
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Development of lithium attachment mass spectrometry – knudsen effusion and chemical ionisation mass spectrometry (KEMS, CIMS)†
A. Murray
Booth
a,
Thomas J.
Bannan
 *a,
Med
Benyezzar
b,
Asan
Bacak
a,
M. Rami
Alfarra
ac,
David
Topping
a and
Carl J.
Percival‡
a
*a,
Med
Benyezzar
b,
Asan
Bacak
a,
M. Rami
Alfarra
ac,
David
Topping
a and
Carl J.
Percival‡
a
aSchool or Earth, Atmospheric and Environment Science, University of Manchester, UK. E-mail: Thomas.bannan@manchester.ac.uk
bPhoton Science Institute, University of Manchester, UK
cNational Centre for Atmosphere Science, UK
First published on 7th September 2017
Abstract
Lithium ion attachment mass spectrometry provides a non-specific, non-fragmenting, sensitive and robust method for the detection of volatile species in the gas phase. The design, manufacture and results of lithium based ion attachment ionisation sources for two different mass spectrometry systems are presented. In this study trace gas analysis is investigated using a modified Chemical Ionization Mass Spectrometer (CIMS) and vapour pressure measurements are made using a modified Knudsen Effusion Mass Spectrometer (KEMS). In the Li+ CIMS, where the Li+ ionization acts a soft and unselective ionization source, limits of detection of 0.2 ppt for formic acid, 15 ppt for nitric acid and 120 ppt for ammonia were achieved, allowing for ambient measurements of such species at atmospherically relevant concentrations. In the first application of Lithium ion attachment in ultra-high vacuum (UHV), vapor pressures of various atmospherically relevant species were measured with the adapted KEMS, giving measured values equivalent to previous results from electron impact KEMS. In the Li+ KEMS vapour pressures <10−3 mbar can be measured without any fragmentation, as is seen with the initial electron impact (EI) set up, allowing the vapor pressure of individual components within mixtures to be determined.
Introduction
Mass spectrometry has provided considerable insight into atmospheric science, notably in the fields of gas kinetics, trace gas analysis and aerosol composition elucidation. Mass spectrometry has been used in aircraft measurements, field campaigns1 and laboratory studies2 and typically consists of an ionizer, an analyser and a detector. The choice of ion source for the different applications each have advantages and drawbacks. Chemical ionization (CI),3 for example, has proved popular for trace gas analysis and gas kinetics, providing a source of ions at high pressures (including up to atmospheric pressure) which is highly specific to the target analyte. However, it requires suitable ionization reactions for the analyte to detect and is thus not universal. Electron impact (EI) has proved a consistently popular as an ionization method, through the provision of high sensitivity and non-selectivity, albeit with the twin drawbacks of including the requirements for high vacuum (10−6 torr) and the inherent fragmentation of gas analytes that results in complicated mass spectra.In order to avoid some of the inherent drawbacks of EI and CI ionization, an alternative method (ion attachment) has previously been developed by Fujii et al.4 It offers a non-specific, non-fragmenting ionisation method resulting in mass spectra consisting of adduct ions formed from the gas-phase attachment of a primary ion and neutral gas species. Thermionic emission of a primary ion, such as lithium ions, which exhibit strong affinity to any neutral species in the gas phase produces adducts ions which consist of analytes which have been “captured” by lithium ions at low kinetic energy. This yields unfragmented adducts which are then easily detected. Adduct ion formation is obtained when a lithium ion Li+ attaches to a sample neutral/molecule (M), producing a single stable molecular ion adduct as follows:
| Li+ + M → Li·M+ | (1) |
Under high pressure regimes (10−1 to 10−2 mbar), a neutral bath gas (g) such as nitrogen, argon, or methane is used to carry ions towards the detection region whilst acting as a third body during the ion-sample attachment process to help remove some of the excess heat or collision energy. This leads to a more stable adduct which can be more readily detected and improves sensitivity:
| Li+ + M + g → Li·M+ + g | (2) |
Fujii and Ohta5 investigated several lithium bearing materials (Li2O, Li2SiO3, Li3PO4, zeolite A and β-eucryptite) as potential sources for lithium ion production. β-eucryptite was shown to provide the highest emission current densities (2 × 10−6 A cm−2) and is the material used by Fujii et al.,4 Selvin and Fujii,6 Nakamura et al.,7 and Edwards et al.8
Presented here are initial results from the testing and construction of lithium-ion attachment sources for two different mass spectrometers. First, a Chemical Ionization Mass Spectrometer (CIMS) used for making trace gas phase measurements will be modified. Use of the Li+ ionization source in this application will allow for a non-specific, non-fragmenting ionization method for trace gas detection. Secondly, in the first application of Lithium ion attachment in ultra-high vacuum (UHV), the Knudsen Effusion Mass Spectrometer (KEMS) will be used for measuring vapour pressures. The primary advantage of the Li+ KEMS over the standard EI ionization KEMS is that individual peaks represent the vapour pressure of individual compounds, in comparison to EI where the sum of the total peaks is used to extract data and no single component data from mixtures is available. Li+ therefore will allow the vapor pressure of individual components within mixtures to be probed.
Materials and methods
Filament production
Fujii et al.,4 Selvin and Fujii,6 Nakamura et al.,7 and Edwards et al.,8 have made β-eucryptite from heating a crushed mix of Al2O3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :SiO2:Li2O in a 1:1:1 molar ratio. In this work filaments were using Al2O3:SiO2:Li2O and Al2O3:SiO2:Li2CO3, both with 1:1:1 molar ratios. The basic filament consists of the β-eucryptite material, which has been fused over a metallic conductor or gauze and heating this structure using electrical currents results in the thermionic emission of Li+ ions. The heat source that used in this study is produced by resistively heating the material through use of DC currents at low voltage. The specific design for the filaments for the CIMS and KEMS will be discussed in their respective sections.
:SiO2:Li2O in a 1:1:1 molar ratio. In this work filaments were using Al2O3:SiO2:Li2O and Al2O3:SiO2:Li2CO3, both with 1:1:1 molar ratios. The basic filament consists of the β-eucryptite material, which has been fused over a metallic conductor or gauze and heating this structure using electrical currents results in the thermionic emission of Li+ ions. The heat source that used in this study is produced by resistively heating the material through use of DC currents at low voltage. The specific design for the filaments for the CIMS and KEMS will be discussed in their respective sections.
Chemical ionisation mass spectrometer
The CIMS system was constructed and designed by the Georgia Institute of Technology and has been described in detail by Nowak et al.9 It is designed to sample gases from high pressure (up to atmospheric) using a differentially pumped arrangement and this specific instrument has been used for both ground and airborne studies.10,11 The differential pumping system has three main regions; an ion-molecule region where sample ionisation takes place with a pressure between 10 and 0.1 mbar. This is linked via a biased pinhole to the collision dissociation chamber (CDC) where there is a DC electric field to pull apart weakly bound cluster ions, the pressure in this region is around 10−2 mbar. This is then linked via another biased pinhole and an octopole ion guide to the final region of the instrument which houses the quadrupole mass analyser. The quadrupole filters ions onto a channeltron detector maintained at a pressure of 10−5 mbar.For the CIMS filament, a source was constructed using off the shelf KF40 components. The whole assembly was fitted in the ion-molecule region of the CIMS instrument (Fig. 1). The filament wire, the details of which are discussed later, was mounted using oxygen free high purity copper clamps. The clamps were connected to a linear motion feedthrough via insulated alumina standoffs allowing the source to be moved relative to a biased pinhole. This helps ions to be transmitted to the next section of the instrument. Standard high current electrical KF feedthroughs are used to pass low voltage DC current to the filament. Currents of up to 20 Amperes can be produced using a regulated power supply (Manson EP 907). A neutral gas flow of nitrogen is maintained between the emitter or ion source region and the sample area. This allows for thermalisation and easier transmission of Lithium ions and facilitates the attachment of such ions onto the sample neutral gas species.
 | ||
| Fig. 1 Schematic for CIMS filament holder. | ||
For the CIMS filament production, in the first instance, the filament wire was high purity 0.25 mm diameter tungsten wire from Advent materials formed into a spiral. Tungsten was chosen by virtue of its extremely high melting point (3422 °C). However, in the presence of oxygen it rapidly oxides at high temperatures meaning it can only be used under vacuum or an inert atmosphere. Filaments were also produced using platinum (melting point 1768 °C) and iridium (melting point 2466 °C) wire, both far more prone to burning out in comparison to the tungsten. Platinum in particular is quite brittle and the act of forming a spiral results in sufficient defects in the metal to significantly reduce the lifetime of the filament and thus an inappropriate metal for a filament. All the wire filaments were produced by making a slurry of the β-eucryptite pre mix using some liquid (usually water) and painting this onto the wire with a paintbrush, a small current was then passed through the wire (∼2 amps) to drive off the solvent and dry the β-eucryptite paste onto the wire. Once under vacuum the level of heating required for thermionic emission (∼4–6 amps) is sufficient to melt the β-eurcyptite pre mix paste onto the wire. An improved filament production method was also used consisting of a mesh, rather than a wire to provide a much larger area for lithium emission and hence a large number of emitted lithium ions. A platinum–iridium (Pt90:Ir10) mesh (>99.9% purity 150 mesh, woven from 0.043 mm wire sourced from Alfa-Aesar) was trialed to avoid the mechanical brittleness associated with pure platinum. Dry β-eurcyptite pre mix was placed on the mesh and was then heated sufficiently to produce a ‘glassy mix’. The Al2O3:SiO2:Li2CO3 mix releases excess CO2 upon heating and yields a transparent white/greyish glass structure.12 To avoid filament ‘burnout’ the temperature of the wires and meshes were measured using an optical pyrometer (Land Instruments Cyclops 100) to obtain the surface temperature at various currents and pressures. This yielded a safer method for undertaking measurements and prevented heating any part of the holding metallic wire above its melting point.
The CIMS ion optics were altered and optimised from normal chemical ionization mode to allow for the transmission of primary ions from the source (Table SM1†). The settings for optimisation of the primary ion counts are slightly different from those for adduct counts. The optimum voltages for the first pinhole/aperture (1 mm diameter) towards the exit of the ion-molecule region are strongly dependent on the operating pressure (Table SM2 and Fig. SM1†). A higher pressure requires a higher accelerating voltage to the first pinhole due to the increased mean free path in the bath gas which is balanced against an increased adduct stability from thermalisation with the bath gas (N2 removing the excess energy from the lithium ion – sample molecule collision).
Knudsen effusion mass spectrometer
The KEMS has been described in detail in Booth et al.,13,14 and used recently in Bannan et al.,15 with a schematic of the instrument being shown in Fig. 2. The core of the system is a Balzers-Pffierfer QMA 410 quad analyser with a cross-beam electron impact ion source for sampling molecular beams of organics. The detector is a secondary electron multiplier (SEM) and the base pressure of the system is 10−7 mbar. This is the same analyzer as those on aerodyne's quad-aerosol mass spectrometer (AMS)16 so the source could, in principle, be fitted onto an AMS. By swapping the ionization source in the KEMS from EI to Li+, specific and identifiable individual peaks in the mass spectrum will represent the vapour pressure of individual compounds, in comparison to EI where the sum of the total peaks is used to extract data. The Li+ KEMS will therefore allow the vapor pressure of individual components within mixtures to be probed. Previous discussions on potential for sources of uncertainty in the KEMS using the EI source include varying sample phase state, statistical noise, ionisation cross sections or lack of appropriate calibration standard in multiple volatility ranges, all described in detail by Booth et al. (2009).13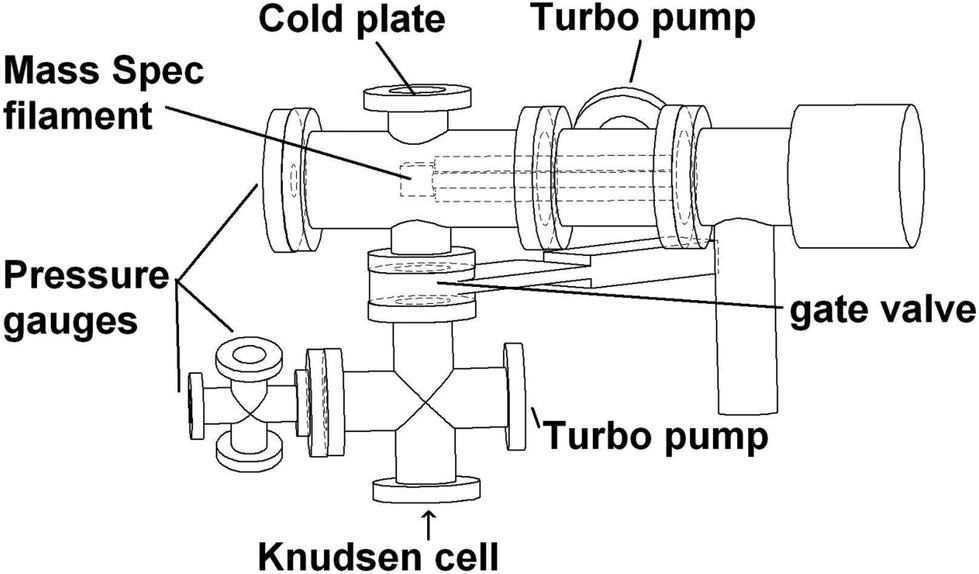 | ||
| Fig. 2 Schematic of the Knudsen effusion mass spectrometer. | ||
For the KEMS filament a standard Balzers-Pffiefer crossbeam electron impact source was adapted. β-Eurcyptite coated tungsten (0.25 mm diameter, >99.9% purity, Sigma Aldritch) and iridium (0.25 mm diameter, >99.9% purity, Advent Metals) wires were spot welded between the two electrodes of the Balzers-Pffiefer crossbeam electron impact source with a Wehnelt repeller around the wire. The assembly is mounted on an alumina holder previously used to hold an electron impact filament. As with the CIMS, the ion optics were first optimized for Li+ primary ions, as detailed in Table SM3.†
Results and discussion
CIMS
Of the filaments tested in the CIMS, the best results were achieved by using a Pt:Ir mesh and the LiCO3 based β-eucryptite synthesis method, giving a factor 500 improvement in primary ion current over the tungsten wire method. The emission current as a function of filament temperature was investigated. Emission current was monitored as a function of peak temperature for a 0.25 mm Tungsten spiral filament and a platinum iridium mesh style filament, shown in the Fig. SM2.† The emission current peaked at around 0.002 μA for the spiral filament at starts to level off as the filament gets past 1350 °C. Above 1350 °C the β-eucryptite in the center of the spiral starts to ablate away whilst material on the sides gets hot enough to emit, leading to a plateau in the emission current. The emission for the mesh increases exponentially to a peak of 0.6 μA before the heat caused uncoated sections of the filament to melt.
Fig. 3 illustrates a background mass scan then a mass scan during an 8 ppb acetone calibration, showing a strong signal at m/z 65. The acetone dimer was also detected at m/z 123 and its relative intensity was controlled by varying the CDC voltages (Table SM2†). Initially while the source was still emitting other alkali metals CH3COCH3·Na and CH3COCH3. K39/41 adducts were observed, as was the H2O·Li+ adduct from residual water vapour in the system. The heavier alkali metals are easier to eject from the filament but constitute a very small fraction (0.01% of the Li) of the material. They rapidly deplete during initial operation (usually within 1 hour).
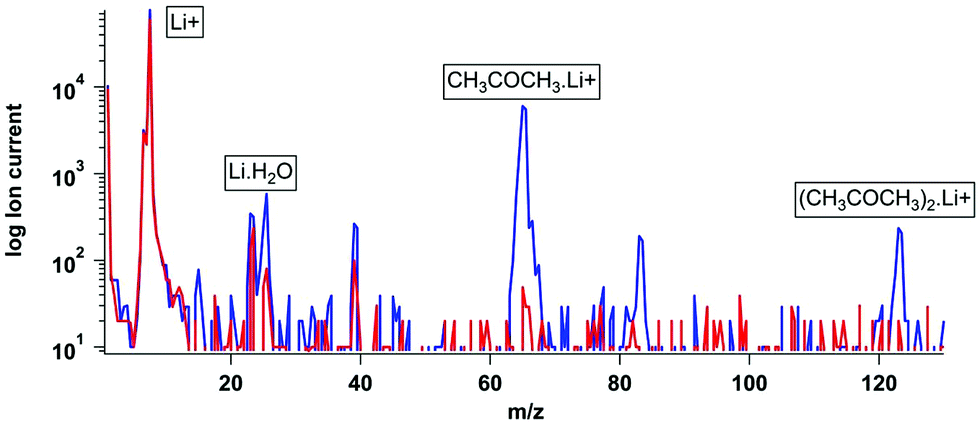 | ||
| Fig. 3 Background and calibration spectra for acetone permeation source. | ||
Formic acid concentrations, at above expected ambient concentrations (up to 0.5% v/v) using the Li+ source were also obtained. This resulted in the production of HCOOH.(H2O)x·Li+ clusters (Fig. SM3†), the intensity of which was highly dependent on the CDC voltage, as was the case with the acetone dimer. The production of formic acid clusters is also observed when measuring formic acid using CIMS with an iodide ionization gas.
The 3σ limit of detections (LODs) using the improved source are 4 ppt for acetone, 0.2 ppt for formic acid, 15 ppt for nitric acid, 0.2 ppt for formic acid, and 120 ppt for ammonia as calculated from permeation tube calibrations. It should be noted that the simultaneous detection of both an acid and a base is not possible for iodide CIMS for example, highlighting the versatility of lithium attachment approach.
A serendipitous reaction of a volatile organic compound stored in a cylinder provided a further test of the Li+ CIMS development, allowing observation of a wide range of reaction products (Table 1). A 1L gas cylinder was filled with 1% v/v of acetone, 1% v/v NO2, and 1% v/v SF6 topped up with N2 and left at room temp for approximately 24 hours before sampling, allowing time for reactions to occur within the mixture. The cylinder was then measured using Li+ CIMS. A number of additional m/z peaks were observed and the conjectured components are shown in Table 3, showing the effectiveness of this ionization method, despite some as yet unidentified peaks. Future tests can be implemented using an instrument with a higher resolving power to improve on species identification.
| m/z | Peak height | Assignment |
|---|---|---|
| 6 | 4.2 × 105 | Li+ primary (minor shoulder) |
| 7 | 1.7 × 106 | Li+ primary |
| 23 | 3 × 103 | Na+ primary |
| 25 | 1.4 × 105 | H2O·Li+ |
| 27 | 1 × 105 | Unknown |
| 30 | 1 × 103 | Unknown |
| 35 | 4 × 102 | N2·Li+ |
| 37 | 3 × 103 | NO·Li+ |
| 39 | 3 × 102 | O2·Li+ |
| 46 | 1 × 103 | Unknown |
| 51 | 1.6 × 103 | CH3NO·Li+ |
| 53 | 1.5 × 103 | NO2·Li+ |
| 65 | 3.2 × 104 | CH3COCH3 (acetone) |
| 67 | 9 × 103 | (NO)2·Li+ |
| 79 | 6 × 102 | Unknown |
| 83 | 6 × 103 | CH3CH3NO2 |
| 85 | 2 × 103 | Unknown |
| 87 | 2 × 102 | Unknown |
| 89 | 4 × 102 | Unknown |
| 97 | 3 × 102 | Unknown |
| 99 | 3 × 102 | N2O4 |
| 101 | 3 × 102 | Unknown |
| 105 | 4 × 102 | CH3CONO2CH3 (nitro acetone) |
| 123 | 3 × 102 | (CH3COCH3)2 (acetone dimer) |
| 125 | 2 × 102 | Unknown |
| 127 | 1.5 × 102 | Unknown |
| 153 | 1 × 103 | SF6 |
Emission current was monitored at constant temperature as a function of time (Fig. SM4†). The longest running filament emission current displayed an exponential decay in emission with a half-life of between 50 to 60 hours, with a filament lifetime of up to 150 hours, however there was a considerable variation in lifetime depending on the quality of manufacture. The filament lifetime in this study, despite a large variation, is a significant improvement over a stated lifetime of between 1–10 hours for Fujii et al.17 In spite of such improvements made to the manufacturing and operation of the filaments and the high sensitivities and low limits of detection achieved, the problem of burnout (from uneven heating and subsequent melting of the wire or mesh) remained persistent, in addition the filament coating was found to be mechanically very brittle and easily shatters resulting in a loss of Li+ emission. Due to the variation in lifetime of the filaments it is unlikely that our Li+-CIMS would be a reliable ionization option for field and aircraft work at this time, however this achieved increase in lifetime makes the Li+ technology a viable option for applications such as laboratory studies and selected measurements where the advantages of such ionisation method could be exploited.
Knudsen effusion mass spectrometry
In comparison to the CIMS method, the attachment probability in the KEMS is much lower and the primary ion signal is orders of magnitude greater than the secondary. This is because under UHV the molecules are travelling in straight lines and only have a single chance to collide. Furthermore the collisions also produce less stable adducts as a result of no additional gas molecules to carry away excess energy from the reaction. With optimised ion optics that allowed adducts above the limit of detection to be detected, primary emission of trace alkali metals such as rubidium, ceasium and francium were also observable. The small size of the opening on the crossbeam ion source meant there was no improvement from using a mesh over a wire. Even a comparatively small amount of lithium (∼2 mm diameter) bearing material on a wire, in comparison to the CIMS, was enough so saturate the entrance to the ionization area. In UHV burnout was less of a problem as Tungsten wire, with its much higher melting point (3422 °C) could be used to make the filament. The lower ionization efficiency of Li+ compare to EI meant that the Knudsen cell in the system had to be moved much closer to the ion source (1 cm rather than 60 cm as was previously the case).Fig. 4 shows a comparison of an electron impact and lithium ion attachment spectra of benzophenone (two benzene rings bridged by a carbonyl), a compound frequently used for calibrations of vapour pressures. The 70 eV electron impact spectra shows the molecular fragment at m/z 182, and major fragments at 105 (benzene ring and CO), 77 (benzene ring) and 55. The Li+ (bottom) only produces a single molecular adduct peak at m/z 189 (e.g. 182 + 7) with a small shoulder for contributions from the minor 6Li+ primary, also visible are the primary emission of Fr85/87.
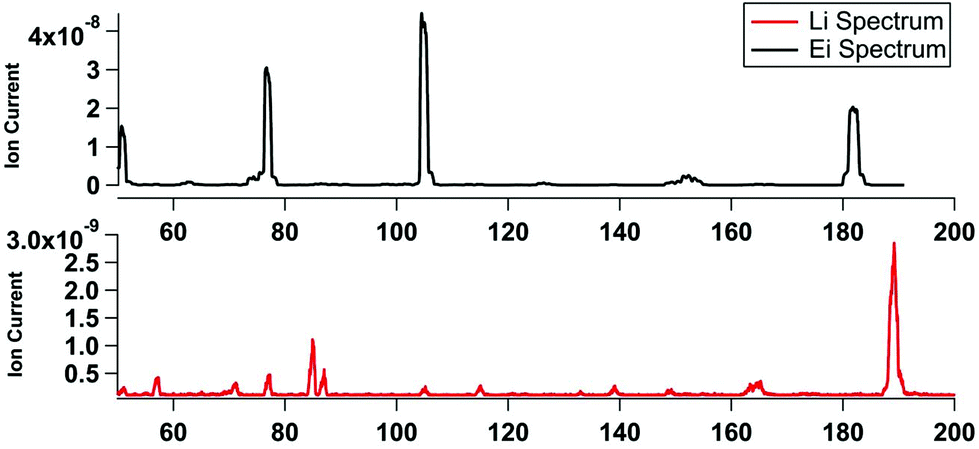 | ||
| Fig. 4 Comparison of EI vs. Li+ KEMS spectra for benzophenone. | ||
Validation of single component and mixture vapor pressure measurements
Work following the Bilde et al.,18 study culminated the in the recent inter-comparison study by Krieger et al.19 The Krieger et al.,19 study defined the homologous series of polyethylene glycols (PEG) ((H–(O–CH2–CH2)n–OH) for n = 3 to n = 8) as the first series of compounds with very good agreement over a wide range of VPs as measured by a range of vapor pressure measurement techniques. PEG samples therefore provide an ideal base mark for characterising the Li+ KEMS. Reported values from the Krieger et al. (2017)19 study show that the VP at 298 k of PEG 3 is 6.68(+1.10/−0.95) × 10−2 (Pa) and PEG 4 is 1.69 (+0.1/−0.10) × 10−2 (Pa). PEG 3 and PEG 4, were both measured with the Li+ KEMS both individually and when mixed. For PEG 3 the Li+ measurement gave a vapour pressure at 298 K of 2.49 × 10−2 (Pa) and for PEG 4 1.08 × 10−2 (Pa). When PEG 3 and 4 were mixed inside of the Knudsen cell, PEG 3 had a measured VP at 298 K of 2.29 × 10−2 (Pa) and for PEG 4 1.04 × 10−2 (Pa). This general good agreement between the reported values of the Krieger et al. (2017)19 study and the measurements of the Li+ KEMS show the validity of using this method from this simple test. Benzophenone was used as the internal reference for these measurements.In CI, a much more specific method than the Li+ ionization, sensitivities of measured species to the reagent ion varies considerably. For example in Bannan et al.,20 sensitivities to different species ranged by over an order of magnitude. The sensitivity of the Li+ KEMS to different reference compounds was therefore probed by calculating vapour pressures of oxalic acid, malonic acid, p-ansic acid, benzophenone and cis-pinonic acid using each of these compounds for calibration for one another. These results are compared to previous published EI measurements with the KEMS, as detailed in Table 2. Fig. 5 shows the measured vapour pressure of malonic acid, with repeats, using different calibrations. Using an average value from all calibrations, extracted values for malonic, P ansic and cis pinonic acid from the Li+ KEMS are over reported by factors of 1.3, 1.4 and 3.3 respectively and oxalic acid is under measured by a factor of 0.75. Oxalic acid as a reference performs most poorly at recreating the measurements with the measured Li+ VP being 42% of that reported by EI, however on average with all calibrations and measurements used, the EI measurements are represented well by the Li+ attachment.
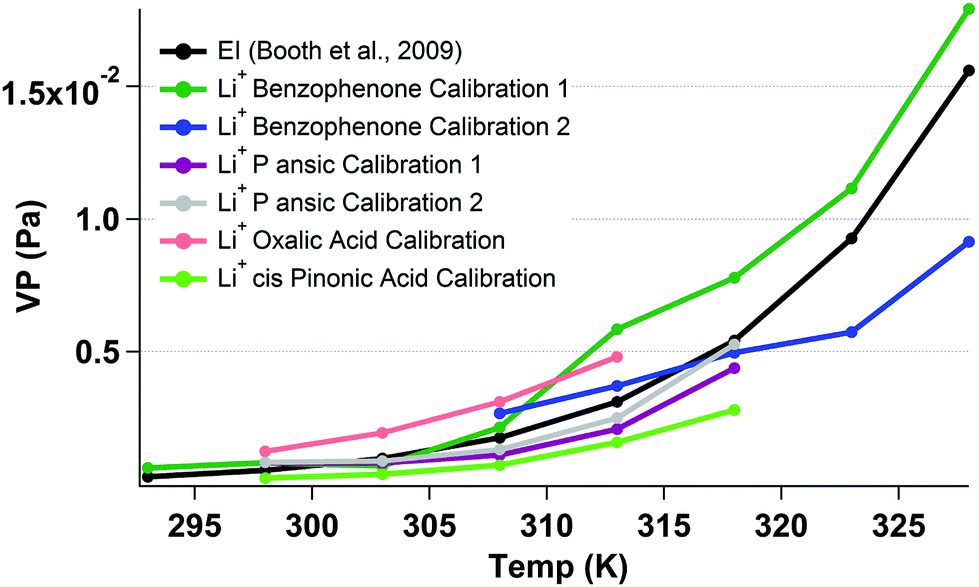 | ||
| Fig. 5 Temperature dependent measurements of malonic acid (VP at 298 K is 5.73 × 10−4) (Booth et al. 2009)13 using different calibrants with the Li+ attachment and EI KEMS. | ||
| EI measurement VP (Pa) | Li+ malonic cal VP (Pa) | Li+ oxalic cal VP (Pa) | Li+ P ansic cal VP (Pa) | Li+cis pinonic cal VP (Pa) | Li+ benzo cal VP (Pa) | |
|---|---|---|---|---|---|---|
| Malonic acid | 5.25 × 10−4 | 1.24 × 10−3 | 8.11 × 10−4 | 2.29 × 10−4 | 3.65 × 10−4 | |
| Oxalic acid | 1.40 × 10−2 | 1.30 × 10−2 | 1.21 × 10−2 | 5.65 × 10−3 | 1.14 × 10−2 | |
| P ansic acid | 5.33 × 10−4 | 5.70 × 10−4 | 1.34 × 10−3 | 2.48 × 10−4 | ||
| cis pinonic acid | 1.51 × 10−4 | 3.47 × 10−4 | 8.18 × 10−4 | 3.24 × 10−4 |
Secondary organic aerosol analysis
Li+ KEMS was also utilised to investigate the vapour pressure of secondary organic aerosol produced from a system of atmospheric relevance, i.e. testing the use of the Li+ KEMS for a mixture of unknown composition. Secondary organic aerosols were generated in the Manchester Aerosol Photochemical Chamber. Briefly, the chamber consists of an 18 m3 FEP, cube shaped, Teflon bag illuminated by a bank of halogen lamps and two 6 kW Xenon arc lamps simulating the solar spectrum (further details can be found in Alfarra et al.21,22). The air charge in the bag was dried and filtered for gaseous impurities and particles, prior to humidification with high purity de-ionised water. The biogenic SOA precursor α-pinene was injected into the chamber with an initial mixing ratio of 250 ppb. NOx and O3 were added with initial mixing ratios of 98 and 40 ppb respectively. The relative humidity was 55% and the temperature was 25 °C. The duration was 97 minutes from lights on to lights off. The total aerosol content of the chamber was then collected on to a filter. The estimated mass of sample on the filter was approximately 1 mg. A background filter was also produced using the same procedure but excluding the reactants (α-pinene, O3 and NOx).In order to provide an internal calibration, the filter samples were loaded into a knudsen cell together with a sample of known vapour pressure (benzophenone). This allowed us to both calibrate in terms of what ion currents corresponded to what vapour pressures, and to normalise the total signal to account for any variations in the lithium emission current between experiment runs. Fig. 6 shows a comparison of Li+ and EI of α-pinene-SOA aerosol from a filter once the background has been subtracted. The Li+ spectra show a much higher contribution from high m/z signals demonstrating conclusively that the EI ionization is fragmenting almost all of the molecules in the sample. Table 3 shows the vapour pressure contribution from the different peaks in the Li+ sample as well as the total vapour pressure for comparison with EI. The vapour pressures of the individual components are within the range expected for compounds capable of partitioning between the particle and gas phase of an aerosol (roughly 0.1–10−5 Pa). The total vapour pressure is quite high for particle phase compounds at 1.6 × 10−1 Pa but this can be explained by the relatively high concentrations in the experimental conditions driving more material into the particle phase than would normally be the case in the atmosphere. The total vapour pressure derived from Li+ measurements is approximately double that derived from EI(1.6 × 10−1vs. 5.3 × 10−2 Pa). This may arise from the necessary assumptions about the ionization cross sections of the molecules in the sample. It is assumed that the ion cross section is identical for all molecules in the sample as well as for the reference compounds. Measurement of the total ion current for the Li+ and EI signals yields an estimate of the relative sensitivity. This shows that EI is ∼75 times more sensitive than Li+.
 | ||
| Fig. 6 EI and Li of aerosol generated in the Manchester Photochemical Aerosol Chamber. | ||
| m/z | 44 | 137 | 155 | 172 | 291 | Total (Li) | Total (EI) |
|---|---|---|---|---|---|---|---|
| Vapour pressure (Pa) | 1.0 × 10−2 | 5.0 × 10−2 | 1.2 × 10−2 | 2.1 × 10−2 | 7.0 × 10−2 | 1.6 × 10−1 | 5.3 × 10−2 |
Conclusions
A lithium ion attachment source for a Chemical Ionisation Mass Spectrometer (CIMS) has been constructed and demonstrated. Its potential uses for a variety of analytes including acids and bases and acetone has been shown. Improvements on this approach have shown up to a 500 fold increase in yield when a gauze is used to hold the lithium ion emitting material (mesh style filament). The source was successfully miniaturized and mounted in a Knudsen Effusion Mass Spectrometer (KEMS). Effused species adducts were detected under UHV conditions with no fragmentation. Spectral signatures were compared between EI and Lithium attachment approaches. High m/z spectral peaks were easily observed using the Lithium attachment method whereas these were absent in the case of the EI method. A direct measurement of α-pinene secondary aerosol volatility has been made and compared with results from EI methods. This showed that in the KEMS the Li+ is less sensitive than EI by a factor of ∼75.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors wish to thank NERC for funding on grant NE/H003061/1.References
- K. A. Pratt and K. A. Prather, Mass spectrometry of atmospheric aerosols—Recent developments and applications. Part I: Off-line mass spectrometry techniques, Mass Spectrom. Rev., 2012a, 31(1), 1–16 Search PubMed.
- K. A. Pratt and K. A. Prather, Mass spectrometry of atmospheric aerosols—Recent developments and applications. Part II: On-line mass spectrometry techniques, Mass Spectrom. Rev., 2012b, 31(1), 17–48 Search PubMed.
- M. S. Munson and F. H. Field, Chemical ionization mass spectrometry. I. General introduction, J. Am. Chem. Soc., 1966, 88(12), 2621–2630 CrossRef CAS.
- T. Fujii, M. Ogura and H. Jimba, Chemical ionization Mass Spectrometry with lithium ion attachment to the molecule, Anal. Chem., 1989, 61(9), 1026–1029 CrossRef CAS.
- T. Fujii and M. Ohta, Filament thermionic sources of Li+ ions in low heating power, J. Phys. D: Appl. Phys., 1995, 28(6), 1268 CrossRef CAS.
- P. C. Selvin and T. Fujii, Lithium ion attachment mass spectrometry: Instrumentation and features, Rev. Sci. Instrum., 2001, 72(5), 2248–2252 CrossRef CAS.
- M. Nakamura, Y. Shiokawa, T. Fujii, M. Takayanagi and M. Nakata, Detection of quasimolecular ion of Cu (hfac)(tmvs) by ion attachment mass spectrometry, J. Vac. Sci. Technol., A, 2004, 22(6), 2347–2350 CAS.
- S. J. Edwards, C. G. Freeman, M. J. McEwan and P. F. Wilson, A selected ion flow tube investigation of the gas phase chemistry of Li+, Int. J. Mass Spectrom., 2006, 255, 164–169 CrossRef.
- J. B. Nowak, J. A. Neuman, K. Kozai, L. G. Huey, D. J. Tanner, J. S. Holloway and F. C. Fehsenfeld, A chemical ionization mass spectrometry technique for airborne measurements of ammonia, J. Geophys. Res.: Atmos., 2007, 112(D10) Search PubMed.
- T. J. Bannan, A. Bacak, J. Muller, A. M. Booth, B. Jones, M. Le Breton, K. E. Leather, P. Xiao, D. E. Shallcross and C. J. Percival, Importance of direct anthropogenic emissions of formic acid measured by a chemical ionisation mass spectrometer (CIMS) during the Winter ClearfLo Campaign in London, January 2012”, Atmos. Environ., 2014, 83, 301–310 CrossRef CAS.
- M. Le Breton, A. Bacak, J. B. Muller, T. J. Bannan, O. Kennedy, B. Ouyang and M. J. Daniels, The first airborne comparison of N 2 O 5 measurements over the UK using a CIMS and BBCEAS during the RONOCO campaign, Anal. Methods, 2014, 6(24), 9731–9743 RSC.
- M. Benyezzar and R. S. Mason, A New Technique for the Preparation of Long Life Lithium Ion Emitters and Application in a Lithium Attachment Mass Spectrometry System, 25th Int. Kinetics Symposium, Manchester, UK, 2008.
- A. M. Booth, T. Markus, G. McFiggans, C. J. Percival, M. R. Mcgillen and D. O. Topping, Design and construction of a simple Knudsen Effusion Mass Spectrometer (KEMS) system for vapour pressure measurements of low volatility organics, Atmos. Meas. Tech., 2009, 2(2), 355–361 CrossRef CAS.
- A. M. Booth, T. Bannan, M. R. McGillen, M. H. Barley, D. O. Topping, G. McFiggans and C. J. Percival, The role of ortho, meta, para isomerism in measured solid state and derived sub-cooled liquid vapour pressures of substituted benzoic acids, RSC Adv., 2012, 2(10), 4430–4443 RSC.
- T. J. Bannan, A. M. Booth, B. T. Jones, S. O'Meara, M. H. Barley, I. Riipinen and D. Topping, Measured Saturation Vapor Pressures of Phenolic and Nitro-aromatic Compounds, Environ. Sci. Technol., 2017, 51(7), 3922–3928 CrossRef CAS PubMed.
- J. T. Jayne, D. C. Leard, X. Zhang, P. Davidovits, K. A. Smith, C. E. Kolb and D. R. Worsnop, Development of an aerosol mass spectrometer for size and composition analysis of submicron particles, Aerosol Sci. Technol., 2000, 33(1–2), 49–70 CrossRef CAS.
- T. Fujii, P. C. Selvin, M. Sablier and K. Iwase, Lithium ion attachment mass spectrometry for on-line analysis of trace components in air: direct introduction, Int. J. Mass Spectrom., 2001, 209(1), 39–45 CrossRef CAS.
- M. Bilde, K. Barsanti, M. Booth, C. D. Cappa, N. M. Donahue, E. U. Emanuelsson and P. Ziemann, Saturation vapor pressures and transition enthalpies of low-volatility organic molecules of atmospheric relevance: from dicarboxylic acids to complex mixtures, Chem. Rev., 2015, 115(10), 4115–4156 CrossRef CAS PubMed.
- U. K. Krieger, F. Siegrist, C. Marcolli, E. U. Emanuelsson, F. M. Gøbel, M. Bilde, A. Marsh, J. P. Reid, A. J. Huisman, I. Riipinen, N. Hyttinen, N. Myllys, T. Kurtén, T. Bannan and D. Topping, A reference data set for validating vapor pressure measurement techniques: Homologous series of polyethylene glycols, Atmos. Meas. Tech. Discuss, 2017, DOI:10.5194/amt-2017-224.
- T. J. Bannan, A. M. Booth, A. Bacak, J. Muller, K. E. Leather, M. Le Breton and S. Visser, The first UK measurements of nitryl chloride using a chemical ionization mass spectrometer in central London in the summer of 2012, and an investigation of the role of Cl atom oxidation, J. Geophys. Res.: Atmos., 2015, 120(11), 5638–5657 CAS.
- M. R. Alfarra, J. F. Hamilton, K. P. Wyche, N. Good, M. W. Ward, T. Carr and G. B. McFiggans, The effect of photochemical ageing and initial precursor concentration on the composition and hygroscopic properties of β-caryophyllene secondary organic aerosol, Atmos. Chem. Phys., 2012, 12(14), 6417–6436 CAS.
- M. R. Alfarra, N. Good, K. P. Wyche, J. F. Hamilton, P. S. Monks, A. C. Lewis and G. McFiggans, Water uptake is independent of the inferred composition of secondary aerosols derived from multiple biogenic VOCs, Atmos. Chem. Phys., 2013, 13(23), 11769 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7an01161j |
| ‡ Current address: Jet Propulsion Laboratory, 4800 Oak Grove Drive, Pasadena, CA 91109. |
| This journal is © The Royal Society of Chemistry 2017 |