
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Kinetically-enhanced DNA detection via multiple-pass exonuclease III-aided target recycling†
Henson L.
Lee Yu
a,
Yinghua
Zhang
b and
I-Ming
Hsing
 *ab
*ab
aDepartment of Chemical and Biomolecular Engineering, The Hong Kong University of Science and Technology, Clear Water Bay, Kowloon, Hong Kong. E-mail: kehsing@ust.hk
bDivision of Biomedical Engineering, The Hong Kong University of Science and Technology, Clear Water Bay, Kowloon, Hong Kong
First published on 6th November 2017
Abstract
One of the promising approaches to address the challenge of detecting dilute nucleic acid analytes is exonuclease III-aided target recycling. In this strategy, the target DNA self-assembles with the reactant DNA probes and displays itself as a reactant and product at the same time. This provides an autonomous mechanism to release and reuse the analyte from each round of reactions for repetitive cycles, which amplifies the signal without amplifying the analyte itself. However, for very low amounts of the analyte, it takes a considerably long time before a detectable signal is generated. Thus, in this paper, we report a kinetically-enhanced target recycling strategy by designing two more target recycling sub-reactions that are triggered by the byproducts of the first reaction involving the target analyte. In this manner, concentrations of up to 0.5 pM of target DNA can be detected in 15 minutes.
A. Introduction
Nucleic acid-based biosensors have been widely used for the detection of infectious diseases,1 cancers,2 or other genetic diseases.3 One advantage of a biosensing device that uses a nucleic acid as the target analyte is its versatility since a primer or probe sequence can be designed to be target-specific.4 Also, DNA detection allows for strain differentiation5 or serotyping,6 tumor genotyping,7 disease risk determination, drug response predictions, and other applications in personalized medicine. Furthermore, using nucleic acids as a bioreceptor allows for the enhancement of the performance of the biosensor by designing appropriate nucleic acid circuitries. These programmable circuitries may be a combination of assembly and disassembly reactions that exploit the predictable nature of Watson–Crick base-pair interactions and the thermodynamics of DNA hybridization.8With the recent advancements in nanotechnology, it is possible to create these biosensors in the form of a portable device that can be delivered at point-of-care. However, currently, most available nucleic acid tests require PCR or other isothermal enzymatic amplification techniques to amplify the target DNA sample prior to its detection.9 The additional step and the need for a thermal cycler in the case of PCR would hinder its widespread use as a point-of-care device. Thus, the enrichment of the signal without the need for direct amplification of the target analyte is an active field of research.
In a simple DNA biosensor platform involving strand-displacement reactions, the kinetics is said to be pseudo first order at low analyte concentrations and sufficiently excess amounts of probes.10 In such a case, the signal produced is proportional to the amount of the analyte, [A]. In order to increase the signal one strategy commonly used is target recycling, wherein the target DNA first participates in a reaction that yields a detectable signal, and then it gets regenerated so that it can undergo a second round of the same reaction. This cycle repeats in an autocatalytic fashion, thereby increasing the signal over time without increasing the amount of the target itself. However, in order to compensate for the concentration of the analyte in very dilute samples, the reaction must be allowed to sit for a long time so that a detectable signal can be obtained.
The use of exonuclease III (exo III), a 3′ recessed end-specific exonuclease, was first used to catalyze the target recycling reactions by Zuo et al.11 The sensitivity reported was 10 pM within 30 minutes, and 20 aM if the assay time is extended to 24 hours. Our group has previously shown that the same technique can be employed in an electrochemical detection platform with a roughly similar detection limit.12
Liu and co-workers have described dual cascade exo III-aided target recycling reactions using two different hairpin probes wherein the second probe is catalyzed by the degradation product of the target and the first hairpin in the presence of exo III. They reported a limit of detection of 0.1 pM for their method.13 Inspired by this method of increasing the sensitivity, we described herein a nucleic acid circuit that involves a dual cascade but with three target recycling reactions, and each reaction uses a double-stranded probe rather than hairpin probes.
In particular, we want to address the issue on kinetics, since aside from sensitivity and selectivity, the total assay time is also considered a hindrance to the development of a useful point-of-care device.14 Thus, in this study, we limit the detection time to 15 minutes, and improve the kinetics of the detection by introducing the two-layer, three-pass exonuclease III target recycling pathway. Through this method, we provide a proof-of-concept that picomolar concentrations of target DNA can be detected within a reasonable time frame for point-of-care devices.
B. Experimental
Apparatus and reagents
All oligonucleotides are sequenced, fluorophore or quencher-labelled, and HPLC-purified from Integrated DNA Technologies, Inc. The sequences are found in Table S1 (ESI†). Exonuclease III and 10× NEBuffer 1 were purchased from New England Biolabs, Inc. Tris base and boric acid were purchased from Sigma. EDTA was purchased from Invitrogen. 1× Tris-EDTA (TE) buffer was prepared by mixing appropriate amounts of each constituent to achieve a final concentration of 10 mM Tris base and 1 mM EDTA; after which, HCl or NaOH was added to adjust the pH to 8.0. 1× Tris-Boric acid-EDTA (TBE) buffer contains 10.8 g L−1 Tris, 5.5 g L−1 boric acid, and 0.45 mg mL−1 EDTA. All consumables used were analytical grade reagents and solutions were prepared in deionized water.Procedures
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.2 molar ratio of the fluorophore to the quencher strand, and similarly annealed as described above. The excess quencher strand assures the complete quenching of the fluorescence to lower the background noise. The three-probe system cannot have a higher concentration of the quencher strand since it can trigger the downstream reactions and would cause a higher background noise.
:1) acrylamide/bisacrylamide solution (BioRad), 1 mL 1× TBE, 5 mL deionized water, 10 μL of ammonium persulfate (Sigma), and 8 μL of N,N,N′,N′-tetramethylethylenediamine (TEMED) (Sigma). 10 μL of each sample supplemented with 2 μL of a 6× gel loading buffer (New England Biolabs, Inc.) was loaded onto the gel. PAGE was performed in 1× TBE at a constant voltage of 100 V for 60 minutes. The UV visualization of DNA fragments was done by pre-staining the gel with an SYBR Gold Nucleic Acid Stain (ThermoFisher Scientific) and visualized using a Gel Doc XR system (BioRad). The fluorescence visualization of the labelled DNA fragments was done using a Typhoon TRIO system (GE Healthcare) without the addition of any chelating dye.
:1.2 molar ratio of the fluorophore to the quencher strand, and similarly annealed as described above. The excess quencher strand assures the complete quenching of the fluorescence to lower the background noise. The three-probe system cannot have a higher concentration of the quencher strand since it can trigger the downstream reactions and would cause a higher background noise.
:1) acrylamide/bisacrylamide solution (BioRad), 1 mL 1× TBE, 5 mL deionized water, 10 μL of ammonium persulfate (Sigma), and 8 μL of N,N,N′,N′-tetramethylethylenediamine (TEMED) (Sigma). 10 μL of each sample supplemented with 2 μL of a 6× gel loading buffer (New England Biolabs, Inc.) was loaded onto the gel. PAGE was performed in 1× TBE at a constant voltage of 100 V for 60 minutes. The UV visualization of DNA fragments was done by pre-staining the gel with an SYBR Gold Nucleic Acid Stain (ThermoFisher Scientific) and visualized using a Gel Doc XR system (BioRad). The fluorescence visualization of the labelled DNA fragments was done using a Typhoon TRIO system (GE Healthcare) without the addition of any chelating dye.
C. Results and discussion
Detection scheme
Miranda-Castro and colleagues described an exonuclease III-aided target recycling reaction with a comparable detection time of 10 minutes which had a detection limit in the nanomolar range.15 Their study used a double-stranded probe with 3′ overhangs which was initially resistant to exo III digestion. The addition of the target displaced one of the strands in the probe and formed a product that had one strand cleaved off by exo III resulting in the target being recycled. However, the other strand in the original probe was not used any further.In this paper, a multiple-pass exonuclease III-aided target recycling pathway is proposed as shown in Fig. 1. Here, instead of the displaced strand becoming a waste strand, we designed a sub-reaction wherein it would act as a pseudo-target strand and could also participate in an independent target recycling circuitry. This was taken one step further by adding two of such pseudo-target strands (labeled A1 and A2 in the figure) and two independent sub-reactions.
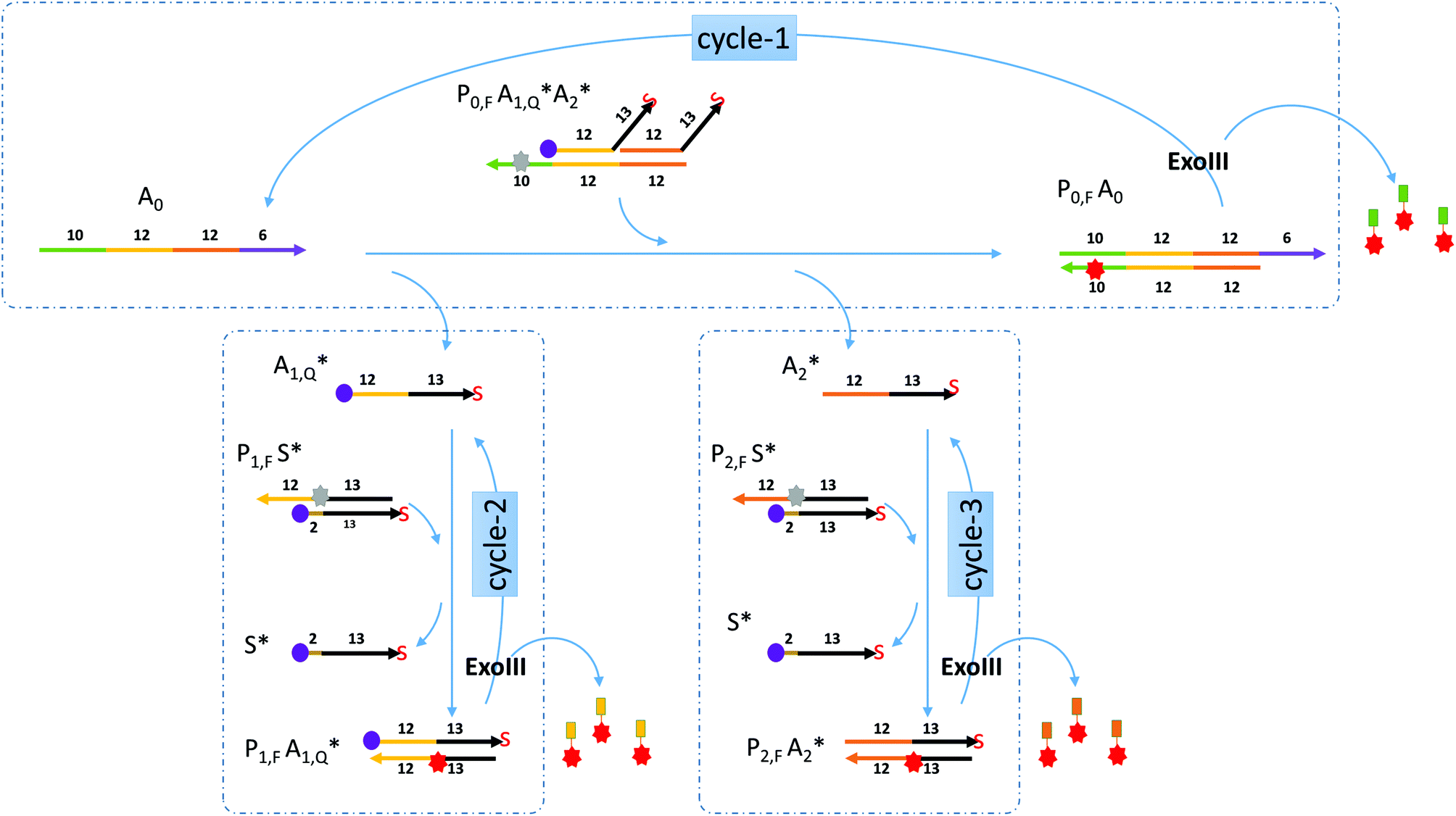 | ||
| Fig. 1 Schematics of the nucleic acid circuitry. The target analyte (A0) binds to P0 and releases A1 and A2 and gives off a fluorescent signal. Exo III then digests Q0 at the 3′ blunt end for target recycling. At the same time A1 and A2 bind to their respective probes P1 and P2 to form Q1 and Q2 which also similarly give off a fluorescent signal and are digested by exo III for two more cycles of target recycling. These three iterative cycles produce a fluorescent signal whenever Q probes are formed. (similar colors represent complementary sequences with the arrowhead signifying the 3′ end. The arrowhead with “S” means that the 3′ end is modified with a phosphorothioate linkage. The numbers correspond to the length of the segment below or above.) | ||
Gel visualization of the target recycling reactions
To demonstrate the feasibility of the nucleic acid circuit as described above, the three target recycling reactions were first visualized as separate reactions through gel electrophoresis. This was important because the non-specific leakage would compound once the three reactions were put together in one reaction vessel. Fig. 2 shows the PAGE results with and without a target, as well as with and without exo III incubation for each cycle. Each gel was run in duplicate, one stained with the SYBR gold stain to visualize all DNA strands present (gels with a gray background), and one unstained and viewed under the excitation wavelength of the fluorophore to visualize the strands containing the fluorophore (gels with a black background). Since the second and third sub-reactions gave similar results, only one is shown in the figure below. Sequences with an asterisk (i.e., A1*, A2*. S*) indicate 3′ phosphorothioate modification. Subscripts F and Q indicate fluorophore and quencher modification, respectively.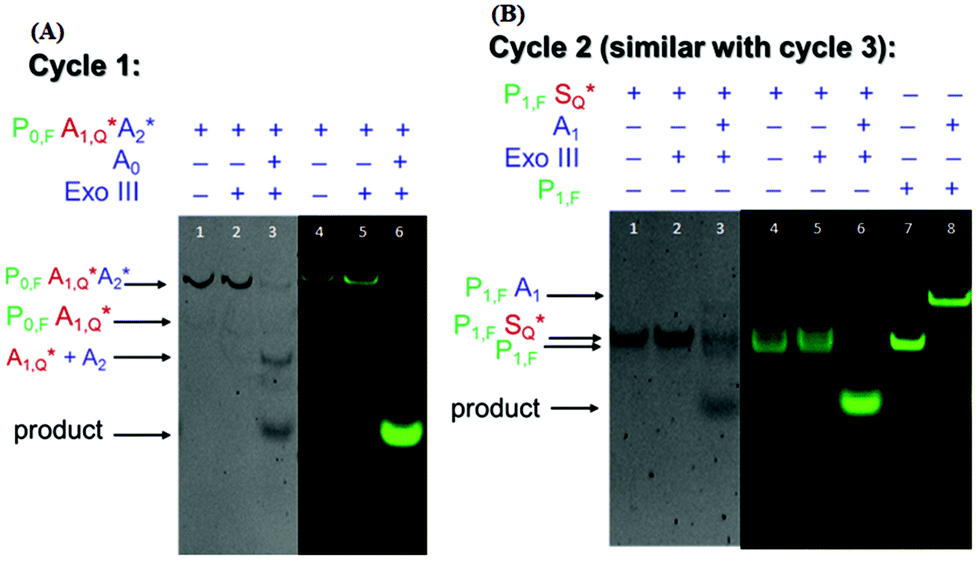 | ||
| Fig. 2 Visualization of (left) reaction 1 and (right) reaction 2 under UV light (gray background) and under green fluorescence (black background). The product is only formed when the target (A0 or A1) is added. In (a), the fluorescence signal of the triplex probe is quenched in the absence of exo III (lane 4), however, there is some residual leakage when exo III is added (lane 5). However, this did not affect the overall concentration of the triplex probe (lanes 1 and 2) signifying that only a few bases were digested in the process. In (b), two separate bands are observed in the lanes corresponding to P1S with or without exo III (lanes 3 and 4). The identity of these bands is confirmed by adding a control lane containing the same concentration of the P1,F strand (lane 7). | ||
By visualizing the reactions separately, we were able to establish the necessity of phosphorothioate modification, otherwise the duplex (P1S) and triplex (P0A1A2) probes would have been digested when exo III is added, even without the addition of their targets, A0 and A1, respectively (data not shown). But upon the introduction of phosphorothioate linkages in the last three nucleotides, there was negligible non-specific digestion (Fig. 2a lanes 2 and 5; Fig. 2b lanes 2 and 5). However, because the strands P0, P1, and P2 need to be digested by exo III for the target recycling, the 3′ end cannot be modified with phosphorothioate. Instead, it was appended with a 10 nt overhang so that despite being cleaved by two to three bases due to some residual, non-specific digestion of exo III, there would be a sufficient length of toehold for the target strand to anchor on. Although exo III is said to be specific to 3′ recessed or blunt ends, we still observed some activity as shown by an increase in the product band when P0A1A2 is incubated with exo III in the absence of the target A0 (not shown). Because of this, the fluorophore was attached to the 6th base (from the 3′ end), instead of the terminus. This significantly decreased the signal as shown by the negligible band and fluorescence when the target was absent (Fig. 2a, lanes 2 and 5) compared to that when the target was present (Fig. 2a, lanes 3 and 6). In Fig. 2b, the P1S probe appeared as two separate bands, with and without exo III (Fig. 2b, lanes 4 and 5). By adding a P1,F strand as the control, the two bands were identified as the quenched P1S strand (top band) and the unreacted P1,F strand. In the succeeding experiments, P1S strands were always formed with a 1.2× excess of the S strand to increase the formation of the duplex probe.
Optimization of the reaction conditions
Despite minimizing the leakage by designing the probes having phosphorothioate linkages and using an internal fluorophore label, there are still undesired leakages. Thus, the reaction buffer was optimized by adjusting the magnesium ion concentrations in the buffer since it is a known cofactor of exonuclease III16 and due to the presence of EDTA in the reaction mixture, we sought to optimize the ion concentrations. Indeed, the concentration affected the signal-to-noise ratio since it is a cofactor, and the positively-charged species in the solution can not only neutralize the negatively-charged phosphate backbone of DNA, but can also form a more stable three-stranded complex (Fig. S1, ESI†). The temperature and the enzyme concentration of the final reaction conditions used in this study are as follows: 0.1 U μL−1 exonuclease III, 25 °C, and 50 mM Mg2+; this was therefore used in all kinetic experiments henceforth. The temperature was chosen to be around room temperature for the simplicity of the setup conditions in light of its future application in a low-cost biosensor. Furthermore, the yield of P1S and P2S formation decreases due to the melting of the two strands at higher temperatures (Fig. S2, ESI†).Signal enrichment from multiple-pass target recycling
In order to verify that the cascaded scheme indeed increased the kinetics of the signal generation, the fluorescence signal generated when 5 nM target DNA (A0) was monitored using one, two, and three target recycling reactions labeled one-pass (P0A1A2 only), two-pass (P0A1A2 and P1S), and three-pass (P0A1A2, P1S, and P2S) cycles respectively as shown in Fig. 3. The sharp downward spike corresponds to the time when the spectrofluorometer was opened and 20 μL of 1 U μL−1 of exo III was added to the cuvette. Considering a linear relationship between the fluorescence signal and time, which can be assumed for low concentrations of the analyte (details in section S4, ESI†) the addition of target recycling sub-reactions increases the rate by 1.7-fold and 3.7-fold respectively. It is also worth noting that for the two-pass and the three-pass systems, although there seems to be a general linear trend with a good correlation coefficient as determined by using the near unity r2 values, it can also be observed that there is a point where the slope shifted higher. This may be explained due to a delay in the contribution of the second and third target recycling reactions. This can be further inferred from the shift in this inflection point to an earlier time (∼13 minute mark for the two-pass system and ∼10 minute mark for the three-pass system). This is consistent with our inferred parabolic relationship between the signal and time (details in section S4, ESI†), when more than one target recycling reaction is considered.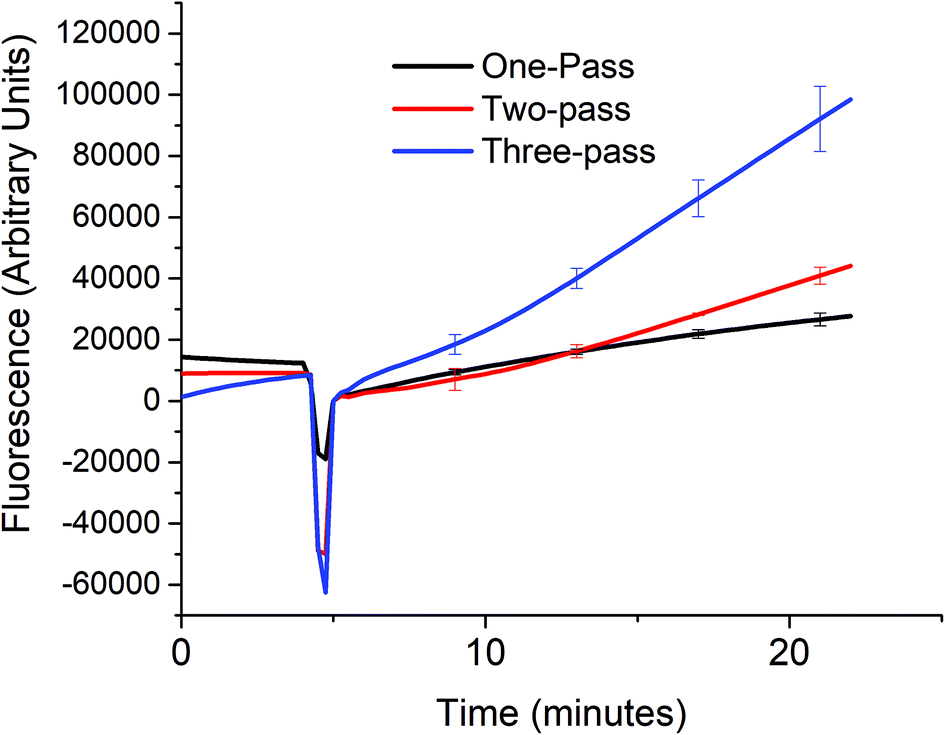 | ||
| Fig. 3 Signal enhancement from multiple-pass target recycling. A 5 nM target DNA was added to a solution containing 50 nM of either only P0A1A2 (one-pass), both P0A1A2 and P1S (two-pass), or P0A1A2, P1S, and P2S (three-pass) and the reaction buffer mixture (1× NEBuffer 1 with 50 mM Mg2+) and 20 μL 1 U μL−1 of exo III. The fluorescence signal was measured every 15 seconds at room temperature. (At t = ∼5, the signal dropped because the spectrofluorometer was opened when exo III was added.) | ||
Sensitivity of a three-pass target recycling nucleic acid circuit
Using the optimized conditions in the aforementioned section, we studied the sensitivity of such a biosensor given a limited timeframe of 15 minutes, with a 5-minute incubation prior to the addition of exonuclease III. The incubation period allows for the signal to be stabilized due to the initial reaction between the target analyte and the probe, and for the temperature of the solution to reach 25 °C. The fluorescence signal generated was measured every 15 seconds, and the standard deviations of three trials shown as error bars are indicated in intervals for simplicity.Despite the optimized conditions, an increasing fluorescence signal was still observed due to the incomplete formation of the triplex probe P0A1A2 (see Fig. 4). Any unbound A1 and A2 probes could trigger the two sub-reactions. And, upon the addition of exonuclease III, the unwanted signal would also be amplified. However, in the presence of a target up to a concentration of 0.5 pM, the signal is distinguishable from the background noise within 15 minutes after the addition of exo III. The difficulty of being able to bring down the sensitivity even further is not unique to most solution-phase biosensors such as the one described in this paper. That is, the presence of certain species in the matrix that causes non-specific signal generation cannot be removed, because a washing step cannot be introduced between the addition of the target and the detection of the signal, unlike in heterogeneous formats.17
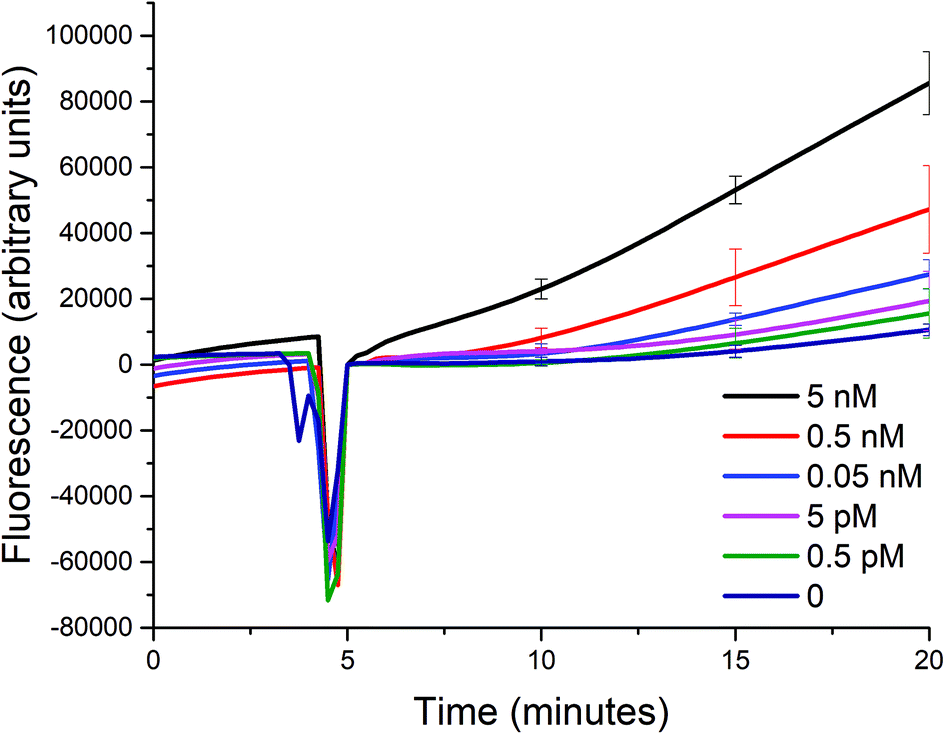 | ||
| Fig. 4 Sensitivity of the biosensor. The fluorescence signal after the addition of different concentrations of A0 to a mixture of P0A1A2, P1S, and P2S (50 nM each) was monitored for 15 minutes in the presence of 0.1 U μL−1 of exo III at room temperature (25 °C). (At t = ∼5, the signal dropped because the spectrofluorometer was opened when exo III was added.) | ||
Selectivity of the biosensor
The selectivity of the biosensor was measured by calculating the discrimination factor (DF) of the target against two single-nucleotide variants (SNV A and SNV B). DF is calculated as:18where F is the average fluorescence signal 15 minutes after exo III was added to the solution of three trials. Fig. 5 shows that the described detection scheme can differentiate the target from SNV A or B.
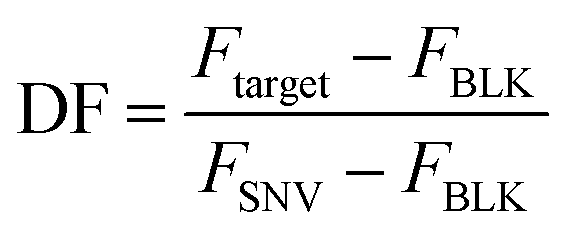
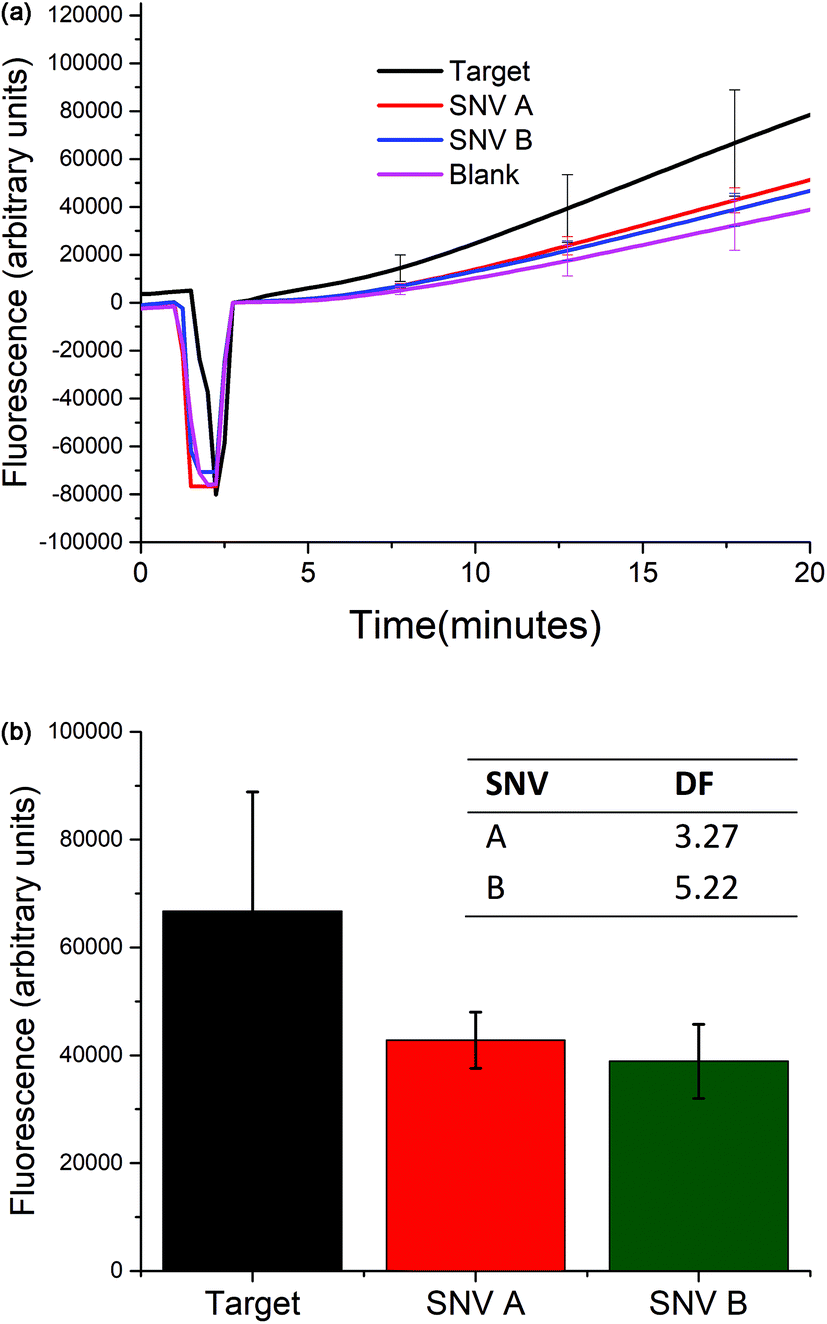 | ||
| Fig. 5 Selectivity studies. (Top) The selectivity of the biosensor was studied by comparing the fluorescence response over time when 5 nM of either the target, SNV A, or SNV B was added. (Bottom) Values of the fluorescence signal 15 minutes after exo III was added. The inset shows the calculated discrimination factor (DF). (At t = ∼5, the signal dropped because the spectrofluorometer was opened when exo III was added.) | ||
D. Conclusions
In this paper, we demonstrated the feasibility of coupling a target recycling reaction with two other independent reactions to amplify the signal. While there is a significant improvement of up to three orders of magnitude as compared to a single-pass target recycling reaction, the number of independent reactions is limited by the compounded background signal that is generated mainly due to the incomplete formation of the multiplex probe. Despite this challenge, we herein present a proof-of-concept to achieve detectable signals for a fluorescence-based nucleic acid biosensor within a reasonable amount of time. Further work is warranted to improve the selectivity of the biosensor by lowering the background noise and by improving the discrimination factor between the target and SNVs. The first can be done by maximizing the yield of the multiplex probe in order to increase the number of pseudo-target strands that can be hybridized to a long fluorescently-labelled strand. A thermodynamically stable multiplex probe needs to overcome the repulsive forces of the negatively charged DNA backbone by increasing the length of the double-stranded region for each pseudo-analyte and possible single nucleotide spacers in between them. Secondly, the selectivity towards the target over spurious targets can be improved by extending the current design to include competitive sink probes to better discriminate the target and the SNVs such as those described in the studies of Chen et al.19 and Wang et al.20 With this optimized and thermodynamically stable multiplex probe, an even greater improvement is expected in the sensitivity and the speed of the biosensor.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge the Research Grants Council of the Hong Kong SAR Government (GRF# 16301817) for the funding support. HLY would like to acknowledge the fellowship support from the Hong Kong PhD Fellowship Scheme.References
- E. A. Mothershed and A. M. Whitney, Clin. Chim. Acta, 2006, 363, 206–220 CrossRef CAS PubMed.
- H. Dong, J. Lei, L. Ding, Y. Wen, H. Ju and X. Zhang, Chem. Rev., 2013, 113, 6207–6233 CrossRef CAS PubMed.
- W. Laureyn, T. Stakenborg and P. Jacobs, Handb. Biosens. Biochips., 2008 DOI:10.1002/9780470061565.hbb111.
- M. R. Hartman, R. C. H. Ruiz, S. Hamada, C. Xu, K. G. Yancey, Y. Yu, W. Han and D. Luo, Nanoscale, 2013, 5, 10141–10154 RSC.
- L. T. H. Kao, L. Shankar, T. G. Kang, G. Zhang, G. K. I. Tay, S. R. M. Rafei and C. W. H. Lee, Biosens. Bioelectron., 2011, 26, 2006–2011 CrossRef CAS PubMed.
- N. V. Zaytseva, R. A. Montagna and A. J. Baeumner, Anal. Chem., 2005, 77, 7520–7527 CrossRef CAS PubMed.
- L. V. Sequist, R. S. Heist, A. T. Shaw, P. Fidias, R. Rosovsky, J. S. Temel, I. T. Lennes, S. Digumarthy, B. A. Waltman, E. Bast, S. Tammireddy, L. Morrissey, A. Muzikansky, S. B. Goldberg, J. Gainor, C. L. Channick, J. C. Wain, H. Gaissert, D. M. Donahue, A. Muniappan, C. Wright, H. Willers, D. J. Mathisen, N. C. Choi, J. Baselga, T. J. Lynch, L. W. Ellisen, M. Mino-Kenudson, M. Lanuti, D. R. Borger, A. J. Iafrate, J. A. Engelman and D. Dias-Santagata, Ann. Oncol., 2011, 22, 2616–2624 CrossRef CAS PubMed.
- P. Yin, H. M. T. Choi, C. R. Calvert and N. A. Pierce, Nature, 2008, 451, 318–322 CrossRef CAS PubMed.
- A. Niemz, T. M. Ferguson and D. S. Boyle, Trends Biotechnol., 2011, 29, 240–250 CrossRef CAS PubMed.
- A. J. Genot, D. Y. Zhang, J. Bath and A. J. Turberfield, J. Am. Chem. Soc., 2011, 133, 2177–2182 CrossRef CAS PubMed.
- X. Zuo, F. Xia, Y. Xiao and K. W. Plaxco, J. Am. Chem. Soc., 2010, 132, 1816–1818 CrossRef CAS PubMed.
- F. Xuan, X. Luo and I. Hsing, Anal. Chem., 2012, 84, 5216–5220 CrossRef CAS PubMed.
- S. Liu, Y. Lin, L. Wang, T. Liu, C. Cheng, W. Wei and B. Tang, Anal. Chem., 2014, 86, 4008–4015 CrossRef CAS PubMed.
- Y.-H. Hsieh, C. A. Gaydos, M. T. Hogan, O. M. Uy, J. Jackman, M. Jett-Goheen, A. Albertie, D. T. Dangerfield, C. R. Neustadt, Z. S. Wiener and A. M. Rompalo, PLoS One, 2011, 6, e19263 CAS.
- R. Miranda-Castro, D. Marchal, B. Limoges and F. Mavre, Chem. Commun., 2012, 48, 8772–8774 RSC.
- J. A. Cowan, BioMetals, 2002, 15, 225–235 CrossRef CAS PubMed.
- H. L. Lee Yu, A. Maslova and I. Hsing, ChemElectroChem, 2017, 4, 795–805 CrossRef.
- T. Wu, X. Xiao, Z. Zhang and M. Zhao, Chem. Sci., 2015, 6, 1206–1211 RSC.
- S. X. Chen and G. Seelig, J. Am. Chem. Soc., 2016, 138, 5076–5086 CrossRef CAS PubMed.
- J. S. Wang and D. Y. Zhang, Nat. Chem., 2015, 7, 545–553 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7an01423f |
| This journal is © The Royal Society of Chemistry 2017 |