
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSecondary electrospray ionization proceeds via gas-phase chemical ionization
Alberto Tejero
Rioseras
 abc,
Martin Thomas
Gaugg
a and
Pablo
Martinez-Lozano Sinues
*ad
abc,
Martin Thomas
Gaugg
a and
Pablo
Martinez-Lozano Sinues
*ad
aDepartment of Chemistry and Applied Biosciences, ETH Zurich, Zurich, Switzerland. E-mail: pablo.mlsinues@ukbb.ch
bSEADM, S.L., Spain
cDepartment of Analytical Chemistry, University of Cordoba, Spain
dUniversity Children's Hospital Basel, University of Basel, Switzerland
First published on 3rd July 2017
Abstract
Our main goal was to gain further insights into the mechanism by which gas-phase analytes are ionized by interaction with plumes of electrospray solvents. We exposed target vapors to electrosprays of either water or deuterated water and mass analyzed them. Regardless of the solvent used, the analytes were detected in protonated form. In contrast, when the ionization chamber was humidified with deuterated water, the target vapors were detected in deuterated form. These observations suggest that either there is no interaction between analytes and electrospray charged droplets, or if there is any, a subsequent gas-phase ion–molecule reaction governs the process. Implications in practical examples such as breath analysis are discussed.
Introduction
The concept of “ambient ionization-mass spectrometry” essentially involves the exciting possibility of direct analysis with minimal sample preparation.1,2 Well before a plethora of acronyms describing ambient ionization methods emerged during the second half of the 2000's, a number of pioneering papers describing similar concepts were presented. We feel that this pioneering work should be acknowledged in this special issue. For example, back in the 70's and early 80's Sciex's Trace Atmospheric Gas Analyzer (TAGA) was used to investigate human breath metabolites in real-time3–6 and for environmental monitoring.7 In these cases, chemical ionization coupled to atmospheric pressure ionization-mass spectrometry was at the core of such impressive early examples of the potential of what we call today ambient mass spectrometry.Similarly, during the early days of electrospray ionization development, John B. Fenn and co-workers noted the remarkable ability of electrosprays to ionize not only analytes in the liquid-phase,8 but also pre-existing species in the gas-phase.9–11 Similar observations on the potential of electrospray to ionize vapors were noted contemporarily by other groups.12–14 Eventually, this technique was referred to as Secondary Electrospray Ionization (SESI) by Hill and co-workers to describe the use of a pure solvent electrospray to efficiently ionize gas-phase analytes.15,16 Ever since, SESI has been used to detect trace vapors in a wide range of applications by different groups.17–24
While it is generally accepted that SESI coupled to modern atmospheric pressure mass spectrometry is a sensitive method to detect gases at trace concentrations in real-time, the lack of full understanding of the SESI mechanism prevents making rational choices for the optimal parameters. In addition, with the advent of several ambient mass spectrometry techniques with overlapping features, fundamental aspects of such techniques have been blurred or confused.25 Our goal in this study was to further previous work on the mechanism by which strictly vapor species are ionized in contact with electrospray plumes of pure solvents.26,27
Methods
SESI-MS
A commercially available SESI source (SEADM, Spain) was used in all experiments.28 It was interfaced with an Orbitrap mass spectrometer (Thermo) and on one occasion (data shown in Fig. 5) with a TripleTOF 5600+ (Sciex).The SESI was operated in the nanoliter range (∼100 nL min−1). We flushed the electrospray region with compressed air at a flow rate of 1 L min−1. Compressed air contained around 5% relative humidity. Between the mass flow controller (Bronkhorst) and the ionization chamber, we placed a three-neck flask with rubber stoppers. Different liquids were injected into the flask to seed the carrier air with their corresponding vapors. Injected ambient vapors included water (H2O), deuterated water (D2O), ethanol (EtOH) and deuterated ethanol (EtOD). Relative humidity within the flask was recorded with a sensor (Alborn Almemo 2590A).
Breath analysis
SESI-MS breath analysis was accomplished by exhaling through the sampling tube of the ion source at a constant exhalation pressure of 10 mbar, as measured by a digital manometer.The study was approved by the local ethical committee (EK 2012-N-49) and the subject gave written informed consent to participate.
Results and discussion
Deuterated spray solvent
Two (simplified) ionization scenarios are plausible in SESI: (i) the neutral species dissolve into the charged electrospray droplets to eventually be re-ejected in ionized form into the gas-phase; (ii) electrospray droplets eject primary reactant ions that undergo an ion–molecule reaction with neutral species. Obviously, both mechanisms lead to very different scenarios to optimize the ionization efficiency. For the former mechanism, solubility of the analyte on the charging spray solvent would be crucial to maximize ionization efficiency. In the latter mechanistic scenario, analyte detection would be ultimately governed by thermochemistry.In order to determine which of the two potential scenarios apply in SESI we exposed neutral vapors to electrosprays of water and deuterated water. The exchange of hydrogen/deuterium has provided very valuable mechanistic insights since the initial developments of chemical ionization/mass-spectrometry.29–32 We hypothesized that, if the neutral vapors interact with the electrospray charged droplets to be re-emitted as gas-phase ions, they should be detected as [M + D]+ ions.
Typical SESI-MS spectra has a rich chemical background in the range 50–500 Da.33,34 For example, volatile polydimethylcyclosiloxanes in ambient laboratory air are well-known interfering species in mass spectrometry.35,36 Similarly, phthalates have also been reported to off-gas from vacuum o-rings.37 Thus, we took advantage of the presence of these ubiquitous gas-phase contaminants to gain insights into how nano-electrosprays of solvent eventually lead to the ionization of such species. Fig. 1 shows the mass spectra resulting of flushing the electrospray chamber with compressed air and using as spray solvent either 0.15% formic acid in H2O (a) or 0.15% deuterated acetic acid (i.e. CD3-COOD) in D2O (b). The resulting spectra are identical, including the known polysiloxanes and phthalates volatile species. Thus, regardless of the use of protonated or deuterated solvents, all species were detected in protonated form. This suggests that, even if the gas-phase species interact with the droplets to acquire a deuterium, eventually they exchange the deuterium with a proton in the gas-phase. In this case, with ambient water vapor, as the carrier gas contained ∼5% of relative humidity.
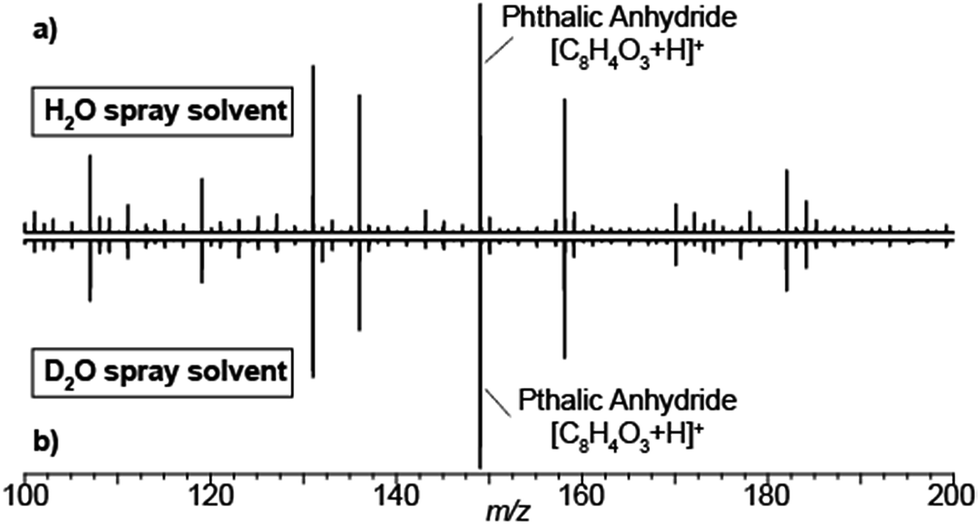 | ||
| Fig. 1 SESI mass spectra are independent of whether electrospray solvent is water or deuterated water. SESI chemical noise mass spectra with different electrospray solutions: (a) 0.15% formic acid in H2O; (b) 0.15% deuterated acetic in D2O. In both cases, the analytes were detected in protonated form. For reference, the dominating peak of phthalic anhydride is labelled. | ||
Doping carrier gas with deuterated vapors
The data shown in Fig. 1b strongly suggest that a gas-phase proton reaction takes place in SESI. To further confirm this hypothesis, we humidified the carrier air with either water or deuterated water. As a result of humidifying with deuterated water, the entire chemical background mass spectrum shifted by 1 Da. Fig. 2 shows a zoomed view of the mass spectrum in the region of one representative example of a phthalate (C16H22O4) and a polysiloxane (C12H36Si6O6) with H2O-humidified air (bottom) and D2O-humidified air (top). The fact that these species deuterate when the air is seeded with deuterated water vapor, but not when deuterated water is used in the spray solvent, strongly indicates that gas-phase ion chemistry plays a critical role in SESI, if not the only one.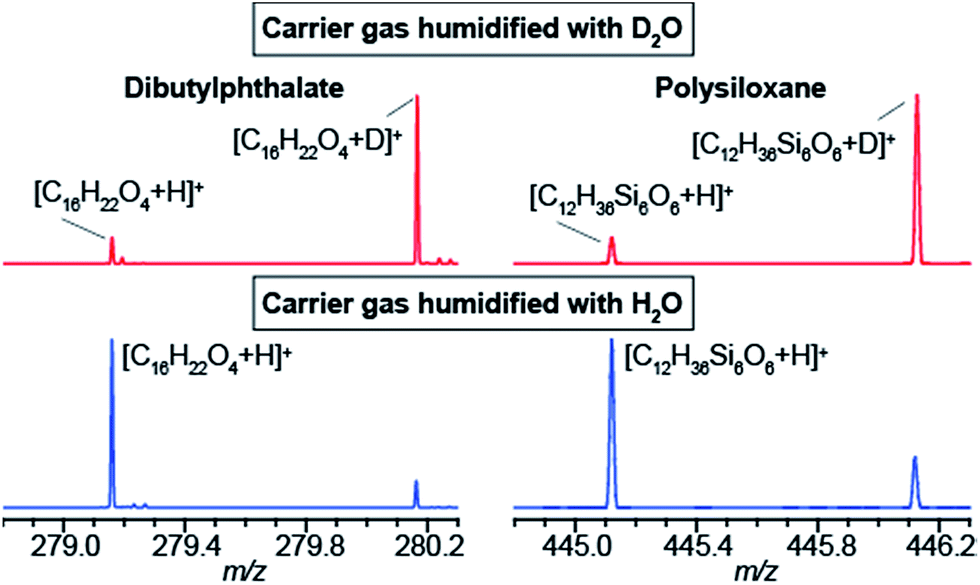 | ||
| Fig. 2 Humidifiying the carrier air with deuterated water leads to deuterated ions in SESI-MS. Electrospray solvent was 0.15% formic acid in H2O. Mass spectra of dibutylphthalate (left) and a polysiloxane (right). As expected, humidifying with H2O leads to protonated species (i.e. [M + H]+; bottom) and humidifying with D2O leads to deuterated species (i.e. [M + D]+; top). | ||
Further insight was gained by seeding the carrier gas with ethanol instead of water. Ethanol has a greater gas-phase proton affinity than water (776.4 vs. 691 kJ mol−1). Thus, it would be expected that, if SESI is governed by thermochemistry of the water and ethanol proton transfer reaction, it would be detected in the presence of the former but not the latter. Fig. 3a and b illustrate the behavior of another representative compound (benzothiazole; C7H5NS).38Fig. 3a shows the [M + D]+/[M + H]+ ratio before and after humidifying with D2O. Clearly, at the time point of D2O injection, the protonated species decays, while simultaneously the deuterated adduct rises. Fig. 3b shows the result of a similar experiment, but instead of D2O, we seeded the SESI chamber with vapors of EtOD. Likewise, the deuterated adduct rose sharply in parallel with the relative EtOD concentration in air, suggesting a direct correlation between EtOD levels and [C7H4NS + D]+ signal intensity. In contrast, the polysiloxane experienced the same response upon exposure to D2O vapors (Fig. 3c), but the signal dropped abruptly to 0 after introducing EtOD (Fig. 3d). Hence, Fig. 3c and d suggests that the polysiloxane has a gas-phase proton affinity greater than water, but lower than ethanol.
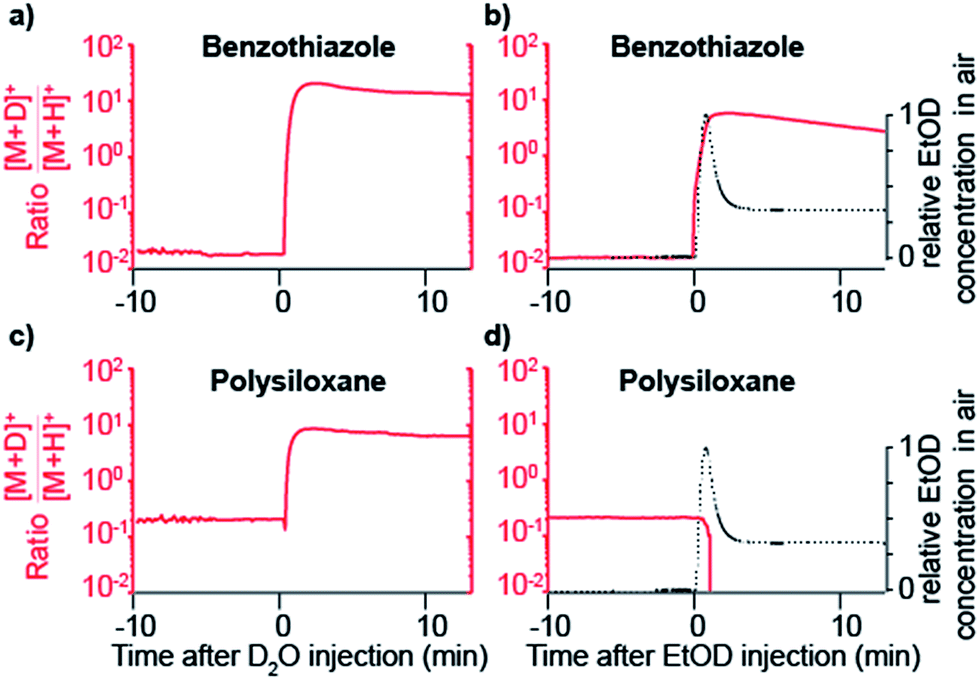 | ||
| Fig. 3 Gas-phase proton affinity of seeding vapors have an effect in SESI analyte detection. Benzothiazole deuterates after adding vapors of D2O (a) and EtOD (b) in the carrier gas. In contrast, the deuterated form of the polysiloxane was detected with D2O (c), but depleted upon exposure to EtOD vapors (d). Relative concentrations of EtOD in the carrier gas are shown for reference. | ||
To confirm that gas-phase thermochemistry ultimately dictates which analytes are detectable by SESI, we chose 2,2,2-trifluoroethanol as target analyte vapor. This particular compound has gas-phase proton affinity of 700.2 kJ mol−1,39 which is in between that of water and ethanol. As expected, this compound was detected as deuterated ion upon doping the carrier gas with D2O, but not when EtOD was used.
Fig. 4a shows how the [M + D]+/[M + H]+ ratio rises sharply immediately after injecting D2O. For reference, relative humidity levels, as measured with the sensor, are overlaid. When the same experiment is performed but instead of D2O, EtOD is injected in the carrier gas, 2,2,2-trifluoroethanol is no longer detected (Fig. 4b).
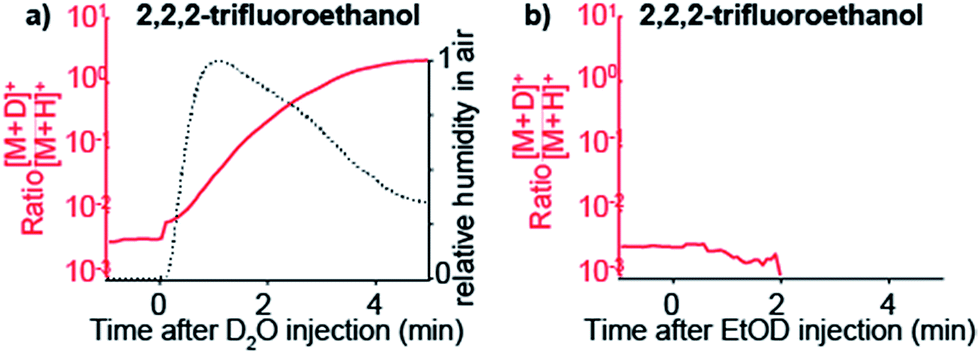 | ||
| Fig. 4 SESI selectivity towards target vapors is governed by the proton affinity of the dopant seeded in the carrier gas. Response of 2,2,2-trifluoroethanol vapors in the presence of D2O (a) and EtOD (b) in the carrier gas. When D2O is introduced in the system the protonated ion declines in favor of the deuterated species (a). In contrast, when EtOD is introduced in excess in the carrier gas the signal of [C2H3F3O + H]+ drops quickly to zero, but the [C2H3F3O + D]+ ion is not formed (b). This is explained by the fact that the gas-phase proton affinity of 2,2,2-trifluoroethanol is greater than water, but smaller than ethanol. | ||
We have just shown compelling evidence indicating that SESI leads to protonated species, even if the spray solvent is deuterated, as long as some water vapor is present in the carrier gas of the sample (in our case ∼5% relative humidity). This directly leads to a gas-phase chemical ionization scenario, at least in the final step of the process. Our hypothesis to explain this observation is that, initially, [(D2O)n + D]+ clusters are ejected from the electrospray droplets by ion evaporation.40–44 These primary ions rapidly exchange deuterium by protons with ambient water, which is in excess in ambient air, leading to eventually dominating [(H2O)n + H]+ clusters. These water clusters would then undergo a proton transfer reaction with neutral vapor species, provided the reaction is thermodynamically favorable. Of course, kinetically-controlled reactions are also conceivable for slow proton transfer, which indeed is known to be the case for many reactions in chemical ionization.44–46 For example, under our particular experimental conditions in Fig. 1b, we formed the electrospray by infusing D2O at ∼100 nL min−1. If we assume that all D2O is immediately vaporized, and considering that the ambient air flushing the SESI chamber at 1 L min−1 contained 5% relative humidity, the expected ratio H2O/D2O in the ionization chamber is in the order of 10. This excess of H2O would then shift the equilibrium of reactant ions from [(D2O)n + D]+ towards [(H2O)n + H]+. This hypothesis is supported by the data shown in Fig. 5. It shows a SESI mass spectrum using water–formic acid as electrospray solvent and the carrier gas bathing the SESI chamber humidified with a 50/50 mixture of H2O and D2O. It shows two binomial distributions for the water trimer and tetramer with all possible combinations of hydrogen and deuterium atoms.
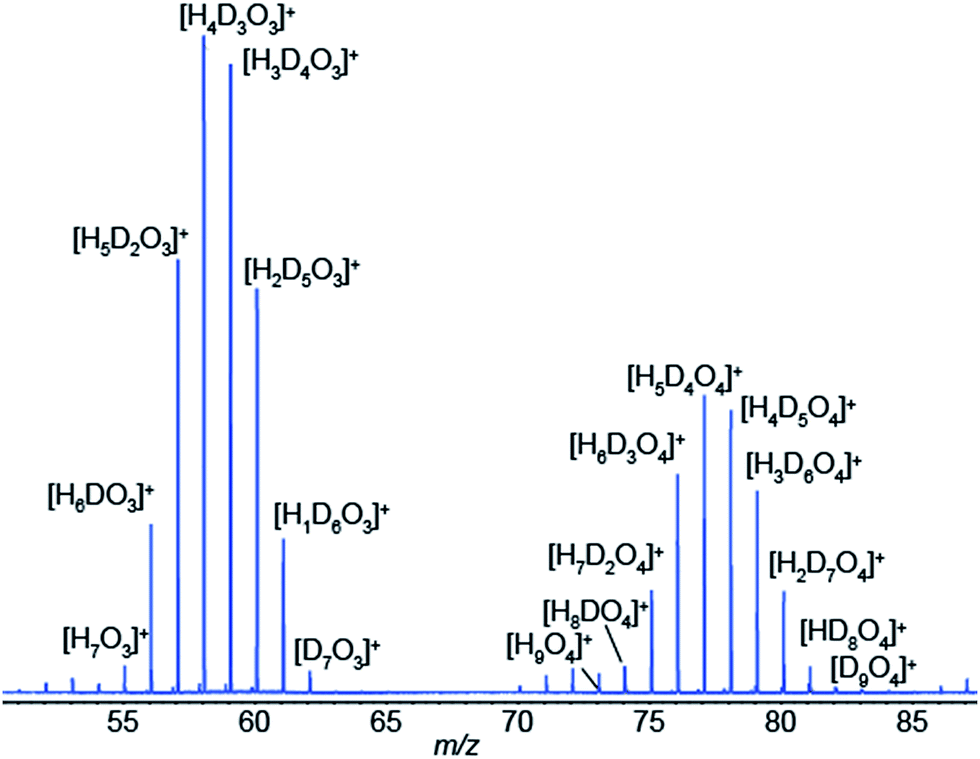 | ||
| Fig. 5 SESI gas-phase reactant ions in the presence of humidity are water clusters. Example of an electrospray of 0.15% formic acid in water flushed with a 50/50 mixture of H2O/D2O vapors, leading to all possible [HxDyO3]+ (x + y = 7) combinations for the trimer (left-hand-side) and [HxDyO4]+ (x + y = 9) for the tetramer (right-hand-side). | ||
Warnings about the importance of gas-phase ion chemistry in the standard electrospray process have been given in the literature.47 For example, Enke and co-workers convincingly showed that analytes present in solution are very likely to be ultimately depleted in the gas phase by the presence of molecules that are stronger gas-phase bases, resulting in ion suppression.39 Our data support this view, and extend it to SESI. Hence, we have advocated that one should be cautious when distinguishing the real-time analysis of gas- vs. condensed-phase analytes with sprays of pure solvent.26 The main reason is that a number of studies have suggested that for samples delivered in aerosol form (i.e. extractive electrospray ionization), solubility of the analytes and interaction with the charging spray solvent is the limiting factor to be considered.48,49 We argue here that, in the case of vapor samples (i.e. secondary electrospray ionization), gas-phase ion chemistry considerations are ultimately governing the detection of such analytes. For this reason, we claim that water electrosprays operated in the cone-jet mode50 are preferred in positive ion mode SESI over widely used H2O/MeOH mixtures, as MeOH will probably deplete species detectable otherwise.
Breath analysis
The analysis of exhaled metabolites in breath is an attractive approach to monitor health status and therapeutic intervention.51 Selected ion flow tube mass spectrometry (SIFT-MS)52 and proton transfer reaction mass spectrometry (PTR-MS)53 are two well established methods for real-time detection of volatiles in breath. One of the main strengths of SIFT-MS is its capability to provide absolute quantifications of trace gases in the parts-per-million to parts-per-billion range.54 PTR-MS provides semi-quantitative information with limits of detection down to parts-per-trillion. Exhaling onto an electrospray plume in front of an atmospheric pressure ionization MS is another highly efficient way to perform such analyses. A wide range of species have been reported using this method; however chemical identification and absolute quantification of the species detected remain a challenge. Given that, low-volatility species such as urea have been detected using this approach,55,56 there has been some debate regarding whether the metabolites detected in breath following this approach come in aerosol form or simply the method is sensitive enough to detect low-volatility vapors.17,55,56 The debate is of importance because aerosols and volatiles have different physiological origins and therefore offer different diagnostic opportunities. Aerosols in breath are thought to be generated by the turbulence-induced aerosolization mechanism of the fluid film in the respiratory tract.57 This leads to a high variability on particle size distribution and number depending on the inhalation and exhalation maneuvers57 and hence difficulties in reproducibility. In contrast, the origin of gas-phase species is predominantly blood borne and therefore enable monitoring different metabolic processes.58 We argue that breath analysis by SESI-MS detects volatile and non-volatile gas-phase species following that: (i) truly non-volatile macromolecules such as cytokines have been reported in exhaled breath condensate,59 but are not detectable by SESI-MS; (ii) exhaling through a particle filter leads to essentially the same mass spectrum as without such a filter;55 (iii) analytical27 and numerical28 models capture experimental observations with reasonable accuracy in systematic mechanistic studies26 and (iv) we have shown how small amounts of injected drugs in mice can be detected in breath shortly afterwards and correlate with plasma levels.60,61 For example, ketamine and its main metabolites, which have molecular weights in the range of 250 Da and a relatively low estimated vapor pressure in the order of 8.5 × 10−5 mmHg at 25 °C (SPARC online calculator).62To further shed light on this mechanistic issue we performed SESI-MS breath analysis using H2O and D2O as spray solvents. Aerosolized samples have been shown to interact in the liquid-phase between charged electrospray droplets and neutral sample aerosols.48 Hence we hypothesized that, if the detected compounds would come in aerosol form, they would necessarily pick up a deuterium from the D2O charged droplets and a proton from H2O sprays. Fig. 6 shows the breath mass spectrum of one subject exhaling onto a spray of 0.15% formic acid in water (a) and 0.15% acetic acid-d4 in D2O (b). In both cases, the spectra were identical, with the vast majority of the species detected in protonated form. This may also explain the noted sensitivity enhancement of SESI in the presence of water vapor in breath.55,64 Admittedly, these data cannot completely exclude the possibility of exhaled aerosols and vapors dissolving in the charged droplets, generation of [M + D]+ gas-phase ions and subsequently exchanging H–D in the gas phase with ubiquitous water vapor. However, it clearly shows that, even if this is the case, gas-phase ion chemistry ultimately plays a critical role in the ionization of breath metabolites.
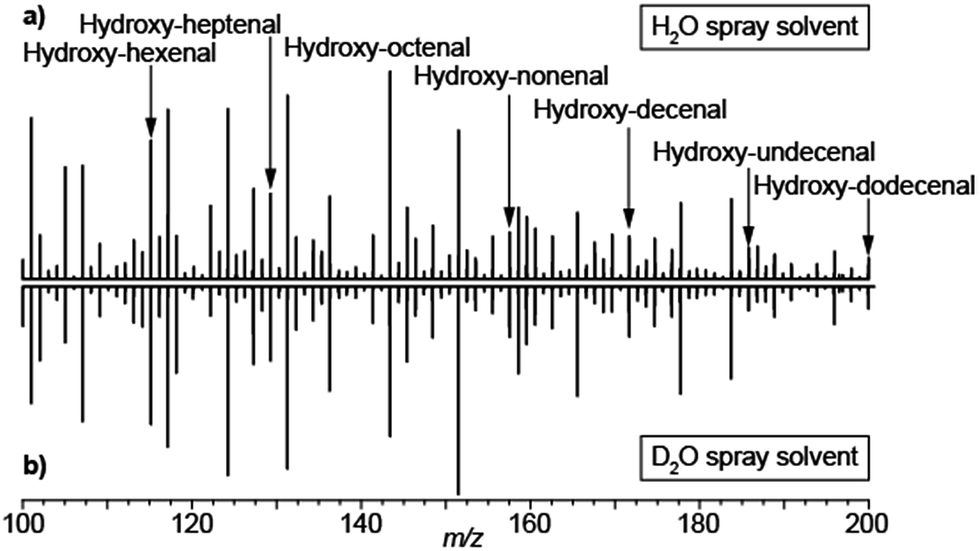 | ||
| Fig. 6 Breath metabolites are ultimately protonated via gas-phase ion–molecule reactions in SESI: (a) breath mass spectrum using 0.15% formic acid in water; (b) breath mass spectrum using 0.15% acetic acid-d4 in heavy water. Breath metabolites were detected in protonated form regardless of the solvent used to generate the electrospray. For reference, a series of hydroxy-alkenals previously identified by García-Gómez63 are indicated. | ||
Conclusions
We conclude that (i) neutral vapors carried in an air stream (5% relative humidity) and exposed to an electrospray of deuterated water leads to protonated ions; (ii) inversely, neutral vapors carried in an air stream seeded with deuterated solvents (e.g. D2O), and exposed to an electrospray of water leads to deuterated ions; (iii) gas-phase proton affinity of the target analytes plays a crucial role in whether or not they are finally detected in SESI; (iv) breath metabolites are detected in protonated form regardless of whether the solvent used is H2O or D2O. These observations lead us to conclude that, in the final stage of SESI, gas-phase ion–molecule reactions govern the mechanism by which electrosprays ionize vapor species.Acknowledgements
We are thankful to Prof. Renato Zenobi for hosting the ACID project at ETH Zurich and for critically reading this manuscript. We gratefully thank Dr Juan Zhang (Novartis AG) for the donation of the LTQ Orbitrap instrument used in this study and Prof. Malcolm Kohler (University Hospital Zurich) for giving access to the Sciex mass spectrometer. The research leading to these results has received funding from the European Community's Seventh Framework Programme (FP7-2013-IAPP) within the project “Analytical Chemistry Instrumentation Development” (609691). This work is dedicated to Prof. John B. Fenn (1917–2010) on the occasion of the centennial celebration of his birth.References
- M. E. Monge, G. A. Harris, P. Dwivedi and F. M. Fernández, Chem. Rev., 2013, 113, 2269–2308 CrossRef CAS PubMed.
- M. Z. Huang, S. C. Cheng, Y. T. Cho and J. Shiea, Anal. Chim. Acta, 2011, 702, 1–15 CrossRef CAS PubMed.
- F. M. Benoit, W. R. Davidson, A. M. Lovett, S. Nacson and A. Ngo, Int. Arch. Occup. Environ. Health, 1985, 55, 113–120 CrossRef CAS PubMed.
- F. M. Benoit, W. R. Davidson, A. M. Lovett, S. Nacson and A. Ngo, Anal. Chem., 1983, 55, 805–807 CrossRef CAS.
- B. A. Thomson, W. R. Davidson and A. M. Lovett, Environ. Health Perspect., 1980, 36, 77–84 CrossRef CAS PubMed.
- A. M. Lovett, N. M. Reid, J. A. Buckley, J. B. French and D. M. Cameron, Biomed. Mass Spectrom., 1979, 6, 91–97 CrossRef CAS PubMed.
- D. A. Lane and B. A. Thomson, J. Air Pollut. Control Assoc., 1981, 31, 122–127 CrossRef CAS.
- J. B. Fenn, M. Mann, C. K. Meng, S. F. Wong and C. M. Whitehouse, Science, 1989, 246, 64–71 CAS.
- S. Fuerstenau, P. Kiselev and J. B. Fenn, ESIMS in the Analysis of Trace Species in Gases, Dallas (TX), 1999 Search PubMed.
- S. Fuerstenau, PhD thesis, Yale University, 1994.
- C. M. Whitehouse, F. Levin, C. K. Meng and J. B. Fenn, Further Adventures with an Electrospray Ion Source, Cincinnati, OH, 1986 Search PubMed.
- Y. H. Chen, H. H. Hill and D. P. Wittmer, J. Microcolumn Sep., 1994, 6, 515–524 CrossRef CAS.
- C.-M. Hong, F.-C. Tsai and J. Shiea, Anal. Chem., 2000, 72, 1175–1178 CrossRef CAS PubMed.
- C. Y. Lee and J. Shiea, Anal. Chem., 1998, 70, 2757–2761 CrossRef CAS PubMed.
- M. Tam and H. H. Hill, Anal. Chem., 2004, 76, 2741–2747 CrossRef CAS PubMed.
- C. Wu, W. F. Siems and H. H. Hill, Anal. Chem., 2000, 72, 396–403 CrossRef CAS PubMed.
- X. Li, L. Huang, H. Zhu and Z. Zhou, Rapid Commun. Mass Spectrom., 2017, 31, 301–308 CrossRef CAS PubMed.
- J. C. Wolf, M. Schaer, P. Siegenthaler and R. Zenobi, Anal. Chem., 2015, 87, 723–729 CrossRef CAS PubMed.
- M. J. Aernecke, T. Mendum, G. Geurtsen, A. Ostrinskaya and R. R. Kunz, J. Phys. Chem. A, 2015, 119, 11514–11522 CrossRef CAS PubMed.
- J. C. Reynolds, M. A. Jimoh, C. Guallar-Hoyas, C. S. Creaser, S. Siddiqui and C. L. P. Thomas, J. Breath Res., 2014, 8, 037105 CrossRef PubMed.
- L. Meier, C. Berchtold, S. Schmid and R. Zenobi, J. Mass Spectrom., 2012, 47, 1571–1575 CrossRef CAS PubMed.
- L. A. Dillon, V. N. Stone, L. A. Croasdell, P. R. Fielden, N. J. Goddard and C. L. Paul Thomas, Analyst, 2010, 135, 306–314 RSC.
- J. Zhu, H. D. Bean, Y. M. Kuo and J. E. Hill, J. Clin. Microbiol., 2010, 48, 4426–4431 CrossRef CAS PubMed.
- N. Brenner, M. Haapala, K. Vuorensola and R. Kostiainen, Anal. Chem., 2008, 80, 8334–8339 CrossRef CAS PubMed.
- A. J. Ingram, C. L. Boeser and R. N. Zare, Chem. Sci., 2016, 7, 39–55 RSC.
- P. Martinez-Lozano Sinues, E. Criado and G. Vidal, Int. J. Mass Spectrom., 2012, 313, 21–29 CrossRef CAS.
- J. F. de la Mora, Int. J. Mass Spectrom., 2011, 300, 182–193 CrossRef.
- C. Barrios-Collado, G. Vidal-de-Miguel and P. Martinez-Lozano Sinues, Sens. Actuators, B, 2016, 223, 217–225 CrossRef CAS.
- D. F. Hunt, C. N. Mcewen and R. A. Upham, Tetrahedron Lett., 1971, 4539–4542 CrossRef CAS.
- D. F. Hunt, C. N. Mcewen and R. A. Upham, Anal. Chem., 1972, 44, 1292–1294 CrossRef CAS.
- W. Blum, E. Schlumpf, J. G. Liehr and W. J. Richter, Tetrahedron Lett., 1976, 17, 565–568 CrossRef.
- A. K. Meher and Y. C. Chen, Anal. Chim. Acta, 2017, 966, 41–46 CrossRef CAS PubMed.
- B. O. Keller, J. Sui, A. B. Young and R. M. Whittal, Anal. Chim. Acta, 2008, 627, 71–81 CrossRef CAS PubMed.
- X. Guo, A. P. Bruins and T. R. Covey, Rapid Commun. Mass Spectrom., 2006, 20, 3145–3150 CrossRef CAS PubMed.
- A. Schlosser and R. Volkmer-Engert, J. Mass Spectrom., 2003, 38, 523–525 CrossRef CAS PubMed.
- J. C. Reynolds, G. J. Blackburn, C. Guallar-Hoyas, V. H. Moll, V. Bocos-Bintintan, G. Kaur-Atwal, M. D. Howdle, E. L. Harry, L. J. Brown, C. S. Creaser and C. L. P. Thomas, Anal. Chem., 2010, 82, 2139–2144 CrossRef CAS PubMed.
- K. M. Verge and G. R. Agnes, J. Am. Soc. Mass Spectrom., 2002, 13, 901–905 CrossRef CAS PubMed.
- D. Garcia-Gomez, L. Bregy, Y. Nussbaumer-Ochsner, T. Gaisl, M. Kohler and R. Zenobi, Environ. Sci. Technol., 2015, 49, 12519–12524 CrossRef CAS PubMed.
- M. a. H. Amad, N. B. Cech, G. S. Jackson and C. G. Enke, J. Mass Spectrom., 2000, 35, 784–789 CrossRef CAS.
- J. V. Iribarne, P. J. Dziedzic and B. A. Thomson, Int. J. Mass Spectrom. Ion Phys., 1983, 50, 331–347 CrossRef CAS.
- B. A. Thomson and J. V. Iribarne, J. Chem. Phys., 1979, 71, 4451–4463 CrossRef CAS.
- J. V. Iribarne and B. A. Thomson, J. Chem. Phys., 1976, 64, 2287–2294 CrossRef CAS.
- V. Znamenskiy, I. Marginean and A. Vertes, J. Phys. Chem. A, 2003, 107, 7406–7412 CrossRef CAS.
- J. Sunner, G. Nicol and P. Kebarle, Anal. Chem., 1988, 60, 1300–1307 CrossRef CAS.
- J. Sunner, M. G. Ikonomou and P. Kebarle, Anal. Chem., 1988, 60, 1308–1313 CrossRef CAS.
- G. Nicol, J. Sunner and P. Kebarle, Int. J. Mass Spectrom. Ion Processes, 1988, 84, 135–155 CrossRef CAS.
- P. Kebarle and U. H. Verkerk, Mass Spectrom. Rev., 2009, 28, 898–917 CrossRef CAS PubMed.
- W. S. Law, R. Wang, B. Hu, C. Berchtold, L. Meier, H. W. Chen and R. Zenobi, Anal. Chem., 2010, 82, 4494–4500 CrossRef CAS PubMed.
- R. Wang, A. Gröhn, L. Zhu, R. Dietiker, K. Wegner, D. Günther and R. Zenobi, Anal. Bioanal. Chem., 2011, 402, 1–11 Search PubMed.
- J. López-Herrera, A. Barrero, A. Boucard, I. Loscertales and M. Márquez, J. Am. Soc. Mass Spectrom., 2004, 15, 253–259 CrossRef PubMed.
- J. D. Beauchamp and J. D. Pleil, in Biomarker Validation, Wiley-VCH Verlag GmbH & Co. KGaA, 2015, pp. 75–93, DOI:10.1002/9783527680658.ch5.
- P. Španěl and D. Smith, in Volatile Biomarkers, ed. A. Amann and D. Smith, Elsevier, Boston, 2013, pp. 48–76, DOI:10.1016/B978-0-44-462613-4.00004-0.
- R. S. Blake, P. S. Monks and A. M. Ellis, Chem. Rev., 2009, 109, 861–896 CrossRef CAS PubMed.
- D. Smith, P. Španěl, J. Herbig and J. Beauchamp, J. Breath Res., 2014, 8, 027101 CrossRef PubMed.
- P. Martínez-Lozano and J. Fernández de la Mora, Int. J. Mass Spectrom., 2007, 265, 68–72 CrossRef.
- H. Chen, A. Wortmann, W. Zhang and R. Zenobi, Angew. Chem., Int. Ed., 2007, 46, 580–583 CrossRef CAS PubMed.
- G. R. Johnson and L. Morawska, J. Aerosol Med. Pulm. Drug Delivery, 2009, 22, 229–237 CrossRef PubMed.
- W. Miekisch, J. K. Schubert and G. F. E. Noeldge-Schomburg, Clin. Chim. Acta, 2004, 347, 25–39 CrossRef CAS PubMed.
- I. Horváth, J. Hunt, P. J. Barnes, K. Alving, A. Antczak, E. Baraldi, G. Becher, W. J. C. van Beurden, M. Corradi, R. Dekhuijzen, R. A. Dweik, T. Dwyer, R. Effros, S. Erzurum, B. Gaston, C. Gessner, A. Greening, L. P. Ho, J. M. Hohlfeld, Q. Jöbsis, D. Laskowski, S. Loukides, D. Marlin, P. Montuschi, A. C. Olin, A. E. Redington, P. Reinhold, E. L. J. van Rensen, I. Rubinstein, P. Silkoff, K. Toren, G. Vass, C. Vogelberg, H. Wirtz, B. Balint, A. Blomberg, J. Freels, M. Goldman, J. Hunt, S. A. Kharitonov, F. Kelly, C. Lehmann, A. Lindstrom, R. Robbins, M. Rothe, H. J. Smith, W. R. Steinhaeusser, W. G. Teague and J. Vaughan, Eur. Respir. J., 2005, 26, 523–548 CrossRef PubMed.
- X. Li, P. Martinez-Lozano Sinues, R. Dallmann, L. Bregy, M. Hollmen, S. Proulx, S. A. Brown, M. Detmar, M. Kohler and R. Zenobi, Angew. Chem., Int. Ed., 2015, 54, 7815–7818 CrossRef CAS PubMed.
- P. Martinez-Lozano Sinues, M. Kohler, S. A. Brown, R. Zenobi and R. Dallmann, Chem. Commun., 2017, 53, 2264–2267 RSC.
- H. P. H. Arp, S. T. J. Droge, S. Endo, W. Giger, K.-U. Goss, S. B. Hawthorne, S. A. Mabury, P. Mayer, M. S. McLachlan, J. F. Pankow, R. P. Schwarzenbach, F. Wania and B. Xing, Environ. Sci. Technol., 2010, 44, 4400–4401 CrossRef CAS PubMed.
- D. García-Gómez, P. Martínez-Lozano Sinues, C. Barrios-Collado, G. Vidal-De-Miguel, M. Gaugg and R. Zenobi, Anal. Chem., 2015, 87, 3087–3093 CrossRef PubMed.
- P. Martinez-Lozano and J. Fernandez de la Mora, Anal. Chem., 2008, 80, 8210–8215 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |