
Development of an enzyme cycling method by a purine nucleoside phosphorylase for assaying inorganic phosphate†
Shigeru
Ueda
 * and
Shin-ichi
Sakasegawa
* and
Shin-ichi
Sakasegawa
R&D Group, Diagnostics Dept., Asahi Kasei Pharma Corporation, 632-1 Mifuku, Izunokuni-shi, Shizuoka 410-2321, Japan. E-mail: ueda.sc@om.asahi-kasei.co.jp; Fax: +81-558-76-7149; Tel: +81-558-76-8593
First published on 3rd October 2017
Abstract
We have developed a novel enzymatic cycling method that uses purine nucleoside phosphorylase (PNP) (EC 2.4.2.1) from Bacillus sp. to measure inorganic phosphate. The method utilizes the reversibility of the PNP reaction, in which the forward and reverse reactions are catalyzed in the presence of an excess amount of inosine and guanine, respectively, a principle similar to that previously demonstrated with creatine kinase (CK). Real-time detection was accomplished by coupling the reaction with commercially available xanthine dehydrogenase (EC 1.17.1.4) in the presence of NAD+. The efficiency of the cycling reaction per unit of enzyme (U ml−1) was remarkably higher than the CK.
Introduction
We recently developed a novel enzymatic cycling method for the quantitative determination of creatine by creatine kinase (CK). The method utilizes both the forward and reverse reactions of the kinase in the presence of excess levels of ATP and IDP, and the production rate of ADP was proportional to the concentration of its substrate. Real-time detection of ADP production was accomplished by including ADP-dependent glucokinase (ADP-GK) and glucose-6-phosphate dehydrogenase as auxiliary enzymes.1Here, we attempted to expand the application of the principle to purine nucleoside phosphorylase (PNP) (EC 2.4.2.1), a ubiquitous enzyme in the purine salvage pathway belonging to the transferase family of enzymes (EC classification code 2; i.e. the same group as kinases). Crucially for the application of our cycling method, the reaction mediated by PNP is reversible and the enzyme accepts at least two natural purine nucleoside substrates, inosine (Ino) and guanosine (Guo).
Our objective was to develop a simple, rapid and highly-sensitive assay method for inorganic phosphate (Pi) by direct photometric measurement. Enzymatic cycling is a sensitive assay method that exploits amplification techniques. Sensitivity is proportional to the concentration of enzyme used in the assay. A key feature of the reaction scheme presented here is that amplification is achieved using a single enzyme and the reaction process can be monitored directly in combination with the auxiliary enzyme.
PNP catalyzes the cleavage of the glycosidic bond of ribo- or deoxyribonucleosides, in the presence of inorganic phosphate (Pi) as a second substrate, to generate the purine base and ribose- or deoxyribose-1-phosphate as shown in the following reaction scheme.
| (Deoxy) purine nucleoside + Pi = purine base + (deoxy) ribose-1-phosphate (R-1-P) |
PNPs are found in both prokaryotes and eukaryotes. In human, a deficiency of PNP activity results in selective T cell immunodeficiency. As such, PNP is an important therapeutic target enzyme. Several PNP inhibitors have been developed to treat cancer, viral infections, and auto-immune diseases.2 Additionally, PNP is used as one of the analytical tools for the determination of serum levels of Pi with xanthine oxidase (XOD) (EC 1.2.3.2). In this methodology, hypoxanthine (Hypo) generated from Ino by PNP is converted into uric acid and hydrogen peroxide.3,4 The method dates back to the study described by HM Kalckar where uric acid was detected directly by measuring the absorbance at around 300 nm (molar extinction coefficient; 9600 M−1 cm−1 at 300 nm).5 Enzyme-based methods are becoming increasingly popular in clinical chemistry because of their high level of specificity. However, biological fluids, such as serum, sometimes contain substances that interfere with the analytical reaction. Development of a highly-sensitive method for Pi would decrease the required amount of specimen, thereby mitigating the burden of blood sampling. Moreover, the interference from the specimen could be reduced. Another application is measurement of enzyme activity such as alkaline phosphatase (ALP) in which Pi is one of the products generated from the phosphorylated-substrate. ALP is widely used as a labelling enzyme in immunoassays because it possesses a relatively high molecular activity, and various highly-sensitive detection methods, such as chemiluminescent assays, have also been developed.6 Pi determination in environmental water is also required for monitoring water quality and eutrophication.7,8
Xanthine dehydrogenase (XDH) (EC 1.17.1.4) in the presence of NAD+ can be replaced with XOD as an auxiliary enzyme. We evaluated the commercially available XDH from microorganisms and confirmed that it did not accept guanine (Gua) as substrate. This finding prompted us to investigate the PNP cycling reaction upon addition of Pi in the presence of excess Ino and Gua in combination with XDH as a novel assay system. In the measurement system, only Hypo, generated from Ino during the cycling reaction, can be detected by measuring the production rate of NADH at 340 nm. It should be noted that 2 moles of NADH are formed from 1 mole of Hypo. Here we report the results of our preliminary study for the development of a potentially highly sensitive system to measure Pi by exploiting the enzymatic cycling reaction of PNP from Bacillus sp.
Materials
PNP from Bacillus sp. and XDH from a microorganism were obtained from Asahi Kasei Pharma Corporation (Tokyo, Japan). Ino, Gua, deoxyIno, guanosine (Guo) and NAD+ were from Sigma-Aldrich (St Louis, MO). Xanthine and dipotassium hydrogen phosphate were from Wako Pure Chemical Co., Ltd (Osaka, Japan).Method
Unless otherwise stated, the PNP cycling reaction was initiated by adding Gua (final concentration 0.5 mM) to 1 ml of solution containing 50 mM Tris–HCl (pH 7.5), 2 mM Ino, 2.5 mM NAD+, 1 U ml−1 XDH at 37 °C in the presence of the given amount of dipotassium hydrogen phosphate as Pi and PNP. The increase in absorbance of the reaction mixture at 340 nm was measured using a Shimadzu UV-2550 or UV-2600 spectrophotometer (Shimadzu, Kyoto, Japan).When the Gua concentration was fixed at 0.5 mM, the dependency of the Ino concentration was evaluated using 1 U ml−1 PNP under the above conditions in the presence of 1 μM dipotassium hydrogen phosphate.
Gel filtration of the PNP was done by using a PD-10 column containing Sephadex G-25 (GE Healthcare Japan) equilibrated in 10 mM Tris–HCl (pH 7.5).
The cycling rate constant of kc (min−1) was calculated using the following equation or converted to that per 1 U ml−1 (ml min−1 U−1).
| kc = ΔAbs/(2εcofl[Pi]0) |
Results
The overall reaction scheme for the enzyme cycling system using PNP is illustrated in Fig. 1. As we have shown in our previous paper,1 the initial concentration of analyte (Pi in this case) is proportional to the production rate of Hypo and Guo generated from excess Ino and Gua.| d[Hyp]/dt = d[Guo]/dt = kc[Pi]0 |
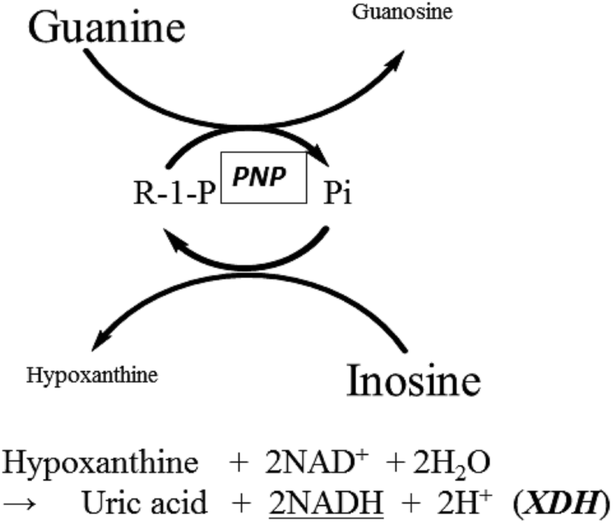 | ||
| Fig. 1 Reaction scheme for the PNP cycling reaction in the presence of XDH. Hypo, generated by the cycling reaction was measured by monitoring NADH at 340 nm. | ||
Initially, we examined the forward reaction (Hypo forming) of PNP using XDH. By adding dipotassium hydrogen phosphate (10 μM) in the presence of 2.5 mM NAD+, 2 mM Ino, 2 U ml−1 PNP, 1 U ml−1 XDH and 50 mM Tris–HCl (pH 7.5), the absorbance at 340 nm increased and reached equilibrium almost instantly, corresponding to the amount of dipotassium hydrogen phosphate. We then added 0.5 mM Gua to the reaction, and observed the re-initiation of NADH. The reaction time course with or without substrate is shown in Fig. 2. We also observed that the NADH production rate in the presence of Gua and Ino increased proportionally with an increase in the initial concentration of dipotassium hydrogen phosphate in the reaction mixture (data in the ESI†).
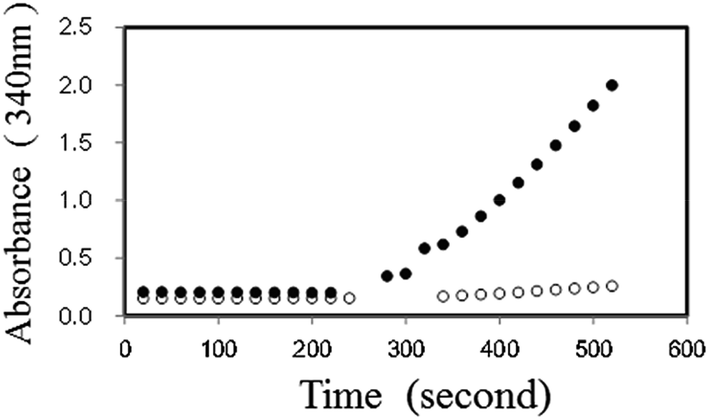 | ||
| Fig. 2 Time course of the PNP reaction before and after the addition of Gua (0.5 mM). The reaction was followed by monitoring the absorbance at 340 nm in the presence (closed circle) or absence (open circle) of 10 μM dipotassium hydrogen phosphate. | ||
In a previous study on the enzymatic cycling of 3α-hydroxysteroid dehydrogenase, the ratio of thio-NAD+ and NADH, rather than the absolute values of their concentrations, had a significant effect on the cycling rate constant (kc (min−1)).9 Thus, we examined the kc with 1 U ml−1 PNP in the presence of varying concentrations of Ino (0.25 to 2 mM) and a fixed concentration of Gua (0.5 mM). With 2 μM dipotassium hydrogen phosphate, the kc value increased depending on Ino (Fig. 3). Further experiments for the cycling reaction were thus conducted using 2 mM Ino and 0.5 mM Gua or 1 mM Ino and 0.25 mM Gua.
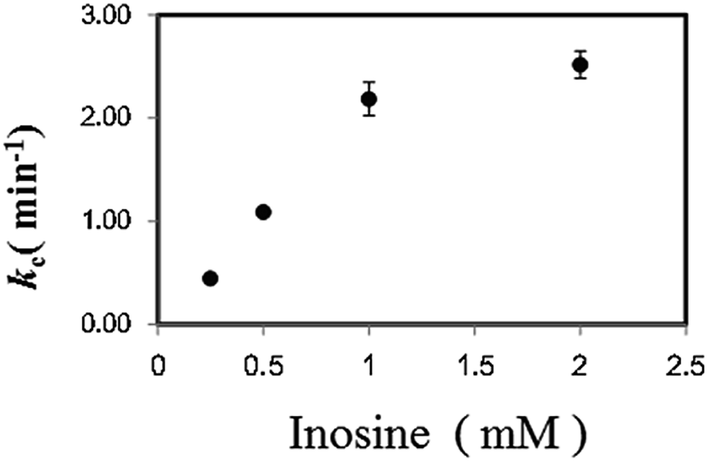 | ||
| Fig. 3 Effect of Ino concentration on the cycling rate constant (kc) (min−1) with 1 U ml−1 PNP in the presence of 2 μM dipotassium hydrogen phosphate and 0.5 mM Gua. | ||
Next, the linearity of response was evaluated as follows. To 1 ml of reagent containing 50 mM Tris–HCl (pH 7.5), 2.5 mM NAD+, 2 mM Ino, 1 U ml−1 XDH and 1 U ml−1 PNP in a cuvette was added 0.02 ml of 0, 25, 50, 100, 150, 200 or 250 μM of dipotassium hydrogen phosphate solution (0, 0.5, 1.0, 2.0, 3.0, 4.0 and 5.0 μM in the reaction mixture). The reaction mixture was then pre-incubated for 3 min at 37 °C. The cycling reaction was initiated by addition of Gua (0.5 mM) to the solution and the rate of NADH formation measured between 3 and 4 min later. We confirmed the concentration range in which signals were linear at least up to 250 μM of dipotassium hydrogen phosphate using 1 U ml−1 PNP. We compared the linearity range with the previously reported enzymatic spectrometric assay using PNP and XOD (0.2–2.5 mM),4 where the limit of detection (0.2 mM) was an eight times higher than our results (25 μM) in the sample. When Ino was replaced with deoxyIno, the cycling reaction was also observed, but at only ∼40% of the rate seen with Ino (Fig. 4). The cycling rate constant per 1 U ml−1 of PNP with Ino and Gua was calculated as ∼3 (ml min−1 U−1) (3.03 (mean) ± 0.74 (SD)), and compared to our previous result in terms of CK. The value of the PNP was more than 400 times higher than that of CK (0.00682) from rabbit muscle. The results reveal that a given cycling rate constant can be attained with less PNP. Thus, the detection of a much lower concentration of Pi was expected by increasing the amount of PNP, as it was known that kc increases proportionally, depending on the enzyme amount.1,9 However, the reagent blank became larger almost in proportion to the amount of PNP. This observation may arise either due to contamination of unknown substances, or endogenous Pi in the reagent. We speculate that Pi may be present in the reagents, which can be generated from the hydrolysis of various phosphate compounds such as pyrophosphate and substances possessing a high-energy phosphate-bond. Thus, the assay method is reliable providing endogenous Pi can be eliminated beforehand. For example, pyrophosphate, one of the products of the reaction mediated by DNA polymerase, can be transferred to Pi by the action of pyrophosphatase (EC 3.6.1.1). In this regard, development of a highly sensitive Pi assay method would be feasible. We suspected the PNP itself may contain some Pi and attempted to remove it by gel filtration chromatography. When 5 U ml−1 PNP was used in the assay, the reagent blank rate (min−1) was approximately one fourth that prior to treatment (from 0.0632 to 0.0178). The signal to noise ratio with or without 2.5 μM dipotassium hydrogen phosphate in the sample (i.e. 0.05 μM in the reaction mixture) improved from 1.15 to 1.47. The lower limit of detection was evaluated in both with or without the treatment, using the mean values and three standard deviations (AVG ± 3SD),10 in which each measurement was carried out five times using 0, 1.0, 2.5 and 5.0 μM of dipotassium hydrogen phosphate (Fig. 5). At least, 2.5 μM of Pi in the sample was detectable in the treatment of the PNP (0.0178 ± 0.0025 (0 μM) and 0.0262 ± 0.0025 (2.5 μM)). However, still remaining of the reagent blank suggested that it was not only attributed to endogenous Pi in the PNP but also in the other chemicals of the reagents. Further procedures to thoroughly remove endogenous Pi will be needed in order to detect much lower amounts of Pi. One possible resolution to mitigate against the problem of Pi contamination in the reagents might be to use phosphodeoxyribomutase (EC 2.7.5.6) together with PNP. This enzyme combination would convert R-1-P, the product from Pi, into ribose-5-phosphate before initiation of the cycling reaction. Indeed, such a strategy has been discussed in the literature.11
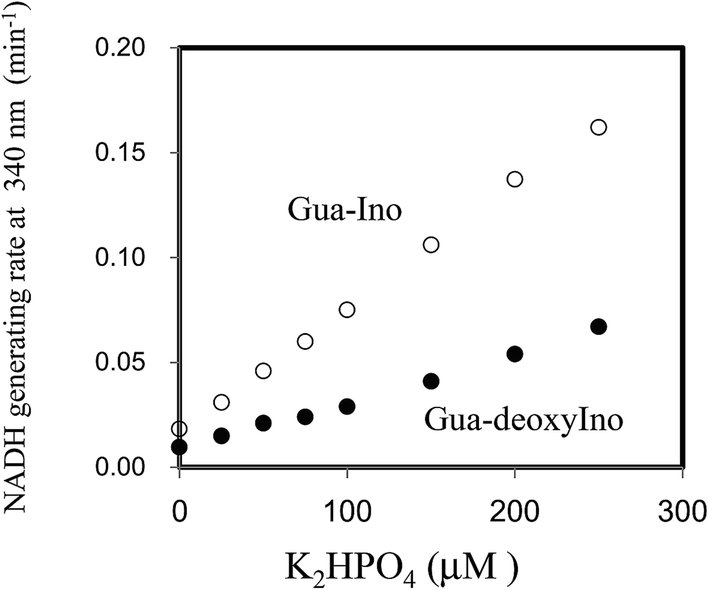 | ||
| Fig. 4 Linearity of the dipotassium hydrogen phosphate assay with 1 U ml−1 PNP in a concentration range of 25 to 250 μM. The rate of change in absorbance at 340 nm was monitored 3–4 min after addition 0.5 mM Gua in the presence of 2 mM Ino (upper plot) or deoxyIno (lower plot). The measurements at each concentration were carried out in triplicate. | ||
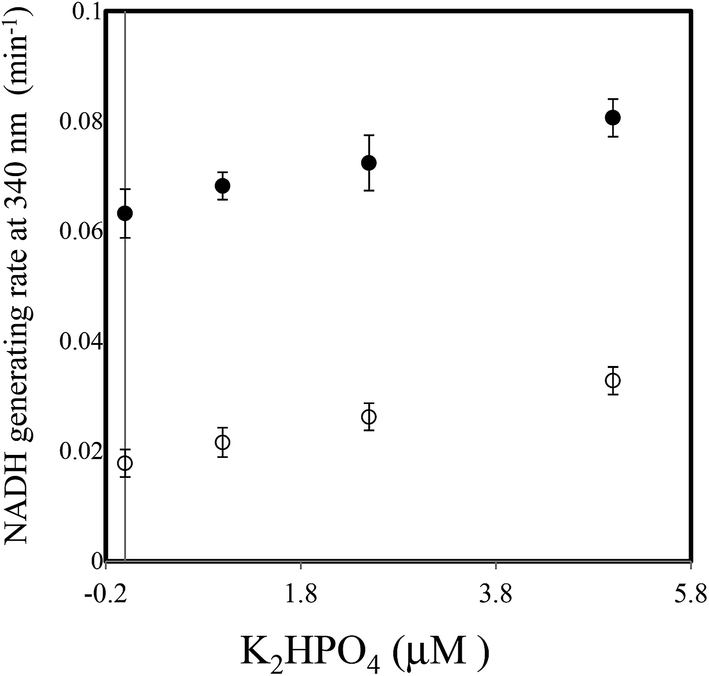 | ||
| Fig. 5 The limit of detection (LOD) after the treatment of the PNP (open circle) was at least 2.5 μM according to the 3SD method where the absorbance change rate at 340 nm was monitored for 0, 1.0, 2.5 and 5.0 μM dipotassium hydrogen phosphate. All measurements were carried out five times. Closed circle showed the results before the treatment. The error bars represent three standard deviations from the average. | ||
Discussion
Here we have developed a novel method for the quantitative determination of Pi via an enzyme cycling reaction using PNP in the presence of an excess amount of Ino and Gua. Real-time detection of Pi was accomplished using XDH as a coupling enzyme. Spectrophotometrically, we can detect at least 2.5 μM of Pi in the sample. However, the presence of endogenous Pi in the reagents was an obstacle to the practical use of this approach for more highly sensitive assays.We previously reported the development of a similar assay system using kinase such as CK. It was notable that the cycling rate constant per 1 U ml−1 of PNP was much greater than for the previously described assay.
In the reaction involving PNP shown in Fig. 1, the presence of an excess amount of both Ino and Gua over Pi is essential for the assay to work successfully. Hence, this cycling reaction is a part of the (deoxy) ribose exchange reaction between (deoxy) Ino and Gua mediated by Pi and (deoxy) R-1-P shown in the following scheme.
| (Deoxy) Ino + Gua ↔ Hypo + (deoxy) Guo |
Alternatively, the (deoxy) ribose exchange reaction can be depicted as shown in Fig. 6. The reaction itself was observed as deoxyribosyl exchange activity12 and could proceed reversibly depending on the concentration gradient. Nonetheless, the quantitation of Pi using this reaction has not been reported.
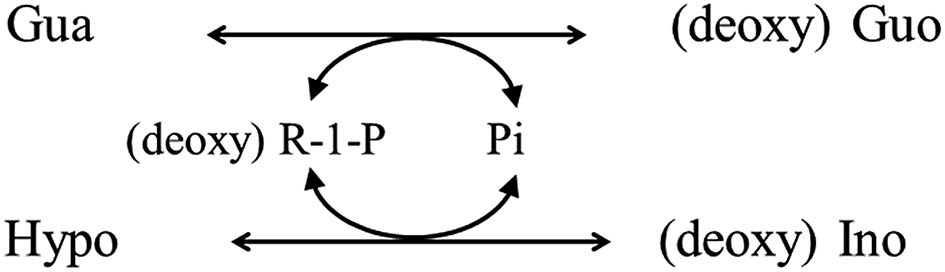 | ||
| Fig. 6 Schematic illustration of the (deoxy) ribose exchange reaction mediated by PNP via Pi and (deoxy) R-1-P. | ||
As a key enzyme in the purine salvage pathway, hypoxanthine phosphoribosyltransferase (HPRT) (EC 2.4.2.8) is well known and can be found in bacteria, yeast and mammals.13 HPRT catalyzes the following reaction.
| Hypo + 5-phosphorybosyl pyrophosphate (PRPP) = inosine-5-phosphate (IMP) + pyrophosphate (PPi) |
Hypo, the purine moiety of Ino, is converted to IMP by HPRT. HPRT also converts Gua to GMP. In human, a lack of HPRT causes Lesch–Nyhan syndrome.14 For HPRT from human, it was reported that the pyrophosphorolysis reaction (Hypo forming) was accelerated by Gua and the exchange reaction of the ribosylphosphate between IMP and Gua has been observed.13,15 Such an exchange reaction catalyzed by HPRT can be viewed as the same as the reaction mediated by PNP described above.
Given that Hypo and Gua are common substrates for both HPRT and PNP, the network between them should be possible. PNP from calf spleen displayed allosteric regulation by Pi.16,17 Curiously, the Hypo forming reaction catalyzed by the PNP was strongly inhibited by PRPP (Ki = 5 μM), which is a substrate of HPRT.16 However, feedback regulation of human HPRT has not been reported.14 Thus, this Pi and R-1-P mediated ribose exchange reaction as an extended reaction of the PNP cycling reaction, prompted us to envisage involvement in the nucleoside metabolism of salvage pathway, given that Pi exists ubiquitously in vivo. In this regard, how far the PNP cycling reaction is observed for other PNPs, especially those from human or vertebrates, is a matter of interest. Because the measurement system for this assay is very simple, integration with other analytical techniques, such as biosensors and dry chemistry, will be awaited.
Conclusion
We have developed a novel enzymatic cycling method catalyzed by PNP, which utilizes the reversibility of the reaction in the presence of an excess amount of Ino and Gua. Spectrophotometric real-time detection was accomplished by using XDH as an auxiliary enzyme. However, the relatively high reagent blank rate accompanying the increase in the amount of PNP limited assay sensitivity. Subjecting PNP to gel filtration chromatography to remove trace amounts of Pi reduced the blank rate by one fourth, increasing the S/N ratio and enabling the detection of 2.5 μM of Pi. The detection of Pi at lower levels will necessitate the incorporation of additional procedures to thoroughly eliminate endogenous Pi.Conflicts of interest
None.Abbreviations
| PNP | Purine nucleoside phosphorylase |
| Ino | Inosine |
| Gua | Guanine |
| Guo | Guanosine |
| Hypo | Hypoxanthine |
| Pi | Inorganic phosphate |
| R-1-P | Ribose-1-phosphate |
| XOD | Xanthine oxidase |
| XDH | Xanthine dehydrogenase |
| CK | Creatine kinase |
| HPRT | Hypoxanthine phosphorybosyltransferase |
| PRPP | 5-Phosphorybosyl pyrophosphate |
| IMP | Inosine-5-phosphate |
| PPi | Pyrophosphate |
Acknowledgements
We thank M. Iwasaki (Asahi Kasei Pharma Corporation) for technical assistance.References
- S. Ueda and S. Sakasegawa, Anal. Biochem., 2016, 506, 8–12 CrossRef CAS PubMed.
- A. Bzowska, E. Kulikowska and D. Shugar, Pharmacol. Ther., 2000, 88, 349–425 CrossRef CAS PubMed.
- P. Fossati, Anal. Biochem., 1985, 149, 62–65 CrossRef CAS PubMed.
- J. P. Ungerer, H. M. Oosthuizen and S. H. Bissbort, Clin. Chim. Acta, 1993, 223, 149–157 CrossRef CAS.
- H. M. Kalckar, J. Biol. Chem., 1947, 167, 429–443 CAS.
- I. Bronstein, J. C. Voyta, G. H. Thorpe, L. J. Kricka and G. Armstrong, Clin. Chem., 1989, 35, 1441–1446 CAS.
- J. H. Ryther and W. M. Dunstan, Science, 1971, 171, 1008–1013 CAS.
- H. Cao, Z. Chen and Y. Huang, Talanta, 2015, 143, 450–456 CrossRef CAS PubMed.
- S. Ueda, M. Oda, S. Imamura and M. Ohnishi, Anal. Biochem., 2004, 332, 84–89 CrossRef CAS PubMed.
- D. A. Armbruster, M. D. Tillman and L. M. Hubbs, Clin. Chem., 1994, 40, 1233–1238 CAS.
- A. E. Nixon, J. L. Hunter, G. Bonifacio, J. F. Eccleston and M. R. Webb, Anal. Biochem., 1998, 265, 299–307 CrossRef CAS PubMed.
- R. Abrams, M. Edmonds and L. Libenson, Biochem. Biophys. Res. Commun., 1965, 20, 310–314 CrossRef CAS PubMed.
- Y. Xu, J. Eads, J. C. Sacchettini and C. Grubmeyer, Biochemistry, 1997, 36, 3700–3712 CrossRef CAS PubMed.
- T. Yamaoka, M. Yano, M. Kondo, H. Sasaki, S. Hino, R. Katashima, M. Moritani and M. Itakura, J. Biol. Chem., 2001, 276, 21285–21291 CrossRef CAS PubMed.
- C. Salerno and A. Giacomello, J. Biol. Chem., 1979, 254, 10232–10236 CAS.
- P. A. Ropp and T. W. Traut, Arch. Biochem. Biophys., 1991, 288, 614–620 CrossRef CAS PubMed.
- P. A. Ropp and T. W. Traut, J. Biol. Chem., 1991, 266, 7682–7687 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ay02016c |
| This journal is © The Royal Society of Chemistry 2017 |