
PAHs in animal tissues – the analytics of PAHs in new reference materials and their homogeneity
Anna
Kiełbasa
and
Bogusław
Buszewski
*
Chair of Environmental Chemistry and Bioanalytics, Faculty of Chemistry, Nicolaus Copernicus University, 7 Gagarina St, Kuyavian-Pomeranian District, 87-100 Toruń, Poland. E-mail: bbusz@chem.umk.pl; Fax: +48 56 6114837; Tel: +48 56 6114308
First published on 1st December 2016
Abstract
From among a number of matrices, tissues are the most complex and difficult to prepare for the determination of analytes. Trace amounts of numerous substances in tissues must be determined with adequate precision and accuracy. The QuEChERS technique was used for the extraction of six PAHs from four kinds of tissues (porcine, avian, cod, and herring). HPLC coupled with a fluorescence detector and GC/MS were used for the final analysis. In each tissue, moisture and fat content were determined. The method recovery rate was 84–101% (SD = 0.06–0.12) for the pork tissue, 88–107% (SD = 2.6–6.9) for the cormorant tissue, and R = 89–102% (SD = 4.9–8.9) for the cod tissue. The HPLC/FLD determination of pyrene in the herring tissue was the most problematic. Pyrene was determined by GC/MS. The recovery was 93% (SD = 5.5). For three tissues (i.e. pork, cormorant, and fish), homogeneity and the certified values were determined. The above-mentioned tissues were candidates for new certified reference materials.
Introduction
Tissues are composed of an organized set of many cell types. There exist nerve, muscle, and epithelial tissues. The tissues consist of cells and the extracellular matrix. The cells vary in size, shape, and function. They range from 4 to 100 μm in size, but cells that are several centimeters long are also observed. The extracellular matrix is a liquid or gel composed of water, macromolecules, and/or formed structures (e.g. fibers). Proteins are in the form of peptides of defined amino acid sequences. Carbohydrates are in the form of monosaccharides, polysaccharides, glycoproteins, and complex polysaccharides as well as glycolipids. Lipids are generated as triglycerides, cholesterol, and phospholipids. Glycolipids are composed of polysaccharides and lipids. Glycolipids include cerebrosides and gangliosides. Tissues consist of water and inorganic and organic compounds. The water content is about 70%. The inorganic compounds are cations (such as Na+ and K+) and anions (i.e. HCO3−, HPO42−, SO42−, and Cl−). Glycoproteins, proteins, carbohydrates, lipids, and nucleic acid (DNA and RNA) are the organic components.1–5PAHs are xenobiotics. They are widely distributed in the environment in soil, water, air, flora, and fauna.6–10 They are subject to metabolic processes in the cell. These compounds are carcinogenic, teratogenic, embryotoxic, and mutagenic. They form derivatives covalently bonded to the DNA and negatively influence the cells' replication in the body. PAHs are introduced into the human body through the gastrointestinal tract, respiratory tract and skin. These xenobiotics permeate through protein-lipid membranes very easily. They accumulate in the fatty tissue of the body or the mammary glands.10–14 The process of xenobiotics' metabolism is shown in Fig. 1.
 | ||
| Fig. 1 The process of metabolism of xenobiotics in a living organism. | ||
The analysis of xenobiotics must be quantitative at a very low concentration (trace analysis) and complex matrix (tissue). For such an analysis, it becomes particularly important to ensure the quality and obtain reliable results. For this purpose, reference materials and certified reference materials are used. A certified reference material must meet four basic criteria: stability, homogeneity, analyte content determined with required precision and accuracy. Each certified reference material is accompanied by a certificate.15–22 A reference material must be essentially compatible with a real-life sample. Stability and homogeneity of reference material are significant parameters. Stability is monitored by the definite parameters of the material as a function of time.18,23 Homogeneity is determined for one batch of the material, and the results must be consistent with the values of the same parameters for a different material batch.17,23,24 The requirements for reference materials are collected in the ISO Guides.25 Reference materials are used for validation, instrument calibration, comparison of analytical methods, validation of an analyst's and laboratory's competence and skills, and uncertainty estimation and they are subject of interlaboratory analyses.16,18,22 Many international organizations e.g. the International Organization for Standardization (ISO), International Union of Pure and Applied Chemistry (IUPAC), Cooperation on International Traceability in Analytical Chemistry (CITAC), and the International Laboratory Accreditation Cooperation (ILAC) require the use of certified reference materials.22,25 Laboratories which want to apply for an accreditation certificate should use reference materials. When they base their operations on the same international standard, their analytical and calibration results should be regarded as equivalent.26
Demand for reference materials is still growing and their use is very wide. They should meet the needs of today's analysts, chemists and toxicologists. It is necessary to develop new reference materials with new analytes and matrices. The commercial reference materials for the analysis of PAHs in biological matrices are presented in the International Database for Certified Reference Materials.27
According to the Commission of the European Communities Regulation of 19 August 2011 (no. 835/2011) amending the Commission of the European Communities Regulation of 19 December 2006 (no. 1881/2006), benzo(a)pyrene is not a suitable marker for the occurrence and effects of carcinogenic PAHs in food. The most suitable indicator is the sum of four compounds such as benzo(a)pyrene, chrysene, benz(a)anthracene, and benzo(b)fluoranthene. Products containing the above-mentioned PAHs (one or more) exceeding the maximum levels should not be placed on the market. They are toxic and harmful for public health. The maximum level is set from 1.0 μg kg−1 (for infant products) to 35.0 μg kg−1 (for Bivalve molluscs). Raw fish and fishery products are contaminated with PAHs due to environmental pollution.28
There are many PAH extraction methods from various matrices. The most widely used ones include liquid–liquid extraction (LLE), solid phase extraction (SPE), and accelerated solvent extraction (ASE).7,9,29–35 PAHs were extracted with the use of Soxhlet extraction, sonication extraction, microwave-assisted extraction, dispersive liquid–liquid microextraction, membrane extraction, and solid-phase microextraction (SPME) with modification, e.g. direct immersion cold fiber (DI-CF) or headspace cold fiber (HS-CF).31,36–49 Another method is the QuEChERS technique (Quick Easy Cheap Effective Rugged and Safe). This method was used for the measurement of polycyclic aromatic hydrocarbons (PAHs) associated with particulate matter from ambient air or a combustion process,50 and biological matrices such as fish,51–53 wild and commercial mussels,54 sea urchin roe,55 oysters,56 shrimp,57 seafood,58 different types of meat,59–62 black, green, red and white tea,63 and milk.64
This article contains a description of PAH extraction from biological matrices using the modern extraction technique – QuEChERS. This is a very fast and easy method characterized by satisfactory results. This method was used for isolating PAHs from fish and avian tissue. This method was validated and used for the determination of analyte content in inter-laboratory studies and homogeneity for candidates for new reference materials.
Materials and methods
Reagents and materials
The certified standard of the six PAHs (pyrene, benzo(a)pyrene, benzo(b)fluoranthene, benzo(k)fluoranthene, benz(a)anthracene and indeno(1,2,3-cd)pyrene) in acetonitrile was supplied by LGC Standards. The concentration of each compound was declared as 10 ng mL−1. This solution was stored at −4 °C. Dichloromethane, hexane and acetonitrile were used. They were supplied by Avantor Performance Materials Poland S.A. They were of HPLC grade. The water used was obtained from a MilliQ system. Nitrogen was of a purity higher than 99%. The sample was filtered through syringe filters (PTFE, 13 mm diameter and 0.22 μm pore size).Instruments and equipment
A Moisture Analyzer (MA-35, Sartorius Poland Sp. z o.o.) was used for moisture analysis. QuEChERS (Extract Tubes and Dispersive SPE for fatty samples, Labstore Polska Sp. z o.o.) and an ultrasonic homogenizer type UW 2070 (Bandelin Electronic GmbH & Co. KG, Germany) were used for the extraction of PAHs. An accelerated solvent extraction (ASE 100, Dionex) was used for lipid determination.A liquid chromatograph with a fluorescent detector (Agilent Technologies 1100 Series and 1260 Infinity) was used. The chromatography was performed on a non-polar column YMC PAH (250 mm × 3.0 mm; 5 μm), purchased from Bujno Chemicals. The analytes were determined by gradient elution with an acetonitrile–water binary system and subsequent fluorescence detection set at the appropriate excitation and emission wavelengths. The following gradient program is recommended: 0–5 min, 50% acetonitrile; 5–20 min, 50–100% acetonitrile; 20–28 min, 100% acetonitrile; 28–32 min, 100–50% acetonitrile; and 32–45 min, 50% acetonitrile. The mobile phase flowed at the rate of 1 mL min−1. The column temperature was set to 30 °C. 20 μL of the extract was injected.
A gas chromatograph was fitted with a selective mass detector (Agilent Technologies 6890 N and 5975 Series, respectively) and a 30 m × 0.25 mm × 0.25 μm capillary column (ZB-5MS, Phenomenex). A GC oven was temperature-programmed for an initial temperature of 50 °C, maintained for 1 min, increased to 190 °C (20 °C min−1), maintained for 2 min, and then raised to 300 °C (8 °C min−1), and finally maintained for 6 min. The injector was set to 280 °C. A splitless injection mode was used in the analysis. An aliquot (1 μL) of the acetonitrile extract was injected. The helium carrier gas flow rate was 1.1 mL min−1. The source and quadrupole temperature were set to 300 °C and 150 °C, respectively. The results were obtained with MS in the selected ion monitoring (SIM) mode.
Tissue samples and their characteristics
Four animal tissue sample types were used in this study: herring (M-3 HerTis), cod (M-5 CodTis), cormorant (M-4 CormTis) and pork muscle (ERM-BB384; LGC Standard). The fish and cormorant samples (new reference materials) were supplied by the “MODAS” consortium. According to their documentation the herring was obtained from the North Sea, the cod was obtained from the Baltic Sea, and the cormorant was from the Czech Republic. The samples were stored at −4 °C. The pork muscle tissue was PAH-free. The herring, cod and cormorant tissues were natural and they were not fortified. The tissues were lyophilized, powdered and homogenized. The herring, cod and cormorant tissues were placed in closed dark-glass bottles with a lot number. In each tissue the lipid content and moisture were determined.In each of these tissues the lipid content was gravimetrically determined by the procedure described by Dodds et al.65 with modifications. A 500 mg portion of tissues was extracted using a Dionex ASE 100. The sample was carefully mixed with roast sand and placed in an extraction cell. Dichloromethane was used as a solvent. The extraction ran at 100 °C in two static cycles, 5 min each. Next, the solvent was removed under a nitrogen stream. The mass of the residue was measured to the nearest 0.1 mg. The moisture content was determined with the moisture analyzer. The 500 mg sample was placed on an aluminium weighing pan. The sample was dried at 105 °C to establish a constant weight. The results are shown in Table 1.
| Parameters | Cormorant tissue | Herring tissue | Cod tissue | Pork tissue |
|---|---|---|---|---|
| Lipid g/100 g sample (SD) | 17.2 (1.32) | 10.2 (0.34) | 3.84 (0.62) | 9.21 (0.18) |
| Moisture% (SD) | 6.01 (0.43) | 6.67 (0.21) | 10.3 (0.78) | 4.00 (0.57) |
Extraction of PAHs from tissues
One of the criteria of reference materials is to determine the content of the analytes, the so-called certified values, with adequate precision and accuracy using validated analytical methods. Therefore, the first step in research was to find a method of extraction of PAHs from tissues, which will be characterized by the best recovery and repeatability. For this purpose a certified material of pork muscle was used. The certified standard of the six PAHs was added to the 1.0 g sample. The final concentration was about 0.60 μg L−1. The mixture was stirred and left for 1 hour. The solvent was evaporated in a mild stream of nitrogen.The ultrasound-assisted extraction was the first method used. The sample was placed in a Teflon container and 10 mL of hexane were added. The extraction was carried out for 10 min with an 80% duty cycle. The applied tapered microtip ultrasonic probe was 6 mm (1/4-inch) thick. The process was repeated three times and a new portion of the solvent was used each time. The extracts were collected. The solvent was concentrated under a mild stream of nitrogen at a temperature not exceeding 30 °C. The final volume of each sample was 1 mL and the solvent used was acetonitrile. Prior to carrying out the chromatographic analysis, each sample was filtered by using Teflon syringe filters. In the first case, the extract was not purified. In the second case, the extract was purified with a PSA, C18EC and magnesium sulfate mixture. The last extract was purified using diatomite. The obtained results are shown in Table 2.
| Compound | Hexane | Hexane + PSA + C18EC + MgSO4 | Hexane + diatomite | |||
|---|---|---|---|---|---|---|
| Recovery% | SD | Recovery% | SD | Recovery% | SD | |
| Pyrene | 34 | 10 | 24 | 11 | 68 | 9.2 |
| Benz(a)anthracene | 40 | 5.6 | 34 | 7.2 | 54 | 8.4 |
| Benzo(b)fluoranthene | 51 | 4.2 | 56 | 4.6 | 62 | 6.5 |
| Benzo(k)fluoranthene | 50 | 7.8 | 56 | 5.9 | 61 | 5.1 |
| Benzo(a)pyrene | 51 | 6.9 | 57 | 8.2 | 61 | 5.8 |
| Indeno(1,2,3-cd)pyrene | 56 | 5.7 | 63 | 7.1 | 62 | 8.1 |
Another method applied was the QuEChERS technique (Fig. 2). A 1.0 g sample of each animal tissue was placed into a 50 mL extraction tube. 8 mL of acetonitrile was added to each of the tubes. The sample tubes were hand shaken vigorously for 1 min and then were shaken using a vortex shaker (1 min). Next, packed extraction salt containing 1.0 g sodium citrate, 0.5 g sodium hydrogen citrate sesquihydrate, 4 g magnesium sulfate and 1 g sodium chloride were added to the samples. The tubes were shaken in the same way as before. Later the samples were centrifuged at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 30 minutes. A 4.0–5.0 mL aliquot of the upper acetonitrile layer was transferred into a 15 mL dispersive SPE tubes. These tubes contained 150 mg PSA, 150 mg C18EC and 900 mg magnesium sulfate. After 2 minutes of shaking (1 min hand shake and 1 min vortex shake), the tubes were centrifuged in the same way as before. Next, 3.0–4.0 mL of each sample was concentrated in a mild stream of nitrogen at the temperature not exceeding 30 °C. The results of the PAH content of 0.60 μg L−1 for each of the six compounds are presented in Table 3.
000 rpm for 30 minutes. A 4.0–5.0 mL aliquot of the upper acetonitrile layer was transferred into a 15 mL dispersive SPE tubes. These tubes contained 150 mg PSA, 150 mg C18EC and 900 mg magnesium sulfate. After 2 minutes of shaking (1 min hand shake and 1 min vortex shake), the tubes were centrifuged in the same way as before. Next, 3.0–4.0 mL of each sample was concentrated in a mild stream of nitrogen at the temperature not exceeding 30 °C. The results of the PAH content of 0.60 μg L−1 for each of the six compounds are presented in Table 3.
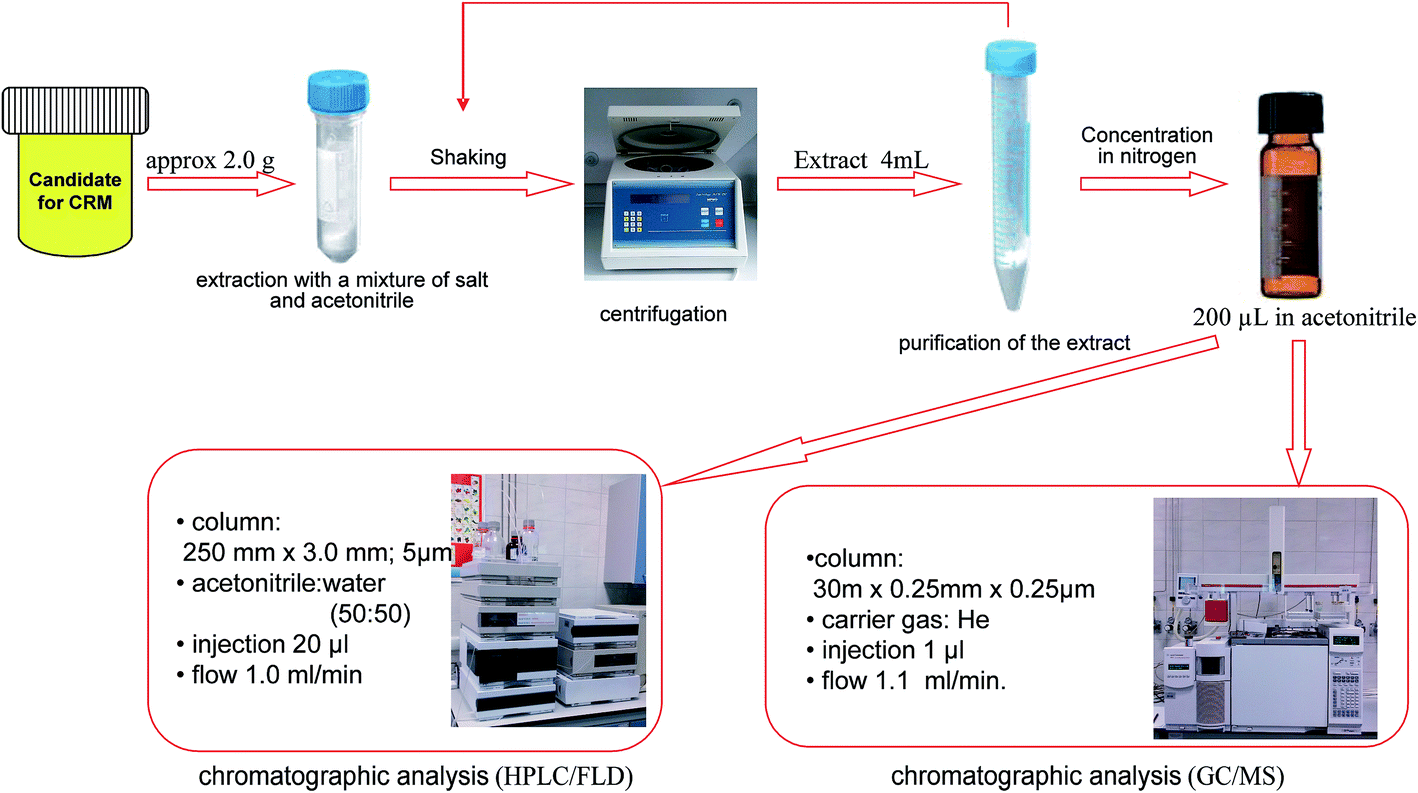 | ||
| Fig. 2 The procedure for the extraction of PAHs from animal tissues. | ||
| Compounds | Recovery% | SD |
|---|---|---|
| Pyrene | 101 | 0.10 |
| Benz(a)anthracene | 94 | 0.10 |
| Benzo(b)fluoranthene | 95 | 0.06 |
| Benzo(k)fluoranthene | 99 | 0.12 |
| Benzo(a)pyrene | 89 | 0.15 |
| Indeno(1,2,3-cd)pyrene | 84 | 0.10 |
Next, the recovery for lower concentration was determined. For this purpose, the QuEChERS technique was also used. The procedure of extraction was as described above. Table 4 presents the results obtained for the PAH content of 0.030 μg L−1 for each of the six compounds in pork muscle.
| Compounds | Recovery% | SD |
|---|---|---|
| Pyrene | 91 | 0.13 |
| Benz(a)anthracene | 102 | 0.57 |
| Benzo(b)fluoranthene | 104 | 0.48 |
| Benzo(k)fluoranthene | 102 | 0.87 |
| Benzo(a)pyrene | 105 | 0.88 |
| Indeno(1,2,3-cd)pyrene | 123 | 0.28 |
Finally, a 2.0 g sample of each tested animal tissue was extracted by the QuEChERS technique and the final volume of each sample was 0.2 mL.
Results and discussion
HPLC/FLD analysis
From the basic standard solution, seven working standard solutions were prepared by dilution to the concentrations of 0.25 (for benz(a)anthracene and benzo(a)pyrene), 0.5, 1.0, 2.5, 4.0, 5.0 and 10.0 μg L−1 (for others PAHs). For each of the compounds examined, an analytical curve was obtained. The detection and determination limits were also calculated. LOD and LOQ were calculated based on the standard deviation of a set of signals and the angle of inclination of the calibration curve. The results are shown in Table 5.| Compounds | LOD [ng g−1] | LOQ [ng g−1] | Equation of the calibration curve | Correlation coefficient |
|---|---|---|---|---|
| Pyrene | 0.040 | 0.12 | y = 2.05x + 0.125 | 0.9999 |
| Benz(a)anthracene | 0.032 | 0.096 | y = 3.15x + 0.045 | 1.0000 |
| Benzo(b)fluoranthene | 0.044 | 0.14 | y = 2.89x + 0.112 | 0.9999 |
| Benzo(k)fluoranthene | 0.040 | 0.12 | y = 4.97x + 0.150 | 0.9999 |
| Benzo(a)pyrene | 0.033 | 0.10 | y = 5.49x + 0.201 | 0.9999 |
| Indeno(1,2,3-cd)pyrene | 0.060 | 0.18 | y = 2.96x + 0.249 | 0.9998 |
GC/MS analysis
From the basic standard solution, five working standard solutions were prepared by dilution to the concentrations of 1.0, 2.5, 5.0, 10.0 and 20.0 μg L−1. The analytical curves as well as detection and determination limits were determined. LOD and LOQ were calculated based on the standard deviation of a set of signals and the angle of inclination of the calibration curve. The equations of analytical curves, the correlation coefficients, LOD, and LOQ for six PAHs are presented in Table 6.| Compounds | LOD [ng g−1] | LOQ [ng g−1] | Equation of the calibration curve | Correlation coefficient |
|---|---|---|---|---|
| a LOD and LOQ obtained by HPLC/FLD were significantly lower than values obtained by GC/MS. | ||||
| Pyrene | 0.28 | 0.82 |
y = 87429x + 4874 |
0.9992 |
| Benz(a)anthracene | 0.24 | 0.71 |
y = 48697x − 10538 |
0.9994 |
| Benzo(b)fluoranthene | 0.25 | 0.76 |
y = 48406x − 1342 |
0.9999 |
| Benzo(k)fluoranthene | 0.29 | 0.87 |
y = 62985x − 5063 |
0.9999 |
| Benzo(a)pyrene | 0.26 | 0.79 |
y = 28095x − 2266 |
0.9996 |
| Indeno(1,2,3-cd)pyrene | 0.30 | 0.91 |
y = 25101x − 9190 |
0.9996 |
PAHs in three animal tissues
A 2.0 g sample of each tissue (cormorant, herring, and cod) was taken. Next, 400 mL of a PAH solution was added to obtain a concentration of 10 μg L−1, then thoroughly mixed and extracted by the QuEChERS technique. The extracts were analyzed by HPLC/FLD. The results are shown in Table 7.| Compounds | Cormorant tissue | Herring tissue | Cod tissue | |||
|---|---|---|---|---|---|---|
| Recovery% | SD | Recovery% | SD | Recovery% | SD | |
| Pyrene | 88 | 2.6 | — | — | 102 | 7.2 |
| Benz(a)anthracene | 106 | 6.9 | 90 | 6.0 | 93 | 8.9 |
| Benzo(b)fluoranthene | 94 | 4.5 | 89 | 5.4 | 91 | 5.2 |
| Benzo(k)fluoranthene | 92 | 4.4 | 96 | 2.5 | 90 | 6.0 |
| Benzo(a)pyrene | 89 | 5.3 | 93 | 5.3 | 89 | 4.9 |
| Indeno(1,2,3-cd)pyrene | 107 | 2.8 | 101 | 8.8 | 99 | 7.0 |
Due to interfering compounds, the quantitative determination of pyrene in the herring tissue is possible only by using GC/MS. The recovery rate was 93% and repeatability was 5.5.
Reference materials for fish and cormorant tissues containing PAHs
The herring, cormorant, and cod tissues were candidates for new certified reference materials. The content of the analytes was determined by a validated method which was described above. This research allowed participation in the inter-laboratory tests, the results of which became the basis for determining the certified values of new certified reference materials. Next, the between-bottle (CBB) and within-bottle (CWB) homogeneities of these materials were determined. The content of PAHs in a single bottle – one batch of material – was the basis for the calculation of variation coefficient for within-bottle homogeneity. On the basis of the results for several bottles (different material batches), the variation coefficient for between-bottle homogeneity was calculated. The analyte content obtained for the three samples of each tissue in inter-laboratory tests, and the results of homogeneity are presented in Table 8. The results allow drawing the conclusion that the candidates for the new reference materials are characterized by a high homogeneity coefficient. The analyte content was determined for three bottles of each material. Each analysis was repeated three times.| Compounds | Analyte content [ng g−1] | SD | C WB% | C BB% | |
|---|---|---|---|---|---|
| HPLC/FLD | GC/MS | ||||
| Cormorant tissues | |||||
| Pyrene | 0.225 | <LOD | 0.022 | 23.8 | 17.8 |
| Benz(a)anthracene | 0.197 | <LOD | 0.018 | 20.2 | 11.2 |
|
|||||
| Herring tissue | |||||
| Pyrene | Not detected | 0.322 | 0.032 | 14.8 | 5.5 |
| Benz(a)anthracene | 0.0752 | <LOD | 0.015 | 9.4 | 5.5 |
| Benzo(b)fluoranthene | 0.0466 | <LOD | 0.014 | 12.3 | 4.0 |
|
|||||
| Cod tissue | |||||
| Pyrene | 0.271 | <LOD | 0.015 | 6.2 | 9.5 |
| Benz(a)anthracene | 0.063 | <LOD | 0.012 | 6.2 | 3.4 |
Pyrene in the herring tissue was the only compound whose contents can be determined quantitatively by GC/MS. Determination of benz(a)anthracene, benzo(b)fluoranthene, benzo(k)fluoranthene, benzo(a)pyrene and indeno(1,2,3-cd)pyrene was not possible because the obtained contents of these compounds were below the limit of detection.
In the calculation, ANOVA was used. The above-mentioned parameters were calculated by using the following equations:
 | (1) |
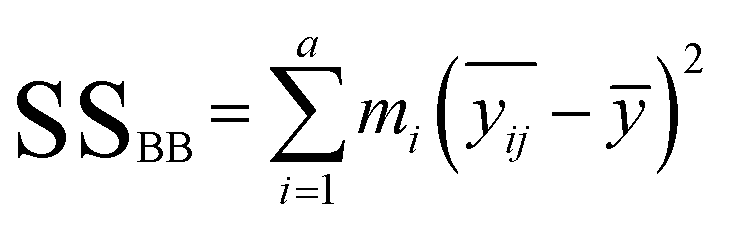 | (2) |
 – the average for the study group; mi – the number of samples in each group; ȳ – overall average. Eqn (3) and (4) refer to the calculation of the number of degrees of freedom (f) taking into account the number of samples (n), and the number of containers (a).
– the average for the study group; mi – the number of samples in each group; ȳ – overall average. Eqn (3) and (4) refer to the calculation of the number of degrees of freedom (f) taking into account the number of samples (n), and the number of containers (a).| fWB = n − a | (3) |
| fBB = a − 1 | (4) |
The between-bottle and within-bottle variations using eqn (5) and (6) were calculated:
 | (5) |
 | (6) |
Next, the standard deviation (s) was determined (eqn (7) and (8)). Finally, the variation coefficients (CV) for each type of homogeneity were determined (eqn (9) and (10)).
 | (7) |
 | (8) |
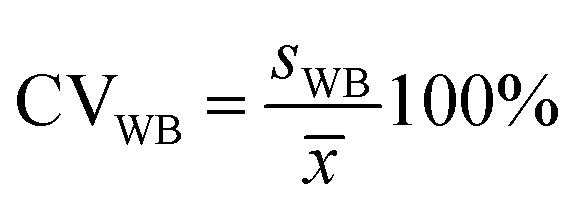 | (9) |
 | (10) |
![[x with combining macron]](https://www.rsc.org/images/entities/i_char_0078_0304.gif) – average of all results.
– average of all results.
Fig. 3 presents the chromatograms of the natural herring, cod, and cormorant tissues obtained with two different chromatographic techniques.
 | ||
| Fig. 3 The chromatograms of PAHs in herring (A), cod (B), and cormorant (C) tissue for HPLC/FLD and GC/MS analysis (1-pyrene, 2-benz(a)anthracene, 3-benzo(b)fluoranthene, 4-benzo(k)fluoranthene, 5-benzo(a)pyrene, and 6-indeno(1,2,3-cd)pyrene). | ||
Conclusion
The rapid development of new scientific disciplines (e.g. proteomics, metabolomics and genomics) involves the development of new reference materials. Developed and well-characterized reference materials can be successfully used to achieve the European Accreditation for testing laboratories. The new reference materials were prepared from natural matrices containing the components of interest rather than preparing them from synthetic mixtures. The prepared material can be used for the determination of trace amounts of PAHs isolated from tissues. The reference materials for tissues may be used by each laboratory for quality and food safety control and environmental analysis, as well as for veterinary purposes. The presented results are a very important part of the research on the preparation and certification of new reference materials. The results of this research were used to complete the creation of reference materials which can be used by different laboratories. The new materials were, in fact, adequately prepared for distribution to other laboratories. They were also accompanied by the appropriate certificate.Acknowledgements
This project was financed in the framework of the grant entitled: “Production and attestation of new types of reference materials crucial for achieving European accreditation for Polish industrial laboratories (MODAS)” provided by the National Center for Research and Development (no. INNOTECH-K1/IN1/43/158947/NCBR/12).References
- Y.-H. Xu, S. Barnes, Y. Sun and G. A. Grabowski, J. Lipid Res., 2010, 51, 1643–1675 CrossRef CAS PubMed.
- P. Chomczyński, BioTechniques, 1993, 15, 532–537 Search PubMed.
- B. Alberts, A. Johnson and J. Lewis, in Molecular Biology of the Cell, Garland Science, New York, 4th edn, 2002, ch. 19 Search PubMed.
- Y. Sato, Cell Struct. Funct., 2001, 26, 9–10 CrossRef CAS PubMed.
- S. G. Lazarowitz, Curr. Opin. Plant Biol., 1999, 2, 332–338 CrossRef CAS PubMed.
- M. Michel and B. Buszewski, Pol. J. Environ. Stud., 2008, 17, 305–319 CAS.
- S.-Y. Jung, J.-S. Park, M.-S. Chang, M.-S. Kim, S.-M. Lee, J.-H. Kim and Y.-Z. Chae, Food Sci. Biotechnol., 2013, 22, 241–248 CrossRef CAS.
- A. Bozcaarmutlu, C. Sapmaz, G. Kaleli, S. Turna and S. Yenisoy-Karakaş, Environ. Sci. Pollut. Res., 2015, 22, 2515–2525 CrossRef CAS PubMed.
- X. Zheng, Y. Yang, M. Liu, Y. Yu, J. L. Zhou and D. Li, Sci. Total Environ., 2016, 557–558, 688–696 CrossRef CAS PubMed.
- L. Singh, J. G. Varshney and T. Agarwal, Food Chem., 2016, 199, 768–781 CrossRef CAS PubMed.
- R. M. Gadzała and B. Buszewski, Pol. J. Environ. Stud., 1995, 4, 5–15 Search PubMed.
- E. Hodgson and R. L. Rose, J. Biochem. Mol. Toxicol., 2007, 21, 182–186 CrossRef CAS PubMed.
- C. J. Omiecinski, J. P. Vanden Heuvel, G. H. Perdew and J. M. Peters, Toxicol. Sci., 2011, 120, S49–S75 CrossRef CAS PubMed.
- B. Ramsauer, K. Sterz, H.-W. Hagedorn, J. Engl, G. Scherer, M. McEwan, G. Errington, J. Shepperd and F. Cheung, Anal. Bioanal. Chem., 2011, 399, 877–889 CrossRef CAS PubMed.
- J. Arunachalam, A. Bleise, R. S. Mahwar, P. Ramadevi and G. V. Iyengar, J. Food Compos. Anal., 2006, 19, 241–249 CrossRef.
- B. King, Accredit. Qual. Assur., 2003, 8, 429–433 CrossRef CAS.
- G. N. Kramer and J. Pauwels, Microchim. Acta, 1996, 123, 87–93 CrossRef CAS.
- K. Kupiec, P. Konieczka and J. Namieśnik, Crit. Rev. Anal. Chem., 2009, 39, 311–322 CrossRef CAS.
- T. P. J. Linsinger, J. Pauwels, A. M. H. van der Veen, H. Schimmel and A. Lamberty, Accredit. Qual. Assur., 2001, 6, 20–25 CrossRef CAS.
- M. Lipp, Accredit. Qual. Assur., 2004, 9, 539–542 CrossRef CAS.
- J. Pauwels and A. Lamberty, Fresenius. J. Anal. Chem., 2001, 370, 111–114 CrossRef CAS PubMed.
- B. Zygmunt and J. Namieśnik, Sci. Total Environ., 1999, 228, 243–257 CrossRef.
- P. McCarron, H. Emtebor, S. D. Giddings, E. Wright and M. A. Quilliam, Anal. Bioanal. Chem., 2011, 400, 847–858 CrossRef CAS PubMed.
- R. Dybczyński, B. Danko and H. Polkowska-Motrenko, Fresenius. J. Anal. Chem., 2001, 370, 126–130 CrossRef.
- ISO Guides, http://www.iso.org, accessed March 2016 Search PubMed.
- International Organization for Standardization, General Requirements for the Competence of Testing and Calibration Laboratories, ISO/IEC 17025, 2005, Geneva, Switzerland, 2005.
- http://www.comar.bam.de, accessed May 2016.
- http://eur-lex.europa.eu/legal-content/EN/ALL/?uri=CELEX%3A32006R1881, accessed August 2016.
- E. Kraleva, V. Karamfilov and G. Hibaum, Ecol. Chem. Eng. S, 2012, 19, 393–403 CAS.
- M. C. Hennion, J. Chromatogr. A, 1999, 856, 3–54 CrossRef CAS PubMed.
- R. Carabias-Martınez, E. Rodrıguez-Gonzalo, J. Domınguez-Álvarez and J. Hernández-Méndez, J. Chromatogr. A, 2000, 869, 451–461 CrossRef.
- J. Hassan, M. Izadi and S. Homayonnejad, J. Braz. Chem. Soc., 2013, 24, 639–644 CAS.
- O. Krüger, G. Christoph, U. Kalbe and W. Berger, Talanta, 2011, 85, 1428–1434 CrossRef PubMed.
- K. Kuosmanen, T. Hyötyläinen, K. Hartonen, J. A. Jönsson and M.-L. Riekkola, Anal. Bioanal. Chem., 2003, 375, 389–399 CrossRef CAS PubMed.
- M. Suranová, J. Semanová, B. Skláršová and P. Simko, Food Anal. Methods, 2015, 8, 1014–1020 CrossRef.
- N. C. Van de Merbel, J. J. Hageman and U. A. Th. Brinkman, J. Chromatogr., 1993, 634, 1–29 CrossRef CAS.
- K. D. Buchholtz and J. Pawliszyn, Anal. Chem., 1994, 66, 160–167 CrossRef.
- A. A. Boyd-Boland, S. Magdic and J. Pawliszyn, Analyst, 1996, 121, 929–937 RSC.
- M. H. Banitaba, S. S. Hosseiny Davarani and S. K. Movahed, J. Chromatogr. A, 2014, 1325, 23–30 CrossRef CAS PubMed.
- H. C. Menezes, B. P. Paulo, M. J. Nunes Paiva, S. M. Resende de Barcelos, D. F. Dias Macedo and Z. L. Cardeal, Microchem. J., 2015, 118, 272–277 CrossRef CAS.
- J. Guo, R. Jiang and J. Pawliszyn, J. Chromatogr. A, 2013, 1307, 66–72 CrossRef CAS PubMed.
- M. Kamankesh, A. Mohammadi, H. Hosseini and Z. M. Tehrani, Meat Sci., 2015, 103, 61–67 CrossRef CAS PubMed.
- S. Hwang and T. J. Cutright, Environ. Int., 2004, 30, 151–158 CrossRef CAS PubMed.
- T. F. Guerin, J. Environ. Monit., 1999, 1, 63–67 RSC.
- N. Ré, V. M. F. Kataoka, C. A. L. Cardoso, G. B. Alcantara and J. B. G. De Souza, Arch. Environ. Contam. Toxicol., 2015, 69, 69–80 CrossRef PubMed.
- A. Retnam, M. P. Zakaria, H. Juahir, A. Z. Aris, M. A. Zali and M. F. Kasim, Mar. Pollut. Bull., 2013, 69, 55–66 CrossRef CAS PubMed.
- I. S. Grover, R. Sharma, S. Singh and B. Pal, Environ. Monit. Assess., 2013, 185, 6459–6463 CrossRef CAS PubMed.
- M. J. Nieva-Cano, S. Rubio-Barroso and M. J. Santos-Delgado, Analyst, 2001, 126, 1326–1331 RSC.
- J. L. Santos, I. Aparicio and E. Alonso, Anal. Chim. Acta, 2007, 605, 102–109 CrossRef CAS PubMed.
- A. Albinet, S. Tomaz and F. Lestremau, Sci. Total Environ., 2013, 450–451, 31–38 CrossRef CAS PubMed.
- M. J. Ramalhosa, P. Paíga, S. Morais, C. Delerue-Matos and M. B. Prior Pinto Oliveira, J. Sep. Sci., 2009, 32, 3529–3538 CrossRef CAS PubMed.
- N. D. Forsberg, G. R. Wilson and K. A. Anderson, J. Agric. Food Chem., 2011, 59, 8108–8116 CrossRef CAS PubMed.
- M. Khorshid, E. R. Souaya, A. H. Hamzawy and M. N. Mohammed, Int. J. Environ. Anal. Chem., 2015, 352610 Search PubMed.
- T. V. Madureira, S. Velhote, C. Santos, C. Cruzeiro, E. Rocha and M. João Rocha, Environ. Sci. Pollut. Res., 2014, 21, 6089–6098 CrossRef CAS PubMed.
- A. Angioni, L. Porcu, M. Secci and P. Addis, Food Anal. Methods, 2012, 5, 1131–1136 CrossRef.
- S.-S. Cai, J. Stevens and J. A. Syage, J. Chromatogr. A, 2012, 1227, 138–144 CrossRef CAS PubMed.
- M. Smoker, K. Tran and R. E. Smith, J. Agric. Food Chem., 2010, 58, 12101–12104 CrossRef CAS PubMed.
- M. Yoo, S. Lee, S. Kim, S. Kim, H. Seo and D. Shin, Int. J. Food Sci. Technol., 2014, 49, 1480–1489 CrossRef CAS.
- T. H. Kao, S. Chen, C. J. Chen, C. W. Huang and B. H. Chen, J. Agric. Food Chem., 2012, 60, 1380–1389 CrossRef CAS PubMed.
- M. Surma, A. S. Rociek and E. Cieślik, Eur. Food Res. Technol., 2014, 238, 1029–1036 CrossRef CAS.
- S. Chen, T. H. Kao, C. J. Chen, C. W. Huang and B. H. Chen, J. Agric. Food Chem., 2013, 61, 7645–7653 CrossRef CAS PubMed.
- T. H. Kao, S. Chen, C. W. Huang, C. J. Chen and B. H. Chen, Food Chem. Toxicol., 2014, 71, 149–158 CrossRef CAS PubMed.
- A. Sadowska-Rociek, M. Surma and E. Cieślik, Environ. Sci. Pollut. Res., 2014, 21, 1326–1338 CrossRef CAS PubMed.
- G. Knobel and A. D. Campiglia, J. Sep. Sci., 2013, 36, 2291–2298 CrossRef CAS PubMed.
- E. D. Dodds, M. R. McCoy, A. Geldenhuys, L. D. Rea and J. M. Kennish, J. Am. Oil Chem. Soc., 2004, 81, 835–840 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2017 |