
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Improvement in the single and simultaneous generation of As, Bi, Sb and Se hydrides using a vapor generation accessory (VGA) coupled to axially viewed inductively coupled plasma optical emission spectrometry (ICP OES)
Maja
Welna
 *,
Anna
Szymczycha-Madeja
and
Pawel
Pohl
*,
Anna
Szymczycha-Madeja
and
Pawel
Pohl
Wroclaw University of Technology, Faculty of Chemistry, Division of Analytical Chemistry and Chemical Metallurgy, Wybrzeze Wyspianskiego 27, 50-370 Wroclaw, Poland. E-mail: maja.welnal@pwr.edu.pl
First published on 6th January 2017
Abstract
Continuous flow (CF) hydride generation (HG) using a vapor generation accessory (VGA) coupled to a simultaneous axially viewed inductively coupled plasma optical emission spectrometer (ICP OES) for the ultrasensitive measurement of As, Bi, Sb and Se was investigated. Hydrides were generated in a gas–liquid phase separation system by mixing acidified aqueous sample, additional HCl and reductant (NaBH4) solutions on-line. Instrumental (emission line wavelength, RF power, and flow rates of the sample and waste solutions, along with the sample read delay and wash times) and chemical (concentrations of NaBH4 and HCl in the sample (S) and additional acid (A) solutions) variables affecting the effectiveness of HG were examined to achieve the optimum conditions for single- and multi-element analysis. The limitations of the HG reaction due to potential interference effects between the hydride-forming elements were identified. Satisfactorily, As and Sb hydrides could be generated in a wide range of HCl concentrations either in the S solution or the A solution, i.e. 1–6 (S) or 5–10 (A) mol L−1 HCl. For Bi and Se, HG depended strongly on the acidity and the optimum HCl concentrations for both elements were completely opposite. HG for Bi was the best at the lowest acidity (1 (S) and 5 (A) mol L−1 HCl), whereas the maximum Se responses were acquired using the highest HCl concentrations (6 (S) and 10 (A) mol L−1 HCl). Compromised conditions for the simultaneous measurement of As + Bi + Sb or As + Sb + Se, without any adverse interactions between the elements, were successfully established with an analysis time of ∼2 min per sample. Under these conditions, the detectability of all the elements was improved greatly (2 orders of magnitude versus ICP OES). Linear concentration ranges (0–20 ng g−1), detection limits from 0.027–0.099 ng g−1 and a precision better than 2% (as RSD) were achieved.
Introduction
Hydride generation (HG) in atomic spectrometry is an analyte introduction technique commonly used for detecting very low levels of hydride-forming elements, typically As, Bi, Sb, Pb, Sn and Se. Reduction of these elements to volatile hydrides in acidic media (mainly HCl) using sodium tetrahydroborate (NaBH4), gas–liquid phase separation, collection of the hydrides and transportation of the evolved gases into an excitation source or an atomization cell increases the sensitivity of the spectrometric methods remarkably as compared to conventional sample introduction by pneumatic nebulization (PN).1,2 Traditional HG is also commonly utilized in the case of Hg, which is converted into a vapor (CV) under the same reaction conditions.3Although a great majority of papers dealing with HG are devoted to its application in combination with atomic absorption spectrometry (AAS)4–6 or atomic fluorescence spectrometry (AFS),7–9 growing attention is being paid to the convenience of HG coupled with different excitation sources for optical emission spectrometry (OES) techniques, e.g. axially and radially viewed inductively coupled plasma (ICP) and microwave-induced plasma (MIP), due to the analytical performance of these techniques in the measurement of hydride-forming elements. Besides the obvious benefits, including separation of the hydride-forming elements from the rest of the sample matrix, very high transport efficiency and enrichment of the analytes, which improve the measurement sensitivities and lower the limits of detection (LODs), a distinctive feature of combining HG with detection by ICP OES comes from the possibility of simultaneously detecting several elements at the same time.10–12 However, it is important to realize that the simultaneous detection of different hydride-forming elements is not an easy task, and several factors that affect the formation of certain hydrides have to be considered. Because of differences in the chemical reaction conditions required for the effective formation of different hydrides, hydride-forming elements are usually detected individually,13–18 with studies concerning As only dominating. In the case of multi-element measurements, compromised conditions that are adequate for every element must be found, as reported in some recent studies, i.e. As + Hg + Sb + Se + Sn,10 As + Bi + Se + Sn + Te,11 As + Bi + Sb + Se,12 As + Bi + Sb,19 As + Se,20 As + Sb21,22 and As + Sb + Se.23 Different instrumental/operational (RF power,10,23 sample,10,20,21,23 HCl20 and NaBH411,21 solution flow rates, Ar carrier flow rate,10,11,20,22 washing/read delay time,20 and choice of analytical line wavelengths11,21) and chemical (concentration of HCl10–12,19–22 and NaBH410–12,22) variables must be examined to ensure the finest sensitivity and stable operation of the ICP excitation source. Spectral interferences between the hydride-forming elements should also be investigated.10
The HG reaction–separation systems used for ICP OES are based on the continuous flow (CF) and flow-injection (FI) manifolds. Usually, commercial assemblies (e.g. P.S. Analytical,12 PerkinElmer,21,22 Thermo Corporation T-PHD10) with or without U-shaped gas–liquid phase separators are used. Home-made HG manifolds with U-type separators have also been applied.11 Briefly, in CF systems, acidified sample and reducing agent solutions are continuously pumped with the aid of peristaltic pumps into a T-junction, a Y-piece or a reaction coil/loop, where the reagents are mixed and reacted immediately. To mix three separate streams of reagents, two T-junctions are used, merging first the sample solution with the acid solution, prior to the reaction with the reducing agent solution. In FI systems, an injection valve is employed to introduce a sample portion into a stream of carrier acid solution.12 The reaction mixture containing the volatile hydrides, other gaseous by-products (H2, water vapor) and the post-reaction solution is then introduced into a gas–liquid phase separator to separate the volatile species from the liquid phase. A stream of carrier Ar can be added to the reaction mixture to strip the hydrides into the gas phase before they enter the gas–liquid phase separator.21 The gaseous products of the HG reaction are then transported directly to the ICP torch in the stream of carrier Ar, while the spent solution is removed via the U-tube of the gas–liquid phase separator11,12 or drained out using a peristaltic pump.21,22 Non-standard constructions utilizing cyclonic spray chambers and nebulizers for the HG reaction have also been applied.19,20 For example, a commercial Jobin Yvon concomitant metal analyzer (CMA) has been used.20 In this manifold, the sample, acid and reductant solutions are delivered separately to a small cavity at the bottom of the cyclonic spray chamber. The separated gaseous products are swept by a carrier Ar stream, introduced through the nebulizer gas inlet, and transported to the plasma torch. The post-reaction solution is drained out with the aid of another peristaltic pump. Alternatively, a non-commercial (home-made), modified cyclone spray chamber (that acts only as a phase separator) and a concentric pneumatic nebulizer can be used.19 In this approach, the reagents are mixed in two Y-junctions, followed by introduction of the resulting reaction mixture through a reaction coil to the U-type end part of the chamber to separate and transport the gaseous reaction products in the stream of nebulizing Ar into the plasma torch. The spent solution is taken out via drains. Recently, a system of unique design for direct HG with a NOVA-2 dual-flow ultrasonic nebulizer (USN) has been reported.23 In this system, the acidified sample and reductant solutions are nebulized via the USN, which has two sample channels, into a cyclonic spray chamber, where the HG reaction takes place.
Considering the commercial assemblies for HG, a Varian/Agilent vapor generation accessory, model VGA-77, featuring CF-HG with gas–liquid phase separation, has also been proposed for (ultra)sensitive detection of hydride-forming elements. This system has the advantage that it can be readily combined with AAS and ICP OES. It is equipped with a pump unit, a reagent module with tubing, adapters and mixing (T-shaped) connectors, a reaction coil, and a glass gas–liquid phase separator. It works in combination with two streams of inert gas, i.e. Ar for ICP OES, added firstly to the reaction coil to strip the evolved gaseous hydrides from the reaction mixture, and secondly introduced into the separator for additional drying of the gas phase from water vapor. The sample and additional acid, e.g. HCl, solutions are mixed before being merged with the reductant solution. The heterogeneous reaction mixture is then introduced into the gas–liquid phase separator to separate the volatile species from the liquid. With respect to ICP OES, the resulting gaseous products are transferred into the plasma through the nebulizer and the spray chamber of the ICP instrument. At present, it seems that studies on HG with the VGA-77 system combined with AAS and ICP OES are not extensively documented, and refer only to single-element analysis, i.e. As24,26,27 and Se28 by HG-AAS, and As,25 Se29 and Hg3 by HG/CV-ICP OES. Therefore, this paper fills the gap related to multi-element analysis, and reports the HG of As, Bi, Sb and Se using VGA-77P in combination with an axially viewed ICP OES spectrometer. The applicability and suitability of the VGA-77P system for ultrasensitive single- and multi-element measurements of hydride-forming elements were examined in detail. The effect of the operating and chemical parameters, which influence the accuracy of the HG-ICP OES measurements, were evaluated. The optimum (compromised) experimental conditions for multi-element measurements of hydride-forming elements, along with the respective analytical figures of merit, were assessed.
Experimental
Reagents and solutions
All chemicals were of analytical grade. A stock standard solution (1000 μg mL−1) of As(III) was prepared from its salt, i.e. NaAsO2 (Sigma-Aldrich, St. Louis, MO, USA). In the case of Bi(III), Sb(III) and Se(IV), Merck (1000 μg mL−1) ICP standard solutions (Merck, Darmstadt, KGaA, Germany) were used. A 37% (v/v) HCl solution from Sigma-Aldrich was used to acidify the sample and standard solutions, and to prepare the additional acid solution for the HG reaction. Working standard solutions were obtained by appropriate stepwise dilution of stock standard solutions. For analysis, single- and multi-element standard solutions (up to 20 ng g−1 for the calibration curves, and 10 or 100 ng g−1 for the optimization investigations) in 1, 3 or 6 mol L−1 HCl were freshly prepared before each set of measurements. Blank sample solutions, i.e. 1, 3 or 6 mol L−1 HCl, were also made, and were considered in the final results. The additional HCl solution for the HG reaction was also prepared and, depending on the experimental conditions, the acid concentration in this solution was varied from 1 to 10 mol L−1. A 0.6% (m/v) solution of NaBH4 (Sigma-Aldrich) was used for the HG reaction. For investigation of the effect of the NaBH4 concentration on the analyte response, solutions containing 0.3, 0.75 and 1.0% (m/v) of the reducing agent in 0.5% (m/v) NaOH were also applied. All of the reductant solutions were prepared daily by dissolving appropriate amounts of powdered NaBH4 in a 0.5% (m/v) NaOH solution for stabilization, and filtered (0.45 μm) before being used. De-ionized water (18.3 MΩ cm) from an EASYpure system (Barnstead, Model D7033) was used in all preparations.Instrumentation
An Agilent bench-top simultaneous optical emission Ar-ICP spectrometer (model 720) with the torch in the axial alignment, in conjunction with an Agilent VGA-77P system (vapor generation accessory), was used in the study. The instrument was equipped with a 4-channel peristaltic pump, a high-resolution Echelle-type polychromator with temperature-controlled optics and a VistaChip II CCD detector. A standard, one-piece, low-flow, extended quartz torch with an injector tube (2.4 mm ID) was used to sustain and operate the plasma. The torch was combined with a glass single-pass cyclonic spray chamber. A high-tech engineering polymer (PFA and PEEK) OneNeb nebulizer was mounted into the spray chamber and applied to introduce the volatile hydrides produced in the VGA-77P system by HG. The same spray chamber and the OneNeb nebulizer were used for pneumatic nebulization (PN) of the sample solutions, to compare the analyte response for both sample introduction systems. The operating parameters for HG using the VGA-77P system and ICP OES detection are given in Table 1. A fitted background mode with 7 points per line profile was applied for background correction. The background corrected intensities (net intensities) of the analytical lines were used in this study.| a PN: pneumatic nebulization with a OneNeb nebulizer/cyclonic spray chamber system; HG: hydride generation with a VGA-77P system. b ID: inner diameter. c I: atomic line. | |
|---|---|
| ICP OES detection | |
| RF power/kW | 1.0–1.2 |
| Ar gas flow rates/L min−1 | Plasma: 15.0 |
| Auxiliary: 1.5 | |
| Nebulizing: 0.75 | |
| Stabilization time/s | 15 |
| Sample delay time/s | 30 (PN), 30–200 (HG) |
| Sample flow rate/mL min−1 | 0.75 (PN), 8.0 (HG) |
| Rinse time/s | 10 |
| Replicate time/s | 1 |
| Number of replicates | 3 (PN), 5 (HG) |
| Instrument pump rate/rpm | 15 (PN), 30−45 (HG) |
| Pump tubingb | Sample/standard – black/black (0.76 mm ID) |
| Wastes: | |
| (PN) blue/blue (1.65 mm ID) | |
| (HG) purple/black (2.29 mm ID) | |
| Fast pump | On (PN), off (HG) |
| Analytical line wavelengthsc/nm | As I 188.9; Bi I 223.1; Sb I 206.8; Se I 196.1 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|
| Hydride generation | |
| Inert gas | Ar |
| Pump tubingb | Sample/standard – purple/white (2.79 mm ID) |
| Additional acid – black/black (0.76 mm ID) | |
| Reductant – black/black (0.76 mm ID) | |
| Solution uptake rate/mL min−1 | Sample: 6.0–8.0 |
| Additional acid: 1.0 | |
| Reductant: 1.0 | |
|
|
| Reagents concentrations | |
| Reductant (R): NaBH4/% (m/v) | 0.3–1.0 (in 0.5% (m/v) NaOH) |
| Sample (S): acidified with HCl/mol L−1 | 1–6 |
| Additional acid (A): HCl/mol L−1 | 5–10 |
Hydride generation
As, Bi, Sb and Se hydrides were generated in a CF mode using the VGA-77P system, working in a gas–liquid phase separation configuration. The reagents, i.e. the sample (S), additional acid (A) and reductant (R) solutions, were simultaneously delivered in three separate streams into the system with the aid of the in-built VGA-77P peristaltic pump unit. The peristaltic pump maintained a constant flow of the solutions, determined by the diameter of the pump tubing. The uptake rates of the S, A and R solutions used met the established specifications for the VGA-77P system supplied by the manufacturer.30 The S and A solutions were merged first in the 1st T-junction, and then the resulting sample solution was mixed with the R solution in the 2nd T-junction. The heterogeneous reaction mixture then immediately entered a reaction coil, facilitated by a carrier Ar stream, and then a glass gas–liquid phase separator, where the evolved hydrides were separated from the liquid into the gas phase. At this point, the 2nd stream of carrier Ar was introduced into the separator to enhance the separation of the hydrides and the drying of the gas phase from water vapor. Finally, Ar enriched with the hydrides passed out of the gas–liquid phase separator and was transported into the plasma through the nebulizer sample inlet. A nebulizing Ar stream, introduced through the gas inlet of the nebulizer, was also used. The post-reaction solutions from the separator were drained using the peristaltic pump of the ICP spectrometer. A schematic diagram of the VGA-77P/ICP OES combined system is shown in Fig. 1. | ||
| Fig. 1 A scheme of the HG-ICP OES system. | ||
Results and discussion
To investigate the feasibility of using the VGA-77P system coupled to the ICP OES spectrometer, the effect of the experimental conditions on the effective HG of As, Bi, Sb and Se using a NaBH4–HCl mixture was studied. The response of the analytes, i.e. the background corrected intensities of their emission lines, was considered and single- and multi-element standard solutions containing As(III), Bi(III), Sb(III) and Se(IV) were used, since these forms guarantee quantitative generation of the respective hydrides. Although recommendations are given by the manufacturer for single-element analysis using the VGA-77P system, the influence of the operative and chemical factors on the analytical performance of the HG-ICP OES method was critically examined and the conditions providing the best results were optimized. A one-at-a-time method, where one parameter was changed while keeping the others constant, was employed for this aim. The optimized parameters included: (1) the NaBH4–HCl reducing conditions, in terms of the NaBH4 concentration in the R solution, as well as the HCl concentrations in the S and A solutions delivered into the VGA-77P system; (2) the S solution flow rate; (3) the flow rate of the waste solution removed from the separator; (4) the sample read delay time (DT) and the wash time (WT) before the measurement run; and (5) the interference effects between the hydride-forming elements of interest under the selected reaction conditions. The best experimental conditions for single- and multi-element detection were discussed.Emission line selection
The commonly applied emission lines of the studied hydride-forming elements were first tested using PN sample introduction. The background corrected (net) intensity of the emission lines (Inet), their signal-to-background ratio (SBR), the repeatability of the measurements, and the LOD of the analyte were the selection criteria. A multi-element working standard solution of As, Bi, Sb and Se (0.5 μg g−1), acidified to 0.5 mol L−1 HCl, was measured to compare the Inet of the emission lines. Standard solutions containing 0.05, 0.10, 0.25, 0.50, 1.00, 2.00 and 5.00 μg g−1 of As, Bi, Sb and Se were used for calibration; the slopes of the resulting calibration curves were measured for calculation of the LOD (ng g−1), which corresponds to three times the standard deviation (3 × SD) of 10 consecutive measurements of a blank sample. The repeatability of the analyte signals was expressed as the relative standard deviation (% RSD) of five replicate measurements of the Inet of the respective emission lines for a 0.50 μg g−1 standard solution of As, Bi, Sb and Se. In addition, after calibration, all of the calibration standard solutions were treated as samples, and the closeness between the measured and the expected concentrations of As, Bi, Sb and Se was evaluated as the recovery of the expected concentration (% Crecovered). This was used to assess the valid concentration ranges for which the hydride-forming elements can be accurately determined.The results achieved for the different emission lines of As, Bi, Sb and Se, including the values of Inet, SBR, LOD and % RSD, are summarized in Table 2. In addition, the relative intensity (Irelative) values, calculated as the ratio between the measured Inet and the Inet of the emission line with the maximum signal (Inet,max), were given. The valid concentration ranges, for which the recoveries of the expected concentrations (% Crecovered) of the elements are quantitative, are also included for each emission line.
| Element | Wavelength/nm | I net (Irelative/%)b | SBRc | LODd/ng g−1 | RSDe/% | Valid concentration range/μg g−1 (Crecovered/%)f |
|---|---|---|---|---|---|---|
| a The analytical lines selected for the HG-ICP OES measurements are underlined; PN: pneumatic nebulization. b Average net intensity of the analytical line for three (n = 3) measurements with the relative intensity (Irelative) in brackets, which is calculated as the ratio between the measured intensity (Inet) and the intensity of the analytical line with the maximum signal (Inet,max). c Signal-to-background-ratio calculated as (I − Ib)/Ib, where I and Ib refer to the intensity of the analytical line (I) and the background intensity (Ib) in the vicinity of this line. d LOD: limit of detection calculated as 3 × SD/a, where SD is the standard deviation of ten (n = 10) measurements of a blank sample solution (0.5 mol L−1 HCl solution), and a is the sensitivity of the analytical line, equal to the slope of the calibration curve. e RSD: relative standard deviation for five (n = 5) measurements of a 0.5 μg g−1 standard solution. f Valid concentration range, for which the measured and expected concentrations of the elements show accurate closeness; accurateness is expressed as the recovery of the expected concentration (% Crecovered in brackets). g ND: not determined (recovery from 150–400%). | ||||||
| As |
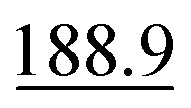
|
940 (100) | 9.3 | 3.5 | 2.4 | 0.05–5.0 (95–102) |
| 193.7 | 900 (96) | 8.5 | 3.3 | 2.1 | 0.05–5.0 (90–105) | |
| 197.3 | 470 (50) | 5.3 | 10 | 4.1 | 0.1–5.0 (87–103) | |
| 0.05 (56) | ||||||
| 228.8 | 640 (68) | 2.2 | 7.9 | 6.0 | 0.05–5.0 (93–104) | |
| 234.9 | 570 (61) | 0.82 | 15 | 3.1 | 0.25–5.0 (97–109) | |
| 0.05–0.1 (70–130) | ||||||
| Bi | 222.8 | 1160 (14) | 1.3 | 4.4 | 3.6 | 0.1–5.0 (86–99) |
| 0.05 (65) | ||||||

|
3150 (38) | 3.2 | 2.0 | 2.0 | 0.05–5.0 (94–100) | |
| 306.8 | 8320 (100) | 0.73 | 5.6 | 3.1 | 0.5–5.0 (99–115) | |
| 0.05–0.25 (ND ) | ||||||
| Sb |

|
1830 (100) | 8.7 | 3.0 | 2.2 | 0.05–5.0 (94–101) |
| 217.6 | 820 (45) | 6.5 | 4.8 | 3.6 | 0.05–5.0 (90–100) | |
| 231.1 | 980 (54) | 2.7 | 9.3 | 3.6 | 0.05–5.0 (95–112) | |
| Se |

|
860 (100) | 7.5 | 4.6 | 2.6 | 0.05–5.0 (91–100) |
| 203.9 | 490 (57) | 3.4 | 10 | 3.1 | 0.1–5.0 (90–105) | |
| 0.05 (76) | ||||||
In the case of As, it was found that the 188.9 and 193.7 nm emission lines behaved similarly, with slightly better analytical performance for the 188.9 nm emission line. For the remaining hydride-forming elements, the 223.1 (Bi), 206.8 (Sb) and 196.1 (Se) nm emission lines provided the highest and most sensitive response (as shown by the SBR), along with the lowest LODs, and the best repeatability (usually with an RSD within 2.0–2.6%, which is adequate for trace element analysis). When choosing the analytical line, it is also important to keep the noise as low as possible, hence the SBRs rather than the Inets of the emission lines were taken into account. Accordingly, among the compared Bi emission lines, the 306.8 nm line was the most intense, but it was also the noisiest, i.e. there was very high background intensity in the vicinity of this line. Additionally, an OH molecular band in the region of 306–312 nm was observed, which interfered with this line. Similarly, the As 197.3 nm emission line seemed to be better than the more sensitive, but noisy 228.8 nm emission line of this element. The recovery experiment showed that the favorable As, Bi, Sb and Se emission lines enabled the sensitivity required for reliable determination of these elements to be attained, i.e. they covered relatively wide concentration ranges. Some of the tested emission lines failed for very low concentrations of As, Bi, Sb and Se (non-quantitative recoveries were obtained), restricting the analytical range for which the analytes could be validly determined (see Table 2). Nevertheless, the calibration curves for all the tested emission lines of As, Bi, Sb and Se were linear up to 5.0 μg g−1 with correlation coefficients better than 0.999. In view of all these results obtained for PN, the As 188.9, Bi 223.1, Sb 206.8 and Se 196.1 nm emission lines were chosen for further HG studies.
Effects of the operative parameters
Before using the VGA-77P system, several operative parameters, including both the S and waste solution flow rates in addition to the DT and the WT, were regulated. The VGA-77P system was configured using the same conditions for all four hydride-forming elements studied. In these experiments, the A and R solutions, i.e. 5 mol L−1 HCl and 0.6% (m/v) NaBH4, respectively, were encountered and pumped at a constant flow rate of 1.0 mL min−1. A multi-element solution of As(III), Bi(III), Sb(III) and Se(IV), containing 100 ng g−1 of each element in 1 mol L−1 HCl, was used. Furthermore, the lowest multi-element calibration standard (20 ng g−1 of each element in 1 mol L−1 HCl) was also tested.To sum up, the optimal operative HG parameters were as follows: reagent solution flow rates: 8.0 mL min−1 (S) and 1.0 mL min−1 (A and R); waste solution flow rate: 30 rpm (as the ICP spectrometer pump speed); DT/WT: 80 s; RF power: 1.2 kW.
Estimation of the effect of the chemical parameters
The chemical conditions based on a combined NaBH4–HCl system were investigated to obtain the best yield of HG for As, Bi, Sb and Se (individually and in a mixture). | ||
| Fig. 2 Effect of acidity related to the HCl concentration in the sample (S) and additional acid (A) solutions during As (a), Bi (b), Sb (c) and Se (d) hydride generation with a fixed NaBH4 concentration (0.6%) in the reducing agent (R) solution. | ||
For As and Sb (Fig. 2a and c), irrespective of the acidity of the S solution, no significant difference in their signals was observed when varying the HCl concentration in the A solution, although 10 mol L−1 HCl yielded the highest responses for these elements. The differences between 5 and 10 mol L−1 HCl in the A solution were 6–13% (As) and 2–7% (Sb). On the other hand, the HCl concentration in the S solution strongly influenced the As and Sb responses. An increase in the acidity of the S solution up to 6 mol L−1 HCl caused growth in their signals by about 45%. The HCl concentration was extremely critical in the case of Se and Bi. The Se signals (Fig. 2d) increased considerably with increasing HCl concentration in the S solution. The highest signal improvement was obtained when HCl was present in the solutions at 6 mol L−1 (S) and 10 mol L−1 (A). Under these conditions, the Se signal was enhanced 6.5-fold as compared to that achieved for 1 mol L−1 HCl (S) and 5 mol L−1 HCl (A). In contrast, generation of the Bi hydride (Fig. 2b) was most effective at relatively low acidity of the solutions, i.e. 1 mol L−1 HCl (S) and 5 mol L−1 (A). With higher acidification of the S solution, the response for Bi started to decrease. For example, the presence of 3 mol L−1 HCl in the S solution led to a reduction of the Bi signal of 1.6 times, while 6 mol L−1 suppressed it totally. The effect of the HCl concentration in the A solution was generally less critical. Above 5 mol L−1 HCl, the responses for Bi were, however, ∼20% lower.
In summary, the optimal chemical conditions for the individual determination of As, Bi, Sb and Se by HG using 0.6% NaBH4 (R) were as follows:
| As(III): 6 mol L−1 (S) and 10 mol L−1 HCl (A) |
| Bi(III): 1 mol L−1 (S) and 5 mol L−1 HCl (A) |
| Sb(III): 6 mol L−1 (S) and 10 mol L−1 HCl (A) |
| Se(IV): 6 mol L−1 (S) and 10 mol L−1 HCl (A) |
It was established that the optimum HCl concentrations for carrying out effective HG for Bi and Se were opposed, which could restrict the simultaneous detection of Bi and Se. The best performance of the HG reaction for Bi took place at low HCl concentrations, while in the case of Se, much greater HCl concentrations were required. It should be noted that under the preferable acid conditions for Se, the Bi signal could not be detected at all. In contrast, under the favorable HG conditions for Bi, the Se signal could be measured. The As and Sb hydrides could be generated in a wide HCl concentration range, importantly including the conditions (both in the S and A solutions) preferable for Bi and Se.
Considering the HG reaction requirements for the studied hydride-forming elements, it appears that in the VGA-77P system employed here, the optimum conditions for As, Sb and Se are the same. Therefore, it was expected that simultaneous detection of these elements should be possible. Based on the results established for the individual elements, the following HG conditions for multi-element analysis were selected:
| I for As(III) + Bi(III) + Sb(III): HCl (S 1 mol L−1, A 5 mol L−1)–NaBH4 (0.6%) |
| II for As(III) + Sb(III) + Se(IV): HCl (S 6 mol L−1, A 10 mol L−1)–NaBH4 (0.6%) |
| III for As(III) + Bi(III) + Sb(III) + Se(IV): HCl (S 3 mol L−1, A 10 mol L−1)–NaBH4 (0.6%) |
Consequently, the selected compromised reaction conditions were used in further investigations.
Interference effects
For the simultaneous detection of As, Bi, Sb and Se in one solution under the compromised HG conditions, the possible inter-element (mutual) interferences during the measurements were initially analyzed. As suggested in the manual of the VGA-77P system,30 loss of the analyte signals due to mutual interference for As, Bi, Sb and Se can vary between 10–50%, being the strongest (>50%) if the Bi hydride is formed in the presence of Se.In reference to the activity of the analytes in the reaction with the 0.6% NaBH4 solution, detection of Bi and Se was found to be complicated when the compromised conditions III were used. Unlike in the case of the individual detection of Bi and Se under their best HG reaction conditions, the response for Bi was decreased considerably (∼45%). For Se, the changes in the analyte signal were minor (∼10% decrease). However, the repeatability of the measurements was reduced. The signals for As and Sb were practically identical to those obtained when the elements were examined alone at the selected HG reaction conditions (compromised conditions III). Also at a lower HCl concentration in the A solution (5 mol L−1), the Bi response was reduced. Finally, lowering the acidification in the S solution to 1 mol L−1 HCl was found to be an alternative for the determination of As + Bi + Sb + Se, but it was only satisfactory for higher Se concentrations in the mixture (≥20 ng g−1). Actually, comparing the results obtained for Se using HG and PN, it can be concluded that in the case of higher concentrations of this element, PN can be used for sample introduction, as the resulting response for Se is similar to that obtained using HG (at the same analyte level). In addition, using high concentrations of Se for HG caused contamination of the HG system, and hence, longer washing times were necessary to clean the system. As a result, the compromised HG reaction conditions for which the Bi and Se hydrides can be simultaneously generated cannot be easily predicted and evaluated. Due to all the difficulties mentioned above, the compromised conditions III were excluded and only the compromised conditions I and II for simultaneous detection of As + Bi + Sb and As + Sb + Se, respectively, were considered in the mutual interference study.
The mutual interference effects were expressed as the relative intensity (Irelative), i.e. the ratio of the Inet acquired for a given analyte in a sample solution with and without the other hydride-elements. In addition, the effect of the presence of Se or Bi on the analyte signals, depending on the compromised HG reaction conditions used, was also checked. In all the studied mixtures, the hydride-forming elements were present at the same concentration. The results of these experiments are given in Table 3. It was established that when using the compromised conditions I and II, none of the studied elements was vulnerable to mutual interference. Changes in the responses of the analytes for single elements versus their mixtures were rather negligible, varying between 1–5% (As), 1–8% (Bi), 1–10% (Sb) and 1–8% (Se). This means that As + Bi + Sb or As + Sb + Se can be measured individually or in mixtures (additionally in the presence of Se or Bi) at the selected compromised HG reaction conditions, without any adverse affect on their signals.
| Compromised conditions I: HCla/mol L−1: S (1), A (5); NaBH4 (0.6%) | Compromised conditions II: HCla/mol L−1: S (6), A (10); NaBH4 (0.6%) | ||||||
|---|---|---|---|---|---|---|---|
| Sampleb | Measured signal | Sampleb | Measured signal | ||||
| As | Bi | Sb | As | Sb | Se | ||
| a A: additional HCl solution; S: sample solution. b At 10 ng g−1 of As(III), Bi(III), Sb(III) and Se(IV) (each element) in a respective mixture. | |||||||
| As + Sb | 0.99 | 0.99 | As + Sb | 1.00 | 1.01 | ||
| As + Bi | 0.97 | 0.96 | As + Bi | 0.99 | |||
| Bi + Sb | 0.99 | 1.04 | As + Sb + Se | 1.00 | 0.94 | 0.99 | |
| As + Bi + Sb | 1.02 | 0.94 | 1.06 | As + Sb + Bi | 1.01 | 1.02 | |
| As + Se | 0.98 | As + Se | 0.98 | ||||
| Bi + Se | 1.08 | Se + Bi | 1.03 | ||||
| Sb + Se | 1.10 | Sb + Se | 0.99 | ||||
| As + Bi + Sb + Se | 1.05 | 0.96 | 0.99 | As + Bi + Sb + Se | 1.05 | 1.07 | 1.08 |
Analytical performance
Considering the selected compromised HG reaction conditions (I and II), the analytical figures of merit for As, Bi, Sb and Se (considered individually and in mixtures) were assessed. This included the LODs, the linearity ranges and correlation coefficients (R2), and the repeatability of the measurements (see Table 4). Six-point calibration curves were constructed for standard solutions containing the analytes at concentrations in the range of 0–20 ng g−1. The LODs (ng g−1) were calculated following the 3 × SD criterion. Repeatability was expressed as the % RSD for five replicated measurements of the analytes at a concentration of 10 ng g−1.| HG conditionsb | Parameter | ||||
|---|---|---|---|---|---|
| S, HCl | A, HCl | R 2 | LOD/ng g−1 | RSDc/% | |
a ![[U with combining low line]](https://www.rsc.org/images/entities/char_0055_0332.gif) ![[n with combining low line]](https://www.rsc.org/images/entities/char_006e_0332.gif) ![[d with combining low line]](https://www.rsc.org/images/entities/char_0064_0332.gif) ![[e with combining low line]](https://www.rsc.org/images/entities/char_0065_0332.gif) ![[r with combining low line]](https://www.rsc.org/images/entities/char_0072_0332.gif) ![[l with combining low line]](https://www.rsc.org/images/entities/char_006c_0332.gif) ![[i with combining low line]](https://www.rsc.org/images/entities/char_0069_0332.gif) = favorable HG reaction conditions.
b A: additional HCl solution; S: sample solution; 0.6% NaBH4 as the reductant solution (R).
c At 10 ng g−1 of the analyte(s) and for five (n = 5) measurements. = favorable HG reaction conditions.
b A: additional HCl solution; S: sample solution; 0.6% NaBH4 as the reductant solution (R).
c At 10 ng g−1 of the analyte(s) and for five (n = 5) measurements.
|
|||||
| Single-element analysis | |||||
| As | 1–![[6 with combining low line]](https://www.rsc.org/images/entities/char_0036_0332.gif) mol L−1 mol L−1 |
5–![[1 with combining low line]](https://www.rsc.org/images/entities/char_0031_0332.gif) ![[0 with combining low line]](https://www.rsc.org/images/entities/char_0030_0332.gif) mol L−1 mol L−1 |
0.9952 | 0.046 | 0.82 |
| Bi | 1 mol L−1 | 5 mol L−1 | 0.9920 | 0.068 | 1.5 |
| Sb | 1– mol L−1 |
5– mol L−1 |
0.9972 | 0.027 | 1.0 |
| Se | 6 mol L−1 | 10 mol L−1 | 0.9974 | 0.087 | 1.7 |
|
|||||
| Multi-element analysis | |||||
| As + Bi + Sb | 1 mol L−1 | 5 mol L−1 | As 0.9954 | 0.087 | 2.0 |
| Bi 0.9961 | 0.061 | 1.9 | |||
| Sb 0.9969 | 0.038 | 1.9 | |||
| As + Sb + Se | 6 mol L−1 | 10 mol L−1 | As 0.9901 | 0.050 | 1.5 |
| Sb 0.9911 | 0.028 | 1.4 | |||
| Se 0.9908 | 0.099 | 2.6 | |||
In all cases, linearity of the response of the analytes up to 20 ng g−1 with high correlation coefficients was established (R2 > 0.99). The repeatability of the measurements of the analyte signals was good (usually within 1–2%), though it was better when the analytes were examined individually. The LODs achieved were very low, below <0.1 ng g−1 and about two orders of magnitude lower than those estimated using PN (As: 3.5 ng g−1; Bi: 2.0 ng g−1; Sb: 3.0 ng g−1 and Se: 4.6 ng g−1). As compared to PN, the respective sensitivity enhancements obtained for HG were 103- (As), 26- (Bi), 105- (Sb) and 61-fold (Se). Satisfactorily, the LODs for each element, calculated using single-element standard solutions, corresponded well with those obtained when mixed standard solutions were used. The LODs for As and Sb assessed under the best HG reaction conditions for Bi (compromised conditions I) were, however, 29–47% higher than those obtained in the presence of a higher acid concentration (compromised conditions II), i.e. the favorable reaction conditions for HG of these elements. Although it is difficult to compare the obtained results with the results reported by other studies, basically due to the different experimental conditions, it can be noted that the analytical feasibility of the VGA-77P system employed for As, Bi, Sb and Se measurements by HG was improved by 1–2 orders of magnitude as compared to other HG systems combined with ICP OES detection and radial viewing of the plasma.12,19,20,23 The LODs achieved here are also better than those reported for HG combined with axially viewed ICPs {Bi (0.16–0.3 ng mL−1),11,14 As (0.1–0.7 ng mL−1),11,21,22,25 Sb (0.23–150 ng mL−1)11,21,22 and Se (0.15–0.5 ng mL−1)11,29}. However, the LODs for As, Sb and Se are higher than those reported by Antolin et al.,10i.e. 0.006 (As), 0.008 (Sb) and 0.005 ng g−1 (Se). The RSDs of the analyte signals achieved here were within the ranges described by other authors (0.6–10%).10,11,14,21
Finally, the effect of adding Se and Bi to the mixtures of As, Bi and Sb (Se) and of As, Sb and Se (Bi) on the response of the analytes was verified. For this purpose, standard solutions (0–20 ng g−1) prepared in 1 and 6 mol L−1 HCl and containing each of the studied elements at the same concentration were measured at the compromised HG reaction conditions I and II. Comparable calibration curves were obtained. The variation in the slopes (as a ratio) of the respective calibration curves was quite low, i.e. for the compromised conditions I: As (0.3%), Bi (9.2%) and Sb (3.0%); for the compromised conditions II: As (5.2%), Sb (6.2%) and Se (6.8%). These results were close to those obtained when the single-element standard solutions were used. This proves again that mutual interference under these conditions can be avoided.
Conclusions
The applicability of the VGA-77P system combined with an axially viewed ICP OES spectrometer for the extremely sensitive and reliable measurement of As, Bi, Sb and Se by HG was demonstrated.The effectiveness of HG for detecting As, Bi, Sb and Se in the form of As(III), Bi(III), Sb(III) and Se(IV) was found to strongly depend on the experimental conditions, i.e. the flow rates of the sample and waste solutions, and the HCl concentration used for sample acidification and in the additional acid solution. This was particularly evident for Bi and Se. NaBH4 reduction of Bi(III) only took place in medium with a low HCl concentration, while at higher HCl concentrations the response for Se was greatly improved. In contrast, the As and Sb hydrides can be generated in a wide range of HCl concentrations, importantly covering the optimal conditions for Bi and Se. Single-element detection of As, Bi, Sb and Se in the form of As(III), Bi(III), Sb(III) and Se(IV), if conducted under appropriate reaction conditions, is rather simple. Unfortunately, due to the dependence of the generation of the Bi and Se hydrides on acidity, simultaneous detection of the studied hydride-forming elements seems to be more complicated, and compromised reaction conditions are strongly required. In addition, mutual interference during multi-element analysis can occur. For example, Bi can suffer from mutual interference, with Se seeming to be the most serious interfering element. The mutual interference between the other hydride-forming elements was rather negligible, and hence, these elements can be measured without any synergistic/antagonistic effects.
This is a preliminary study devoted to application of the VGA-77P system along with ICP OES prior to very sensitive single- and multi-element detection of trace amounts of As, Bi, Sb and Se by HG. As the hydride-forming elements exist in different oxidation states, and because HG is less effective for higher oxidation states, this study was carried out for the As(III), Bi(III), Sb(III) and Se(IV) forms. As a result of wet oxidative digestion of the samples, the higher oxidation states of As, Bi, Sb and Se have to be reduced to lower oxidation states before being detected by HG-ICP OES. For this reason, we are continuing our investigations, and our future work will be focused on developing undemanding procedures for the detection of total inorganic As, Bi, Sb and Se using the VGA-77P system along with ICP OES, with special attention to the pre-reduction step.
Acknowledgements
This study was funded by the National Science Centre of Poland (grant number 2013/09/B/NZ9/00122). In addition, a statutory activity subsidy from the Polish Ministry of Science and Higher Education for the Faculty of Chemistry, Wroclaw University of Technology is acknowledged.References
- J. Dedina and D. L. Tsalev, Hydride generation atomic absorption spectrometry, Wiley, New York, 1995 Search PubMed.
- P. Pohl, TrAC, Trends Anal. Chem., 2004, 23, 87–101 CrossRef CAS.
- J. Hellings, S. B. Adeloju and T. V. Verheyen, Microchem. J., 2013, 111, 62–66 CrossRef CAS.
- J. Moreda-Pineiro, C. Moscoso-Perez, M. Pineiro-Iglesias, P. Lopez-Mahia, S. Muniategui-Lorenzo, E. Fernandez-Fernandez and D. Prada-Rodriguez, Talanta, 2007, 71, 1834–1841 CrossRef CAS PubMed.
- O. Muniz-Naveiro, R. Dominguez-Gonzalez, A. Bermejo-Barrera, J. A. Cocho, J. M. Fraga and P. Bermejo-Barrera, Anal. Bioanal. Chem., 2005, 381, 1145–1151 CrossRef CAS PubMed.
- H. Matusiewicz and M. Mikołajczak, J. Anal. At. Spectrom., 2001, 16, 652–657 RSC.
- R. Miravet, J. F. Lopez-Sanchez and R. Rubio, Anal. Chim. Acta, 2004, 511, 295–302 CrossRef CAS.
- H. Tu and Y. He, Anal. Lett., 2015, 48, 1629–1637 CrossRef CAS.
- M. N. Matos Reyes, M. L. Cervera and M. de la Guardia, Anal. Bioanal. Chem., 2009, 394, 1557–1562 CrossRef CAS PubMed.
- R. Antolin, G. Borge, T. Posada, N. Etxebarria and J. C. Raposo, J. Alloys Compd., 2007, 427, 73–77 CrossRef CAS.
- H. Wiltsche, I. B. Brenner, G. Knapp and K. Prattes, J. Anal. At. Spectrom., 2007, 22, 1083–1088 RSC.
- M. Savio, P. H. Pacheco, L. D. Martinez, P. Smichowski and R. A. Gil, J. Anal. At. Spectrom., 2010, 25, 1343–1347 RSC.
- M. Welna, Aust. J. Chem., 2015, 68, 441–446 CrossRef CAS.
- E. Kilinc and F. Aydin, Anal. Lett., 2012, 45, 2623–2636 CrossRef CAS.
- A. A. Menegario, A. J. Silva, E. Pozzi, S. F. Durrant and C. H. Abreu Jr, Spectrochim. Acta, Part B, 2006, 61, 1074–1079 CrossRef.
- P. C. Hernandez, J. F. Tyson, P. C. Uden and D. Yates, J. Anal. At. Spectrom., 2007, 22, 298–304 RSC.
- M. Welna and A. Szymczycha-Madeja, Food Anal. Meth., 2014, 7, 127–134 CrossRef.
- M. Welna, J. Borkowska-Burnecka and M. Popko, Talanta, 2015, 144, 953–959 CrossRef CAS PubMed.
- M. Welna and W. Zyrnicki, Anal. Lett., 2011, 44, 942–953 CrossRef CAS.
- W. Da Luz Lopes, R. E. Santelli, E. P. Oliveira, M. De Fatima Batista de Carvahlo and M. A. Bezerra, Talanta, 2009, 79, 1276–1282 CrossRef PubMed.
- A. Ilander and A. Vaisanen, Anal. Chim. Acta, 2011, 689, 178–183 CrossRef CAS PubMed.
- E. Pena-Vazquez, A. Bermejo-Barrera and P. Bermejo-Barrera, J. Anal. At. Spectrom., 2005, 20, 1344–1349 RSC.
- A. Tyburska, K. Jankowski, A. Ramsza, E. Reszke, M. Strzelec and A. Andrzejczuk, J. Anal. At. Spectrom., 2010, 25, 210–214 RSC.
- P. K. Petrov, J. Majda Serafimovska, S. Arpadjan, D. L. Tsalev and T. Stafilov, Cent. Eur. J. Chem., 2008, 6, 216–221 CAS.
- P. Masson, T. Prunet and D. Orignac, Microchim. Acta, 2006, 154, 229–234 CrossRef CAS.
- K. Widziewicz and K. Loska, Arch. Environ. Prot., 2012, 38, 11–23 CAS.
- J. R. Behari and R. Prakash, Chemosphere, 2006, 63, 17–21 CrossRef CAS PubMed.
- D. Bohrer, E. Becker, P. C. Do Nascimento, M. Dessuy and L. M. De Carvalho, Food Chem., 2007, 104, 868–875 CrossRef CAS.
- P. Masson, D. Orignac and T. Prunet, Anal. Chim. Acta, 2005, 545, 79–84 CrossRef CAS.
- Agilent Vapor Generation Accessory VGA 77 User's Guide, Agilent Technologies, Inc.10th edn, Publication No 8510104700, April 2012 Search PubMed.
| This journal is © The Royal Society of Chemistry 2017 |