis,40 and nanoparticles on the order of 500 nm and larger are internalized primarily through phagocytosis,7 so inhibitors of these two pathways were used to examine uptake route. Chlorpromazine41 and cytochalasin D42 were used to inhibit clathrin-mediated uptake and phagocytosis, respectively. Concentration optimization was based on maximizing positive control uptake inhibition and minimizing cytotoxicity. Positive controls were chosen based on evidence for exclusive or predominant uptake by a particular pathway – transferrin for clathrin-mediated uptake, and 3 μm polystyrene beads for phagocytosis.43,44 Uptake inhibition fraction was calculated by the following formula: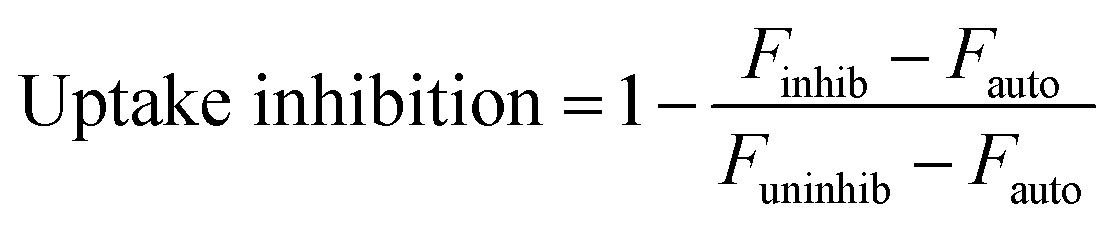 | (1) |
000 cells in the presence or absence of an uptake inhibitor, respectively, and Fauto is the median autofluorescence intensity of 10000 cells. An uptake inhibition of 1 denotes complete inhibition, while an uptake inhibition of 0 denotes no inhibition. Fig. 4 shows uptake inhibition for soluble OVA and PNPs. Both soluble OVA and OVA PNP uptake were strongly inhibited by chlorpromazine, and differences in inhibition were not significant (p > 0.05). Cytochalasin D strongly inhibited soluble OVA uptake, but inhibited OVA PNP uptake to a significantly lesser degree (p < 0.05). No significant differences in uptake inhibition (p > 0.05) were observed among PNP types.
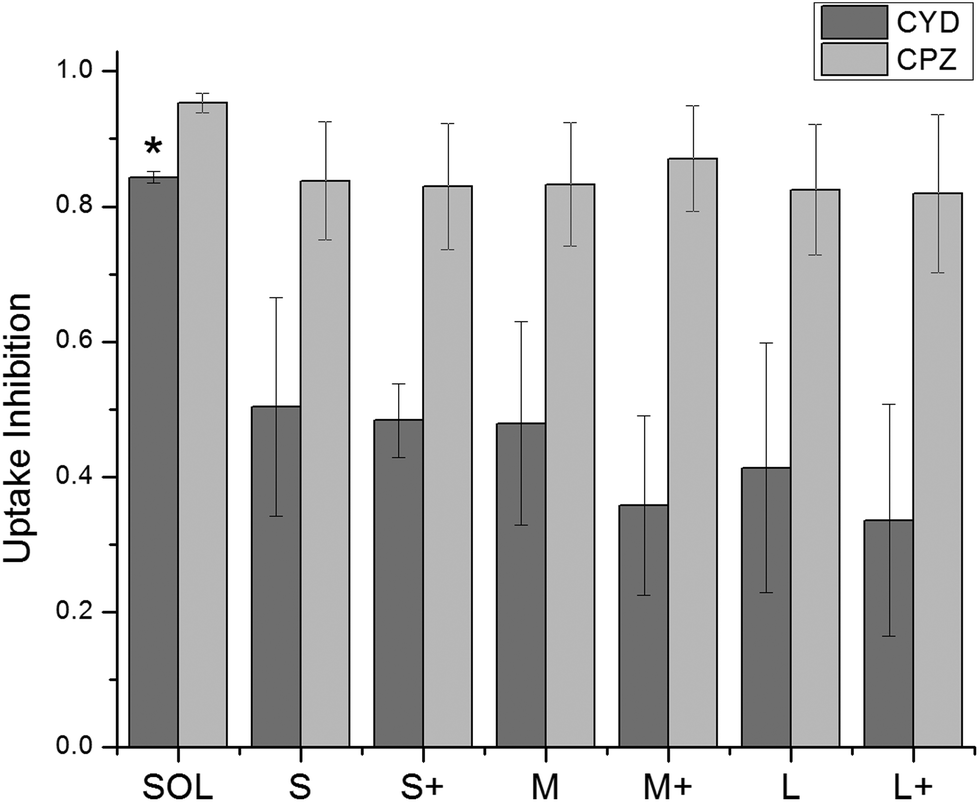 | ||
| Fig. 4 Uptake inhibition of fluorescent PNPs was determined by flow cytometry and normalized by eqn (1). Cytochalasin D = CYD, chlorpromazine = CPZ. Soluble OVA uptake was inhibited by CYD to a significantly greater degree than any PNPs were (*, p < 0.05). CPZ inhibited uptake of PNPs and soluble OVA to a similar degree. Bars show average and standard deviation of three independent replicates (n = 3). | ||
3.5 OVA nanoparticles trigger DC maturation and Fc receptor upregulation
DC upregulation of the maturation marker CD86 was assessed by flow cytometry after stimulation with PNPs (Fig. 5). Isotype controls were used to differentiate between upregulation of CD86 and Fc receptors upon stimulation. PNPs trigger upregulation of CD86, with small, uncoated PNPs causing the greatest upregulation (Fig. 5). However, OVA PNPs also induce upregulation of Fc receptors, which is not observed in response to soluble OVA or lipopolysaccharide positive control.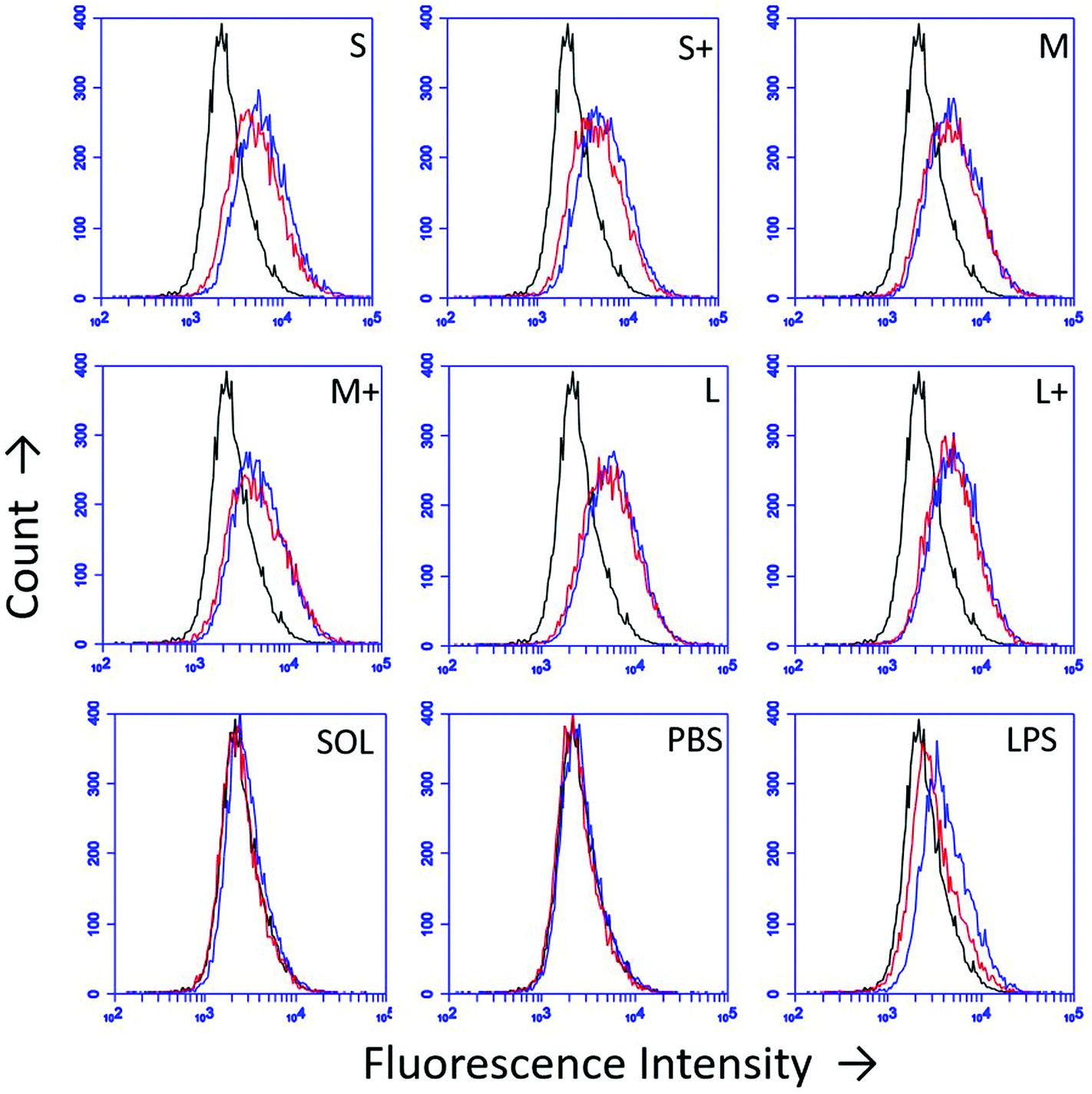 | ||
| Fig. 5 DC CD86 upregulation 24 hours post-PNP treatment was assessed by flow cytometry. Black = unstained control, Red = isotype control, Blue = CD86. Traces are representative of three independent replicates. | ||
3.6 Inflammatory cytokine production is PNP size- and coating-dependent
Production of the pro-inflammatory cytokines TNF-α and IL-1β were examined after PNP and soluble OVA stimulation. IL-1β production was observed in response to soluble OVA and PNPs, but only small and medium coated PNPs induced greater upregulation than soluble OVA (Fig. 6a). Large nanoparticles did not trigger an enhanced IL-1β response regardless of coating. Conversely, TNF-α production in response to PNPs was lower than that of soluble OVA (Fig. 6b). Coating PNPs with additional OVA led to increases in the DC TNF-α responses to medium and large, but not small, PNPs. Although medium coated PNPs triggered the highest levels of TNF-α, they did not reach the levels of TNF-α induced by soluble OVA.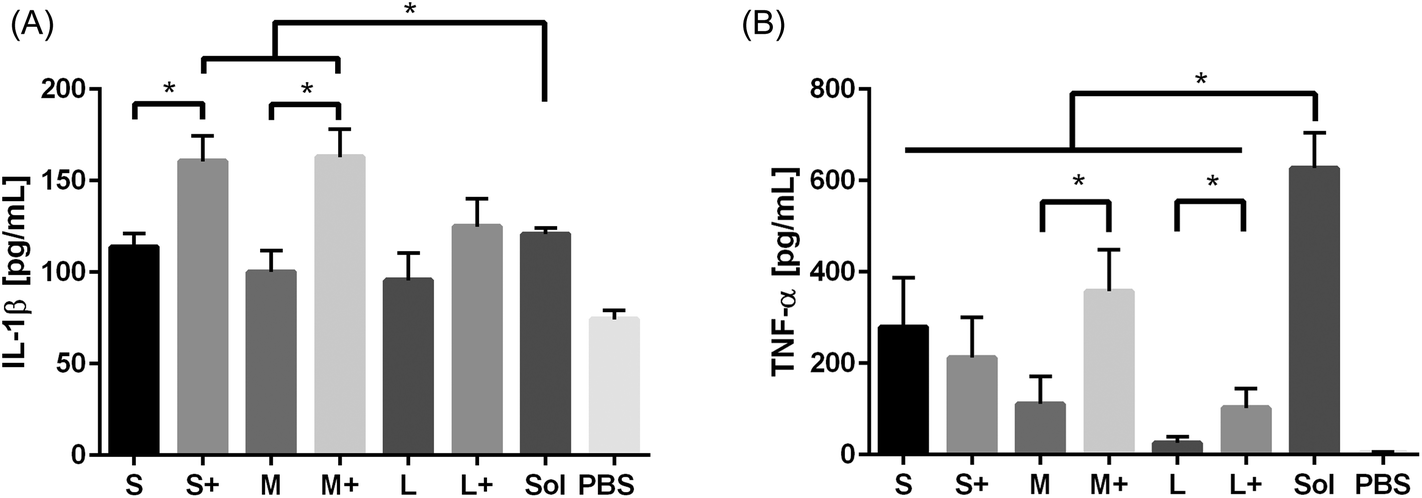 | ||
| Fig. 6 IL-1β (A) and TNF-α (B) production in response to PNPs were assessed by ELISA. Sol = soluble OVA treatment, PBS = negative control. TNF-α averages and standard deviations are of three batches of PNPs, each replicated three times (n = 9). | ||
4. Discussion
Current nanoparticle vaccine research seeks to leverage the encapsulating biomaterial properties to enhance immunogenic responses to component antigens. PNPs, nanomaterials made entirely from crosslinked antigen, demonstrate that altering the presentation of protein antigens from a soluble to a nanoparticulate form is sufficient for enhancing vaccine adjuvancy. Our prior work showed that PNPs made from influenza proteins enhanced antigen immunogenicity and immune protection.35 Our hypothesis that differences in dendritic cell processing of nanoparticulate and soluble antigen led to the different in vivo immune responses motivated the present in vitro study. In addition to examining the differences between soluble OVA and OVA PNPs, we examined the role of two fundamental PNP properties, size and coating, in enhancing DC responses.4.1 Initial PNP uptake and processing is size- and coating-independent
Initial DC uptake and processing was similar among OVA PNPs of different sizes and coatings. PNPs greatly enhanced antigen uptake over soluble antigen (Fig. 2), which has been seen for PNPs made from other proteins for other cell types.34 However, PNP size and coating did not significantly affect antigen internalization in the size range tested. While some studies found a negative correlation between particle size and uptake,30,45 the size range over which uptake differences were observed spanned 50 nm to 3 μm. Still other work saw no influence of size on uptake within this wide size range,46 emphasizing the importance of nanoparticle material on uptake trends. The near-complete inhibition of PNP and soluble OVA uptake by chlorpromazine suggests the importance of clathrin-mediated uptake. Since soluble OVA is known to undergo clathrin-mediated uptake through the mannose receptor,40 the mannose receptor could be involved in OVA PNP uptake as well. The comparable uptake of coated and uncoated PNPs is consistent with mannose receptor-mediated uptake, since glyco-antigens present in OVA would be accessible on either the coated or uncoated PNPs. Similar to our findings of coating-independent degree of uptake, a study by Thomann-Harwood et al. found coating alginate nanoparticles with mannan, a polysaccharide that shares a receptor with OVA, had no effect on particle uptake.37Chlorpromazine inhibited uptake of soluble OVA and PNPs by approximately 90%, indicating clathrin-mediated uptake of both soluble and PNP forms of OVA. Treatment of DCs with cytochalasin D reduced PNP uptake by roughly 50%, but reduced soluble OVA uptake by more than 80%. Given the strong inhibition of cytochalasin D on soluble OVA uptake, it was surprising to see PNPs still taken up despite cytochalasin D treatment. This phenomenon could be explained by the role of actin in clathrin-mediated endocytosis. Actin depolymerization with cytochalasin D treatment is a common strategy for inhibiting phagocytosis,42 but cytochalasin D cannot completely depolymerize F-actin associated with clathrin-coated structures.47 Clathrin-coated vesicle formation from the plasma membrane requires actin polymerization proportional to the maximum circumference of the object being internalized.47 By comparing antigen volume to circumference ratios for spherical soluble proteins and PNPs, we find that PNP uptake requires much less actin polymerization than soluble antigen uptake for the same amount of antigen (Fig. S4†). Given partial inhibition of clathrin-mediated uptake by cytochalasin D, the preceding observation could explain efficient inhibition of soluble OVA uptake but less efficient inhibition of OVA PNP uptake. As with other experiments using endocytosis inhibitors, we cannot discount the impact of off-target effects, including potential compensatory upregulation of other endocytic mechanisms in response to cytochalasin D or chlorpromazine treatment.
Upon antigen internalization, DCs acidified soluble OVA to a significantly greater extent than OVA PNPs. Although uptake inhibition data suggest a common uptake mechanism for soluble OVA and OVA PNPs, the attenuated acidification of PNPs as compared to soluble OVA suggests different internal routing for PNP and soluble antigen. Our data is also consistent with lysosomal colocalization studies that show antigen in nanoparticle cores can achieve cytosolic delivery, while soluble or nanoparticle-adsorbed antigen cannot.18 The pH 4.7 experienced by soluble OVA is characteristic of endosomal fusion with lysosomes, a process promoting proteolytic antigen degradation.48 Attenuated acidification is known to protect antigens from rapid degradation and lead to enhanced cross-presentation of MHC Class I-restricted antigen peptides.30,49 Cross presentation of exogenous antigen on MHC I by dendritic cells is necessary for CD8+ T cell responses, which are essential for providing immunity against intracellular bacterial and viral infections and cancerous host cells.50 Our data showing attenuated PNP acidification allows the possibility of endosomal escape of some internalized PNPs, a phenomenon that would be consistent with the elevated IL-1β levels observed in response to PNP treatment.4
4.2 Inflammatory cytokine secretion is size- and coating-dependent
IL-1β is known to originate from activation of the inflammasome, a heterotrimeric enzyme complex assembled in response to internalization of certain types of particulate antigens.12 IL-1β-production from inflammasome activation allows for the rapid cleavage of pro-inflammatory cytokines into their active form, triggering a local, innate immune response.23 The NLRP3 inflammasome is the only one that does not have a specific ligand associated with it,51 and is implicated in nanoparticle-mediated inflammasome activation.12 Not all nanoparticles can trigger inflammasome activation.12,51 Alum,20 silica,4 LPS-coated PLGA52 and textured polymeric11 particles have all been shown to trigger inflammasome activation, while uncoated PLGA particles52 and lipid cubosomes12 cannot. Recently, soluble ovalbumin was found to activate the inflammasome in a murine model,53 and our stimulation of BMDCs confirms this. Additionally, we see enhanced production of IL-1β in response to small and medium coated PNPs over large PNPs and soluble OVA. This suggests PNP activation of the inflammasome is enhanced by small size and repeated surface antigen display, consistent with a viral-mimetic design strategy.54 Our findings also demonstrate IL-1β production by BMDCs in the absence of pretreatment with LPS or other stimuli, a step commonly used for in vitro inflammasome activation, but known to bias the resulting inflammatory response.51TNF-α enhances local inflammatory responses through activation of the NF-κB pathway in multiple cell types.55 In addition, it directs DCs to begin maturation and migration to lymph nodes.23 Given the strong IL-1β responses observed, it was surprising to see DC TNF-α responses to PNPs lower than soluble OVA. The patterns for TNF-α and IL-1β production are distinct from one another, suggesting different mechanisms of activation despite an overlap in signaling pathways.56 Given soluble OVA's inherent inflammasome-stimulating capabilities, it is possible that epitopes responsible for inflammasome activation also trigger TNF-α production. This would be consistent with our results showing enhanced TNF-α production in response to PNP coating for medium and large particles, but does not explain why coating small OVA PNPs does not enhance TNF-α production. The low TNF-α response of PNPs in comparison to soluble OVA also supports the claim that TNF-α signaling is not characteristic of all types of nanoparticle-mediated adjuvancy.57 Although TNF-α is known to enhance monocyte and neutrophil extravasation at the site of inflammation,23 its role in promoting immunity following vaccination remains to be fully elucidated.
DC responses to coated PNPs may be component-antigen dependent. Given the lack of pathogen-associated molecular patterns (PAMPs) or DC-specific epitopes on ovalbumin, however, the enhanced inflammatory responses seen to coated OVA nanoparticles may be due to proteins other than OVA. Serum protein adsorption to nanoparticles in vitro and in vivo is ubiquitous and dependent on nanoparticle surface properties.58 We found OVA-coated OVA PNPs bind less BSA, a representative serum protein, than uncoated OVA PNPs do (Fig. S5†). The protein corona on nanoparticles has been shown to remain attached during cellular uptake,59 and the uptake of non-immunogenic serum proteins on the PNPs may explain the attenuated DC inflammation observed. In vivo, the protein corona may have immunostimulatory effects if complement or immunoglobulin is adsorbed, and engineering vaccine nanomaterials to preferentially adsorb these proteins is an active area of research.60
While coating PNPs generally enhances DC inflammatory responses, we see deviations from this trend at small sizes for TNF-α. In general, cytokine responses are enhanced in response to coated and smaller-sized PNPs, although the enhancement in response to coating is greater than that of decreasing size. Therefore, we believe particle size plays a lesser role in inflammatory cytokine production than particle coating.
4.3 DC maturation and the relevance to in vivo immunization
All PNP types led to slight CD86 upregulation, indicative of DC maturation, a process necessary for successful antigen presentation. DC maturation markers, including CD86 as well as CD80 and CD40, have been upregulated in response to other types of nanoparticles;46 and while small and small coated PNPs qualitatively appeared to upregulate CD86 the most compared to soluble (Fig. 5), large distributions of maturation marker expression make quantitative comparisons of upregulation difficult. Although protective humoral and cell-mediated immune responses have been observed in response to M2e and hemagglutinin PNPs,35,36 we were unable to observe significant MHC II upregulation in BMDCs after 24 hours of PNP stimulation (Fig. S6†). This discrepancy, coupled with the observed upregulation of MHC II in response to soluble OVA + LPS, suggests that different adjuvants trigger different rates of MHC II upregulation and antigen presentation. Increases in isotype control fluorescence indicate Fc receptor upregulation, a phenomenon observed in response to PNP stimulation of DCs, but not in response to soluble OVA or to the positive control LPS. To our knowledge, this is the first time FcR upregulation has been observed in response to nanoparticle stimulation. While FcR-targeted nanoparticle vaccines are being studied,61 further investigation into the upregulation phenomenon is required to determine if there is a synergistic effect between nanoparticle- and opsonization-mediated adjuvancy.Taken together, our data show that PNPs enhance antigen uptake in DCs, yet enhanced antigen delivery alone does not lead to an across-the-board increase in DC inflammation. The observation that PNPs experience attenuated endosomal acidification in comparison to soluble antigen supports the hypothesis of PNPs enhancing antigen cross-presentation, which was also supported by the successful immunization of mice with M2e peptide PNPs.35 Our incorporation of the full-length proteins OVA and hemagglutinin36 into PNPs suggests that multiple types of protein antigens can be incorporated into immunogenic PNPs. While readouts of PNP uptake, uptake mechanism, and intracellular pH are independent of PNP properties, markers of inflammation are dependent on PNP size and coating. Small and medium PNPs between 270–350 nm in diameter trigger greater inflammatory cytokine responses than 560 nm PNPs, highlighting the importance of small sizes in PNP design. Our data suggest a separation of initial antigen uptake and processing from downstream inflammatory outputs. On a fundamental level, this would imply that size- and coating-mediated nanoparticle adjuvancy does not occur at the point of internalization. Consequently, nanoparticle size can play a complementary role to membrane receptor-mediated adjuvants (i.e. TLR ligands) on the nanoparticle surface for optimal DC responses. In examining these responses, the observed decoupling of TNF-α and IL-1β production in response to soluble or nanoparticulate OVA underscores the importance of identifying in vitro inflammatory correlates to successful in vivo immunization.
5. Conclusion
We have examined differences in dendritic cell responses to PNPs of different sizes and coatings. Comparing soluble OVA and OVA PNPs, we saw differences in uptake amount and route, endosomal acidification, and biomarkers of inflammation and maturation. Among PNP types, we found PNPs of approximately 270 nm in size trigger more inflammatory responses than larger particles, and that the presence of a coating of antigen on nanoparticles triggers stronger inflammatory responses than uncoated nanoparticles. Our work also implicates activation of the inflammasome in response to PNPs, supporting other work showing non-crystalline adjuvant nanoparticles can activate the inflammasome.52 Our results suggest that adjuvant coatings on vaccine nanoparticles should be directed toward activating specific responses rather than only enhancing cellular uptake.Funding statement
This work was supported by the National Institutes of Health (Grant # 1R01AI101047-01), by the National Science Foundation's Graduate Research Fellowship Program (Grant # DGE-1148903 for T. Z. C.) and by the Georgia Institute of Technology's President's Undergraduate Research Award for S. S. S. The funders had no role in study design, data collection, analysis, decision to publish, or preparation of this manuscript. No conflict of interest was found for any of the authors.Acknowledgements
The authors would like to thank Sivabalan Manivasagam for technical assistance, and Drs Jardin Leleux and Jongrok Kim for assistance in collecting mouse BMDCs. This work was performed in part at the GT Institute for Electronics and Nanotechnology, a member of the National Nanotechnology Infrastructure Network, which is supported by the NSF.References
- G. A. Poland, R. B. Kennedy and I. G. Ovsyannikova, Vaccinomics and Personalized Vaccinology: Is Science Leading Us Toward a New Path of Directed Vaccine Development and Discovery?, PLoS Pathog., 2011, 7(12), e1002344 CAS.
- T. Mamo and G. A. Poland, Nanovaccinology: The next generation of vaccines meets 21st century materials science and engineering, Vaccine, 2012, 30(47), 6609–6611 CrossRef CAS PubMed.
- M. Kool, K. Fierens and B. N. Lambrecht, Alum adjuvant: some of the tricks of the oldest adjuvant, J. Med. Microbiol., 2012, 61(7), 927–934 CrossRef CAS PubMed.
- V. Hornung, F. Bauernfeind, A. Halle, E. O. Samstad, H. Kono and K. L. Rock, et al., Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization, Nat. Immunol., 2008, 9(8), 847–856 CrossRef CAS PubMed.
- D. R. Getts, L. D. Shea, S. D. Miller and N. J. C. King, Harnessing nanopartricles for immune modulation, Trends Immunol., 2015, 36(7), 419–427 CrossRef CAS PubMed.
- J. Leleux and K. Roy, Micro and Nanoparticle-Based Delivery Systems for Vaccine Immunotherapy: An Immunological and Materials Perspective, Adv. Healthcare Mater., 2013, 2(1), 72–94 CrossRef CAS PubMed.
- S. D. Xiang, A. Scholzen, G. Minigo, C. David, V. Apostolopoulos and P. L. Mottram, et al., Pathogen recognition and development of particulate vaccines: Does size matter?, Methods, 2006, 40(1), 1–9 CrossRef CAS PubMed.
- A. Stano, C. Nembrini, M. A. Swartz, J. A. Hubbell and E. Simeoni, Nanoparticle size influences the magnitude and quality of mucosal immune responses after intranasal immunization, Vaccine, 2012, 30(52), 7541–7546 CrossRef CAS PubMed.
- P. L. Mottram, D. Leong, B. Crimeen-Irwin, S. Gloster, S. D. Xiang and J. Meanger, et al., Type 1 and 2 immunity following vaccination is influenced by nanoparticle size: Formulation of a model vaccine for respiratory syncytial virus, Mol. Pharmaceutics, 2007, 4(1), 73–84 CrossRef CAS PubMed.
- S. Kumar, A. C. Anselmo, A. Banerjee, M. Zakrewsky and S. Mitragotri, Shape and size-dependent immune response to antigen-carrying nanoparticles, J. Controlled Release, 2015, 220(Pt A), 141–148 CrossRef CAS PubMed.
- C. A. Vaine, M. K. Patel, J. T. Zhu, E. Lee, R. W. Finberg and R. C. Hayward, et al., Tuning Innate Immune Activation by Surface Texturing of Polymer Microparticles: The Role of Shape in Inflammasome Activation, J. Immunol., 2013, 190(7), 3525–3532 CrossRef CAS PubMed.
- S. Neumann, K. Burkert, R. Kemp, T. Rades, P. R. Dunbar and S. Hook, Activation of the NLRP3 inflammasome is not a feature of all particulate vaccine adjuvants, Immunol. Cell Biol., 2014, 92(6), 535–542 CrossRef CAS PubMed.
- B. S. Zolnik, A. Gonzalez-Fernandez, N. Sadrieh and M. A. Dobrovolskaia, Minireview: Nanoparticles and the Immune System, Endocrinology, 2010, 151(2), 458–465 CrossRef CAS PubMed.
- L. Zhao, A. Seth, N. Wibowo, C.-X. Zhao, N. Mitter and C. Yu, et al., Nanoparticle vaccines, Vaccine, 2014, 32(3), 327–337 CrossRef PubMed.
- B. Slutter, L. Plapied, V. Fievez, M. A. Sande, A. des Rieux and Y. J. Schneider, et al., Mechanistic study of the adjuvant effect of biodegradable nanoparticles in mucosal vaccination, J. Controlled Release, 2009, 138(2), 113–121 CrossRef PubMed.
- A. E. Gregory, R. Titball and D. Williamson, Vaccine delivery using nanoparticles, Front. Cell. Infect. Microbiol., 2013, 3 Search PubMed.
- S. Manmohan, Vaccine adjuvants and delivery systems, Wiley-Interscience, Hoboken, N.J., 2007 Search PubMed.
- L. Liu, P. Ma, H. Wang, C. Zhang, H. Sun and C. Wang, et al., Immune responses to vaccines delivered by encapsulation into and/or adsorption onto cationic lipid-PLGA hybrid nanoparticles, J. Controlled Release, 2016, 225, 230–239 CrossRef CAS PubMed.
- M. Kanekiyo, C. J. Wei, H. M. Yassine, P. M. McTamney, J. C. Boyington and J. R. R. Whittle, et al., Self-assembling influenza nanoparticle vaccines elicit broadly neutralizing H1N1 antibodies, Nature, 2013, 499(7456), 102–108 CrossRef CAS PubMed.
- H. F. Li, S. B. Willingham, J. P. Y. Ting and F. Re, Cutting edge: Inflammasome activation by alum and alum's adjuvant effect are mediated by NLRP3, J. Immunol., 2008, 181(1), 17–21 CrossRef CAS.
- F. A. Sharp, D. Ruane, B. Claass, E. Creagh, J. Harris and P. Malyala, et al., Uptake of particulate vaccine adjuvants by dendritic cells activates the NALP3 inflammasome, Proc. Natl. Acad. Sci. U. S. A., 2009, 106(3), 870–875 CrossRef CAS PubMed.
- C. Thery and S. Amigorena, The cell biology of antigen presentation in dendritic cells, Curr. Opin. Immunol., 2001, 13(1), 45–51 CrossRef CAS PubMed.
- K. Murphy, P. Travers, M. Walport and C. Janeway, Janeway's immunobiology, Garland Science, New York, 2012 Search PubMed.
- J. Banchereau and R. M. Steinman, Dendritic cells and the control of immunity, Nature, 1998, 392(6673), 245–252 CrossRef CAS PubMed.
- J. Leleux, A. Atalis and K. Roy, Engineering immunity: Modulating dendritic cell subsets and lymph node response to direct immune-polarization and vaccine efficacy, J. Controlled Release, 2015, 219, 610–621 CrossRef CAS PubMed.
- S. T. Reddy, A. J. van der Vlies, E. Simeoni, V. Angeli, G. J. Randolph and C. P. O'Neill, et al., Exploiting lymphatic transport and complement activation in nanoparticle vaccines, Nat. Biotechnol., 2007, 25(10), 1159–1164 CrossRef CAS PubMed.
- V. Manolova, A. Flace, M. Bauer, K. Schwarz, P. Saudan and M. F. Bachmann, Nanoparticles target distinct dendritic cell populations according to their size, Eur. J. Immunol., 2008, 38(5), 1404–1413 CrossRef CAS PubMed.
- T. Fifis, A. Gamvrellis, B. Crimeen-Irwin, G. A. Pietersz, J. Li and P. L. Mottram, et al., Size-dependent immunogenicity: Therapeutic and protective properties of nano-vaccines against tumors, J. Immunol., 2004, 173(5), 3148–3154 CrossRef CAS.
- J. M. Brewer, K. G. J. Pollock, L. Tetley and D. G. Russell, Vesicle size influences the trafficking, processing, and presentation of antigens in lipid vesicles, J. Immunol., 2004, 173(10), 6143–6150 CrossRef CAS.
- K. K. Tran and H. Shen, The role of phagosomal pH on the size-dependent efficiency of cross-presentation by dendritic cells, Biomaterials, 2009, 30(7), 1356–1362 CrossRef CAS PubMed.
- L. P. H. Estrada and J. A. Champion, Protein nanoparticles for therapeutic protein delivery, Biomater. Sci., 2015, 3(6), 787–799 RSC.
- T. A. Willett, A. L. Meyer, E. L. Brown and B. T. Huber, An effective second-generation outer surface protein A-derived Lyme vaccine that eliminates a potentially autoreactive T cell epitope, Proc. Natl. Acad. Sci. U. S. A., 2004, 101(5), 1303–1308 CrossRef CAS PubMed.
- K. Langer, S. Balthasar, V. Vogel, N. Dinauer, H. von Briesen and D. Schubert, Optimization of the preparation process for human serum albumin (HSA) nanoparticles, Int. J. Pharm., 2003, 257(1–2), 169–180 CrossRef CAS PubMed.
- L. H. Estrada, S. Chu and J. A. Champion, Protein nanoparticles for intracellular delivery of therapeutic enzymes, J. Pharm. Sci., 2014, 103(6), 1863–1871 CrossRef CAS PubMed.
- L. Wang, A. Hess, T. Z. Chang, Y. C. Wang, J. A. Champion and R. W. Compans, et al., Nanoclusters self-assembled from conformation-stabilized influenza M2e as broadly cross-protective influenza vaccines, Nanomed. Nanotechnol., 2014, 10(2), 473–482 CrossRef PubMed.
- L. Wang, T. Z. Chang, Y. He, J. R. Kim, S. Wang and T. Mohan, et al., Coated protein nanoclusters from influenza H7N9 HA are highly immunogenic and induce robust protective immunity, Nanomedicine, 2016, 13(1), 253–262 Search PubMed.
- L. J. Thomann-Harwood, P. Kaeuper, N. Rossi, P. Milona, B. Herrmann and K. C. McCullough, Nanogel vaccines targeting dendritic cells: Contributions of the surface decoration and vaccine cargo on cell targeting and activation, J. Controlled Release, 2013, 166(2), 95–105 CrossRef CAS PubMed.
- P. Pradhan, H. Qin, J. A. Leleux, D. Gwak, I. Sakamaki and L. W. Kwak, et al., The effect of combined IL10 siRNA and CpG ODN as pathogenmimicking microparticles on Th1/Th2 cytokine balance in dendritic cells and protective immunity against B cell lymphoma, Biomaterials, 2014, 35(21), 5491–5504 CrossRef CAS PubMed.
- C. L. Chu, Y. L. Yu, Y. C. Kung, P. Y. Liao, K. J. Liu and Y. T. Tseng, et al., The Immunomodulatory Activity of Meningococcal Lipoprotein Ag473 Depends on the Conformation Made up of the Lipid and Protein Moieties, PLoS One, 2012, 7(7), e40873 CAS.
- S. E. Autenrieth and I. B. Autenrieth, Variable antigen uptake due to different expression of the macrophage mannose receptor by dendritic cells in various inbred mouse strains, Immunology, 2009, 127(4), 523–529 CrossRef CAS PubMed.
- E. M. Plummer and M. Manchester, Endocytic Uptake Pathways Utilized by CPMV Nanoparticles, Mol. Pharmaceutics, 2013, 10(1), 26–32 CrossRef CAS PubMed.
- L. Thiele, B. Rothen-Rutishauser, S. Jilek, H. Wunderli-Allenspach, H. P. Merkle and E. Walter, Evaluation of particle uptake in human blood monocyte-derived cells in vitro. Does phagocytosis activity of dendritic cells measure up with macrophages?, J. Controlled Release, 2001, 76(1–2), 59–71 CrossRef CAS PubMed.
- B. D. Chithrani and W. C. W. Chan, Elucidating the Mechanism of Cellular Uptake and Removal of Protein-Coated Gold Nanoparticles of Different Sizes and Shapes, Nano Lett., 2007, 7(6), 1542–1550 CrossRef CAS PubMed.
- E. J. Brown and T. H. Steinberg, Phagocytosis, in Biomembranes: A Multi-Volume Treatise, ed. A. G. Lee, JAI, 1996, vol. 4, pp. 33–63 Search PubMed.
- C. Foged, B. Brodin, S. Frokjaer and A. Sundblad, Particle size and surface charge affect particle uptake by human dendritic cells in an in vitro model, Int. J. Pharm., 2005, 298(2), 315–322 CrossRef CAS PubMed.
- B. Koppolu and D. A. Zaharoff, The effect of antigen encapsulation in chitosan particles on uptake, activation and presentation by antigen presenting cells, Biomaterials, 2013, 34(9), 2359–2369 CrossRef CAS PubMed.
- A. Collins, A. Warrington, A. Taylor Kenneth and T. Svitkina, Structural Organization of the Actin Cytoskeleton at Sites of Clathrin-Mediated Endocytosis, Curr. Biol., 2011, 21(14), 1167–1175 CrossRef CAS PubMed.
- A. Savina, C. Jancic, S. Hugues, P. Guermonprez, P. Vargas and I. C. Moura, et al., NOX2 Controls Phagosomal pH to Regulate Antigen Processing during Crosspresentation by Dendritic Cells, Cell, 2006, 126(1), 205–218 CrossRef CAS PubMed.
- D. Accapezzato, V. Visco, V. Francavilla, C. Molette, T. Donato and M. Paroli, et al., Chloroquine enhances human CD8(+) T cell responses against soluble antigens in vivo, J. Exp. Med., 2005, 202(6), 817–828 CrossRef CAS PubMed.
- R. Salerno-Goncalves and M. B. Sztein, Cell-mediated immunity and the challenges for vaccine development, Trends Microbiol., 2006, 14(12), 536–542 CrossRef CAS PubMed.
- O. Gross, C. J. Thomas, G. Guarda and J. Tschopp, The inflammasome: an integrated view, Immunol. Rev., 2011, 243(1), 136–151 CrossRef CAS PubMed.
- S. L. Demento, S. C. Eisenbarth, H. G. Foellmer, C. Platt, M. J. Caplan and W. M. Saltzman, et al., Inflammasome-activating nanoparticles as modular systems for optimizing vaccine efficacy, Vaccine, 2009, 27(23), 3013–3021 CrossRef CAS PubMed.
- M. Ritter, K. Straubinger, S. Schmidt, D. H. Busch, S. Hagner and H. Garn, et al., Functional relevance of NLRP3 inflammasome-mediated interleukin (IL)-1 beta during acute allergic airway inflammation, Clin. Exp. Immunol., 2014, 178(2), 212–223 CrossRef CAS PubMed.
- U. Schroeder, A. Graff, S. Buchmeier, P. Rigler, U. Silvan and D. Tropel, et al., Peptide Nanoparticles Serve as a Powerful Platform for the Immunogenic Display of Poorly Antigenic Actin Determinants, J. Mol. Biol., 2009, 386(5), 1368–1381 CrossRef CAS PubMed.
- S. Vallabhapurapu and M. Karin, Regulation and Function of NF-kappa B Transcription Factors in the Immune System, Annu. Rev. Immunol., 2009, 27, 693–733 CrossRef CAS PubMed.
- C. A. Dinarello, Interleukin-1, Cytokine Growth Factor Rev., 1997, 8(4), 253–265 CrossRef CAS PubMed.
- T. D. Karlson, Y. Y. Kong, C. L. Hardy, S. D. Xiang and M. Plebanski, The signalling imprints of nanoparticle uptake by bone marrow derived dendritic cells, Methods, 2013, 60(3), 275–283 CrossRef PubMed.
- M. Lundqvist, J. Stigler, G. Elia, I. Lynch, T. Cedervall and K. A. Dawson, Nanoparticle size and surface properties determine the protein corona with possible implications for biological impacts, Proc. Natl. Acad. Sci. U. S. A., 2008, 105(38), 14265–14270 CrossRef CAS PubMed.
- C. C. Fleischer and C. K. Payne, Nanoparticle-Cell Interactions: Molecular Structure of the Protein Corona and Cellular Outcomes, Acc. Chem. Res., 2014, 47(8), 2651–2659 CrossRef CAS PubMed.
- C. Gunawan, M. Lim, C. P. Marquis and R. Amal, Nanoparticle-protein corona complexes govern the biological fates and functions of nanoparticles, J. Mater. Chem. B, 2014, 2(15), 2060–2083 RSC.
- L. J. Cruz, F. Rueda, B. Cordobilla, L. Simon, L. Hosta and F. Albericio, et al., Targeting Nanosystems to Human DCs via Fc Receptor as an Effective Strategy to Deliver Antigen for Immunotherapy, Mol. Pharmaceutics, 2011, 8(1), 104–116 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6bm00500d |
| This journal is © The Royal Society of Chemistry 2017 |