DOI:
10.1039/C7AN00757D
(Paper)
Analyst, 2018,
143, 133-140
Magnetic bead-based peptide extraction methodology for tissue imaging
Received
7th May 2017
, Accepted 31st October 2017
First published on 3rd November 2017
Abstract
MALDI-TOF imaging mass spectrometry (IMS) is a common technique used for analyzing tissue samples, as it allows the user to detect multiple different analytes simultaneously. However, the detection and analysis of these analytes may sometimes be hampered due to the presence of contaminants in the tissue microenvironment, which leads to ion suppression. This challenge necessitates the development of an active extraction technique to selectively isolate analytes of interest without compromising their spatial localization within a tissue sample. This study proposes a magnetic bead-based active extraction approach to selectively sequester peptides of interest from tissue samples. The technique utilizes a heterobifunctional cross-linker to covalently bind peptides with free primary amine groups to functionalized magnetic beads. The cross-linked peptides can then be collected using a transfer magnet and imaged using MALDI-TOF IMS. We have established that this technique not only successfully isolates peptides both in-solution and on a solid surface, but also extracts peptides from a tissue section without significantly compromising their spatial localization. This novel method provides the means to detect a unique subset of peptides from tissue sections when compared to unextracted tryptically digested tissue, all while minimizing the presence of contaminants and maintaining spatial localization.
Introduction
Tissue imaging is critical in understanding biomolecule localization and deciphering biochemical pathways.1,2 Although many molecular imaging techniques, such as fluorescence-mediated tomography (FMT) and magnetic resonance imaging (MRI) are well established, these techniques require the use of biological probes, limiting the number of possible analytes that can be detected.1,3 In addition, these techniques do not allow the discovery of new analytes when mapping biological molecules in tissues.4 Matrix-assisted laser desorption/ionization time-of-flight imaging mass spectrometry (MALDI-TOF IMS) is an effective method to image biomolecules, as it uses the mass resolution capabilities of a mass spectrometer to generate large sets of multiplexed data, allowing the user to detect many different analytes of varying mass to charge ratios in parallel without the use of biological probes.1,4,5
Studying peptide and protein distributions in tissues using MALDI-TOF IMS has long been of interest, with applications ranging from imaging neuropeptides and precursor peptide production to identifying biomarkers and tracking peptide chemical modifications.4,6–9 The wide dynamic range of MALDI-TOF permits the concurrent detection of a number of different analyte classes such as small molecules, proteins, lipids, carbohydrates, and metal ions.1,10,11 As MALDI readily desorbs and ionizes a wide range of molecules, interfering species are often ionized along with the analyte of interest, leading to analyte ion suppression.12 This problem can be particularly pronounced when imaging tissue specimens.13
To resolve this issue, tissue-blotting has been employed using materials such as C18, nitrocellulose, and conductive polyethylene membranes to extract peptides without compromising their relative locations.7,13,14 A study by Caprioli and coworkers shows that low molecular weight lipid peaks that contribute to the complex microenvironment of tissue are significantly reduced in tissue-blotted C18 resin when compared to images generated directly from the tissue itself.7 In addition, Fournaise and Chaurand successfully demonstrated that some proteins detected on tissue-blotted nitrocellulose coated slides are not observed when doing direct tissue analysis.14 Although these tissue-blotting methods allow detection of intractable peptides by removing interfering analytes, both studies also demonstrate that some subsets of proteins have better ionization when directly desorbed from tissue slices than when compared to those blotted onto a surface. This difference in ionization is possibly due to a difference in affinity for the blotting substrate. As a result, there is an observed difference in abundance of certain proteins after blotting.14 One possible solution to this issue would be to employ an active extraction method when transferring peptides onto the blotting surface, as opposed to using passive diffusion. This active extraction approach would allow sampling of intractable peptides by mitigating ion suppression like other blotting methods, while increasing the detection of peptides that might be hindered by inefficient analyte transfer due to a difference in affinity for the blotting substrate.
Our research group previously demonstrated that a selective, bead-based global peptide capturing method, using a heterobifunctional cross-linker (Fig. 1), can be utilized to selectively isolate peptides15 (Fig. 2). However, the effectiveness of this method with respect to selectivity in a complicated matrix and retention of analyte spatial localization in tissue has yet to be assessed. In this study, we demonstrate that this extraction approach is an effective method for isolating peptides from tissue samples. By selectively cross-linking peptides to magnetic beads, we are able to detect a unique subset of peptides while maintaining the spatial localization of species within the tissue.
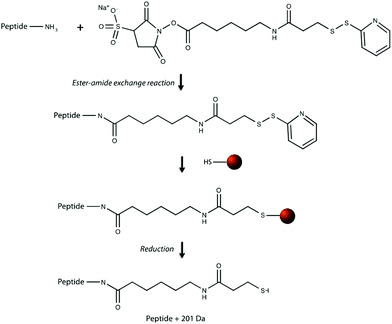 |
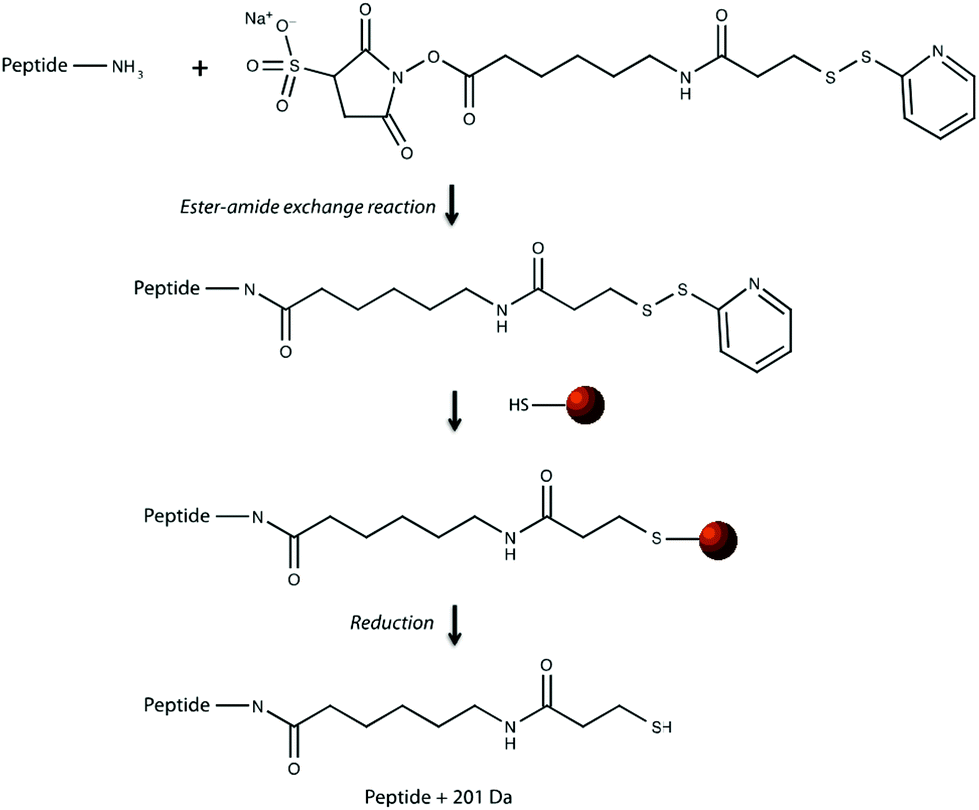
| | Fig. 1 Reaction chemistry of cross linking peptides to sulfinated functionalized magnetic beads. Peptides are first reacted with Sulfo-LC-SPDP heterobifunctional cross linker via an ester-amide exchange reaction with a free primary amine on the peptide. Peptides covalently bound to linkers are then reacted with sulfinated magnetic beads to form a disulfide bond, linking the peptide to the bead until reduction by DTT. | |
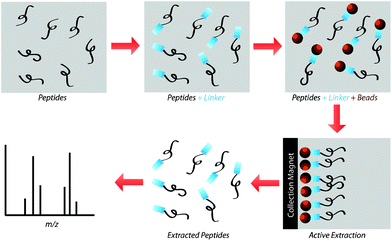 |
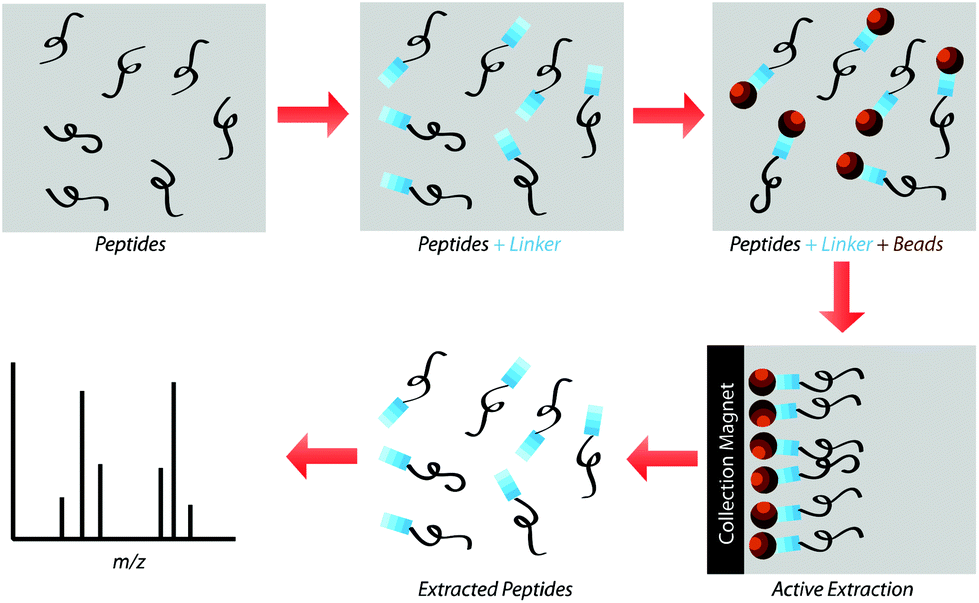
| | Fig. 2 Peptides in solution are reacted with Sulfo-LC-SPDP cross-linker before subsequently reacting with functionalized magnetic beads overnight. Beads are then collected using a magnetic separator and rinsed to wash away any unbound peptide. Disulfide bonds between the beads and the linker are reduced using DTT and the freed peptides are analyzed using MALDI-TOF. | |
Experimental
Materials and reagents
Sulfo-LC-SPDP heterobifunctional cross-linker was purchased from Thermo Scientific (Rockford, IL). Synthetic positive control peptides with a primary sequence of FTATDSHTDTPEALTTK, as well as negative control peptides with a primary sequence of Ac-FDTLYGPVSAEGTM were obtained from GenScript (Piscataway, NJ). BcMag long-arm Sulfhydryl-terminated magnetic beads (approximately 5 μm in diameter) and magnetic separators were purchased from Bioclone, Incorporated (San Diego, CA) and dialysis tubing was obtained from Spectrum Laboratories (Rancho Dominguez, CA). Neodymium transfer magnets were purchased from K&J Magnetics, Incorporated (Pipersville, PA). Proteomics grade trypsin was obtained from Promega (Madison, WI). Peptide calibration standard was obtained from Bruker (Billerica, MA) and all unspecified reagents were purchased from Sigma Aldrich (St Louis, MO). C57BL/6 mouse tissue was acquired by generous donation from the M. Sharon Stack laboratory at the University of Notre Dame. The tissues were unused brains from mice sacrificed under approval from the Institutional Animal Care and Use Committee (IACUC) at the University of Notre Dame.
Mass spectrometry
MALDI-TOF MS spectra were obtained using a Bruker AutoFlex Smartbeam II MALDI mass spectrometer (Billerica, MA) equipped with a frequency-tripled Nd:YAG laser (355 nm). Tissue imaging experiments were conducted using a Bruker UltrafleXtreme MALDI mass spectrometer (Billerica, MA) with a laser spot size of 150 μm at a 1000 Hz sampling frequency. All samples analyzed using MALDI-TOF IMS were coated with 10 μg μL−1 of 2,5-dihydroxybenzoic acid (DHB) matrix solution (50% H2O, 50% acetonitrile, with 0.1% TFA) using an HTX Imaging TM-Sprayer (Chapel Hill, NC). Samples were coated with 8 passes at a 0.1 mL min−1 flow rate, track spacing of 2 mm, pressure of 10 psi, gas flow of 3 L min−1, and 1000 mm min−1 velocity. DHB matrix solution was chosen over CHCA because it produces less background signal from matrix clusters.16 Minimizing background signal is of particular importance when imaging on substrates, which would inherently have some amount of background themselves. In addition, experiments were conducted using a PBS buffer, which would also complicate spectra due to the presence of salt adducts. This also makes reducing background signals particularly important in these experiments. Imaging data was acquired using flexImaging software v4.1. Queried imaging data was analyzed using flexAnalysis v3.4 (Bruker). All experiments were replicated a minimum of three times for reproducibility, though only one replicate is shown in this manuscript for the sake of brevity.
In-solution extraction
Peptide solutions were composed of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 dilution of peptide calibration standard, as well as a 1:40 dilution of 1 μg μL−1 synthetic control peptide, combined in a 2:1 ratio in nanoPure H2O for a total volume of 20 μL (8.33 ng μL−1 final concentration of synthetic peptide). In-solution cross-linking reactions were carried out by adding 20 μL of 10 μg μL−1 Sulfo-LC-SPDP heterobifunctional cross linker (in nanoPure H2O) to each of the peptide solutions for one hour before dialyzing overnight. Following dialysis, samples were dried using a speed vacuum concentrator for 1 hour at 30 °C and pellets were resuspended in 30 μL phosphate buffered saline pH 7.5 (PBS). Magnetic beads (200 μL of 30 mg mL−1) were prepared by rinsing three times with PBS prior to and immediately following cross-linking using a magnetic separator, according to the manufacturer's specifications (Bioclone Incorporated). PBS supernatant was removed from the magnetic beads and dialyzed peptide-linker solutions were added to the separated beads. Cross-linking between the dialyzed peptides and the magnetic beads was carried out by vortexing overnight at 4 °C. Following the cross-linking reaction, residual unbound peptides were rinsed from the beads according to manufacturer's specifications before the beads were reduced with 200 μL of 100 μM dithiothreitol (DTT). Peptides recovered from the beads following DTT reduction were collected, dried using a speed vacuum concentrator for 1 hour at 30 °C, resuspended in 10 μL nanoPure H2O, and desalted using C18 ZipTips (Millipore, Billerica, MA). Following desalting, 1 μL of each sample was plated onto and ITO-coated glass slide and analyzed using MALDI-TOF.
:3 dilution of peptide calibration standard, as well as a 1:40 dilution of 1 μg μL−1 synthetic control peptide, combined in a 2:1 ratio in nanoPure H2O for a total volume of 20 μL (8.33 ng μL−1 final concentration of synthetic peptide). In-solution cross-linking reactions were carried out by adding 20 μL of 10 μg μL−1 Sulfo-LC-SPDP heterobifunctional cross linker (in nanoPure H2O) to each of the peptide solutions for one hour before dialyzing overnight. Following dialysis, samples were dried using a speed vacuum concentrator for 1 hour at 30 °C and pellets were resuspended in 30 μL phosphate buffered saline pH 7.5 (PBS). Magnetic beads (200 μL of 30 mg mL−1) were prepared by rinsing three times with PBS prior to and immediately following cross-linking using a magnetic separator, according to the manufacturer's specifications (Bioclone Incorporated). PBS supernatant was removed from the magnetic beads and dialyzed peptide-linker solutions were added to the separated beads. Cross-linking between the dialyzed peptides and the magnetic beads was carried out by vortexing overnight at 4 °C. Following the cross-linking reaction, residual unbound peptides were rinsed from the beads according to manufacturer's specifications before the beads were reduced with 200 μL of 100 μM dithiothreitol (DTT). Peptides recovered from the beads following DTT reduction were collected, dried using a speed vacuum concentrator for 1 hour at 30 °C, resuspended in 10 μL nanoPure H2O, and desalted using C18 ZipTips (Millipore, Billerica, MA). Following desalting, 1 μL of each sample was plated onto and ITO-coated glass slide and analyzed using MALDI-TOF.
Extraction on a solid-substrate
1 μL of the previously described peptide solutions were spotted onto an Indium–Tin–Oxide (ITO)-coated glass slide (Delta Technologies, Limited) and dried in a desiccator. An equivalent volume of 100 μg μL−1 Sulfo-LC-SPDP cross-linker was spotted onto the dried peptide spots and incubated for two hours at 37 °C in a moist, covered petri dish to prevent sample evaporation while the linker reacts with the peptides. To actively extract peptides, 225 μg of magnetic beads in pH 7.5 PBS were carefully added to each sample using a 10 μL syringe and the samples were incubated overnight at 37 °C in a moist, covered petri dish to prevent sample evaporation while allowing cross-linking between the beads and the linker-bound peptides. Slides were incubated on top of a neodymium magnet to reduce lateral diffusion of the beads in the samples. The samples were then blotted onto DTT-coated C18 tape for 30 seconds using a neodymium transfer magnet (Fig. 3) and rinsed three times by pipetting nanoPure water to remove any remaining linker or beads. No pressure was applied during the blotting process. The C18 tape was prepared using a protocol previously developed by Caprioli, Farmer, and Gile.7 The tape was coated with 100 μM DTT using an HTX Imaging TM-sprayer (80 °C nozzle temperature, 6 passes, 0.1 mL min−1 flow rate, 1200 mm min−1 velocity, 3 mm track spacing, 10 psi nitrogen pressure, 3 L min−1 gas flow rate with 30 seconds drying time). The peptides that transferred to the C18 tape, as well as those left on the slide after blotting, were analyzed using MALDI-TOF IMS.
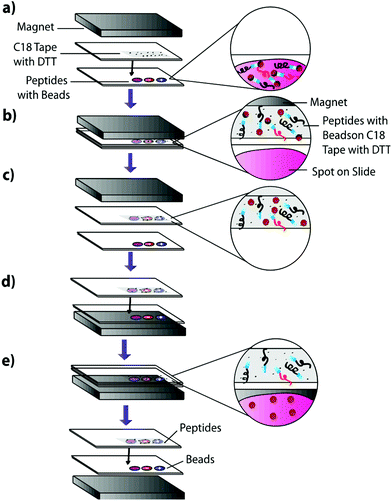 |
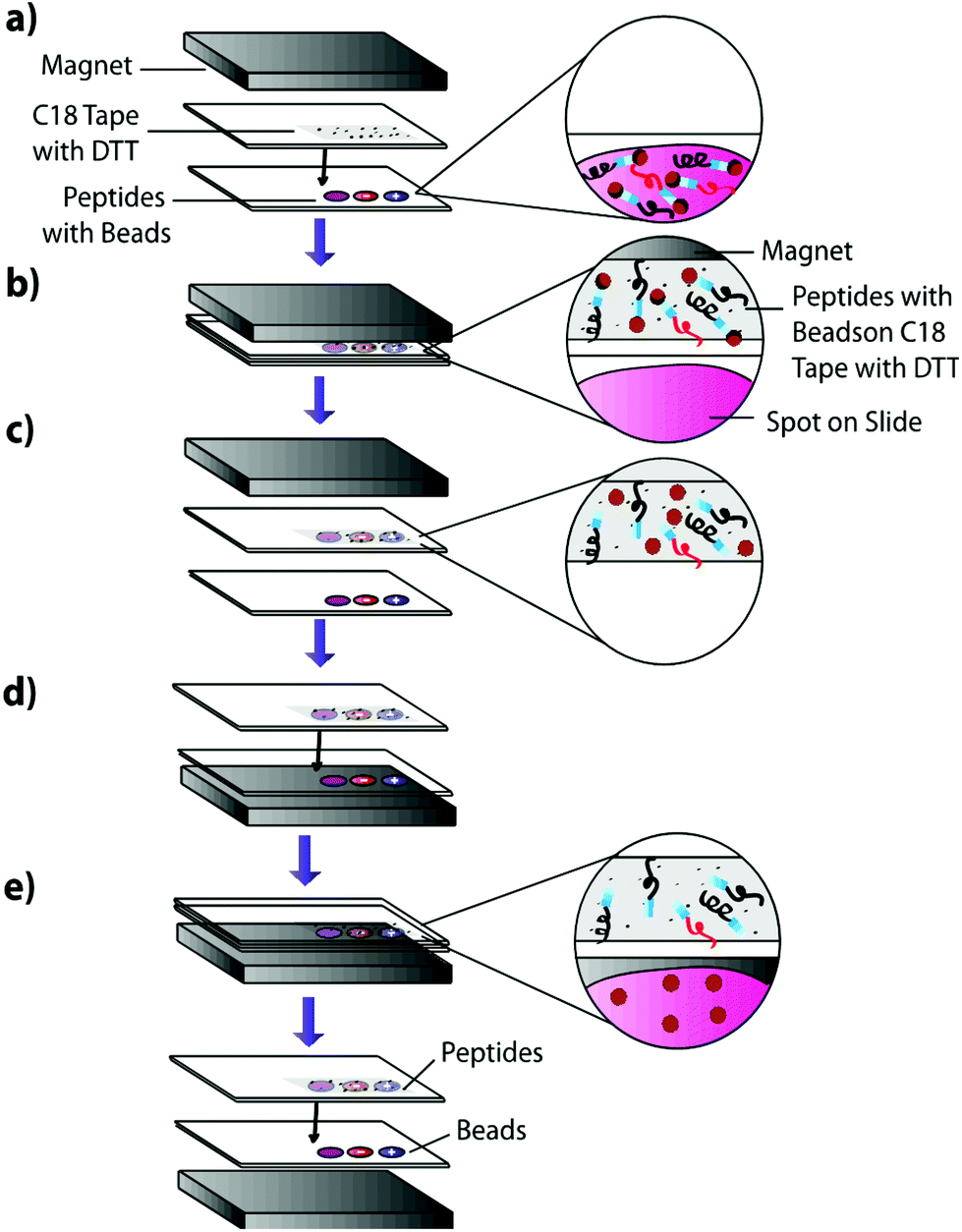
| | Fig. 3 Peptide blotting technique using cross-linking magnetic beads. (a) Standard peptide mixture spiked with either negative control peptide, positive control peptide, or neither, are spotted onto a slide and cross linked to magnetic beads. The slide is positioned under C18 tape coated with DTT and a transfer magnet. (b) The slides are blotted together and the magnetic bead cross-linked peptides are magnetically extracted onto the C18 surface due to the transfer magnet. (c) DTT coating on the C18 tape reduces disulfide bonds, releasing cross-linked peptides from magnetic beads. (d) The transfer magnet is repositioned below the original slide. (e) Free magnetic beads are pulled back onto the original slide due to their attraction to the transfer magnet, leaving freed peptides on the C18 surface for MALDI-TOF IMS analysis. | |
Extraction from mouse brain tissue section
Mouse brain tissue was sectioned into 15 μm-thick coronal sections using a cryostat sectioning device at −20 °C. Sections were first digested using a HTX Imaging TM sprayer with an on-tissue tryptic digest.17 The on-tissue digest protocol was performed according to manufacturers specifications (aqueous solvent with 50 mM ABC and 0.01 mg mL−1 trypsin, 45 °C nozzle temperature, 14 passes, 0.1 mL min−1 flow rate, 1200 mm min−1 velocity, 3 mm track spacing, 10 psi nitrogen pressure, 3 L min−1 gas flow rate with 30 seconds drying time). A tissue section was then coated with heterobifunctional cross-linker and beads and blotted onto C18. Cross-linker coating was conducted using an HTX Imaging TM-sprayer with 45 °C nozzle temperature, 8 passes, 0.1 mL min−1 flow rate, 1200 mm min−1 velocity, 3 mm track spacing, 10 psi nitrogen pressure, 3 L min−1 gas flow rate with 30 seconds drying time. After cross-linker coating, the sample was incubated for two hours at 37 °C in a moist, covered petri dish to prevent sample evaporation while the linker reacts with peptides in the tissue section. Samples were then coated with 225 μg of magnetic beads using a 10 μL syringe and incubated as previously described. 225 μg of beads in a 10 μL volume was chosen as it is the highest concentration that allows deposition using a syringe. Such a high concentration would ensure that the entire tissue surface is in contact with beads. Other deposition methods such as a TM-Sprayer cause the beads to dry out, inhibiting the cross-linking reaction. After incubation, samples were blotted onto DTT-coated C18 tape using the blotting protocol from the previous experiment (Fig. 3). The tissue blot and a digested tissue sample were then analyzed and compared using MALDI-TOF IMS.
Results and discussion
The present study gauged the validity of this active extraction method and its ability to isolate certain subsets of peptides, as well as its ability to operate on a solid-substrate as opposed to in-solution. By doing so, we demonstrated that this active extraction approach provides us with a method to obtain a unique subset of detectable peptides from tissue samples without compromising their spatial localization.
Spiked peptide standard
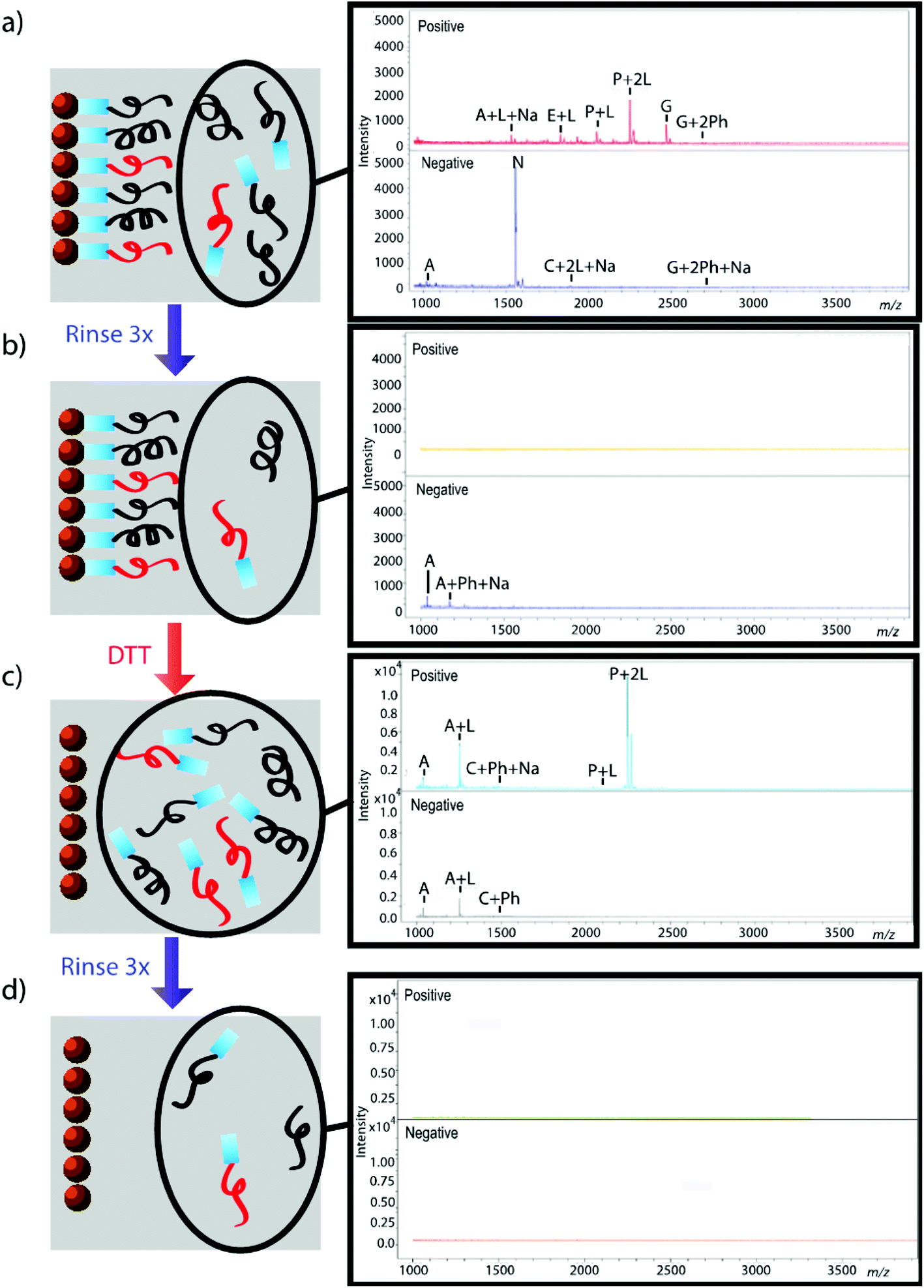
The effectiveness of the magnetic bead-based active extraction method to selectively isolate peptides in a more complicated matrix was assessed using a peptide calibration standard mixture spiked with synthetic positive or negative control peptides. This peptide standard mix contains 7 peptides, ranging in MW from 1000–3150 Da (Table 1). The amounts of each peptide in the standard mixture vary, with angiotensin II being the most abundant peptide species in the mixture. Peptides A through H contain a free primary amine on their N-terminus; and thus, have the ability to conjugate to a linker molecule (Fig. 1). The negative control peptide, which has an acetylated N-terminus does not conjugate to the linker molecule. In addition to a free primary amine at the N-terminus, the positive control also contains a C-terminal lysine, allowing it to bind two linker molecules. Peptides were extracted as previously described and the supernatant, before and after reduction by DTT, was collected for MALDI-TOF analysis (Fig. 4). The majority of the unconjugated peptides were washed off during the three rinsing steps. Upon reduction by DTT, the disulfide bonds between the beads and linker were broken and the peptides were released. Mainly angiotensin II and the positive control peptide were captured by the beads, most likely due to their abundance in the standard mixture of peptides. Because all of the peptides in the standard mixture meet the criteria for selective extraction, it is logical that the most abundant peptides in the solution would bind preferentially. In addition, the absence of the negative control peptide after reduction by DTT indicates its inability to bind to the beads, verifying the selective extraction of peptides with primary amines from a solution.
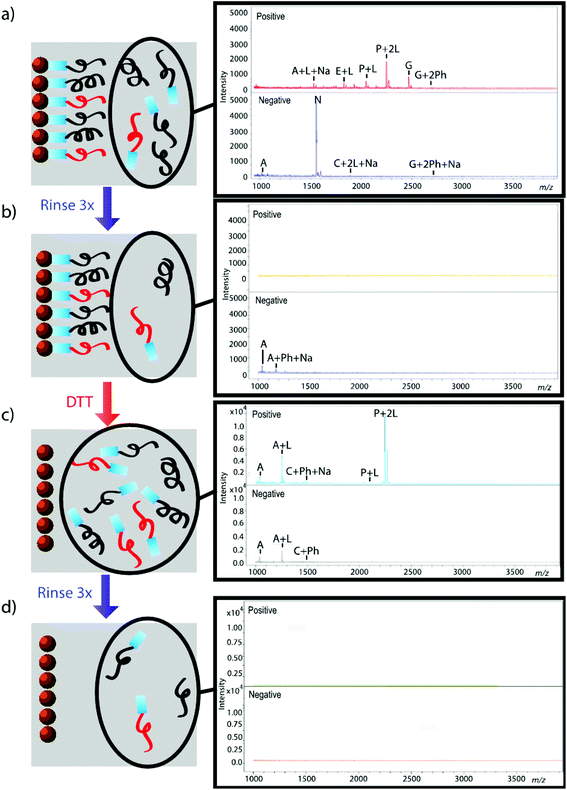 |
| | Fig. 4 Standard peptide mixture spiked with either positive or negative control peptide. Peptides that were not conjugated to beads before (a, b) and after (c, d) reduction by DTT were analyzed using MALDI-TOF. Peptides are denoted with their label from Table 1. Labels Ph, Na, and L denote the presence of a phosphate adduct, sodium adduct, or bound linker, respectively. Absence of signal after rinsing indicates that peptides were successfully removed from solution (b, d); thus, reemergence of signal after reduction by DTT demonstrates that peptides were successfully released from beads (c). | |
Table 1 Peptides from the calibration standard, as well as control peptides that are spiked into the peptide solution. Not all peptides in the solutions are present in equal amounts
| Peptide |
[M + H]+ average (Da) |
Label |
| Angiotensin II |
1047.19 |
A |
| Angiotensin I |
1297.49 |
B |
| Substance P |
1348.64 |
C |
| Bombesin |
1620.86 |
E |
| ACTH clip 1–17 |
2094.43 |
F |
| ACTH clip 18–39 |
2466.68 |
G |
| Somatostatin 28 |
3149.57 |
H |
| Positive control |
1840.00 |
P |
| Negative control |
1554.00 |
N |
Extraction on solid substrate
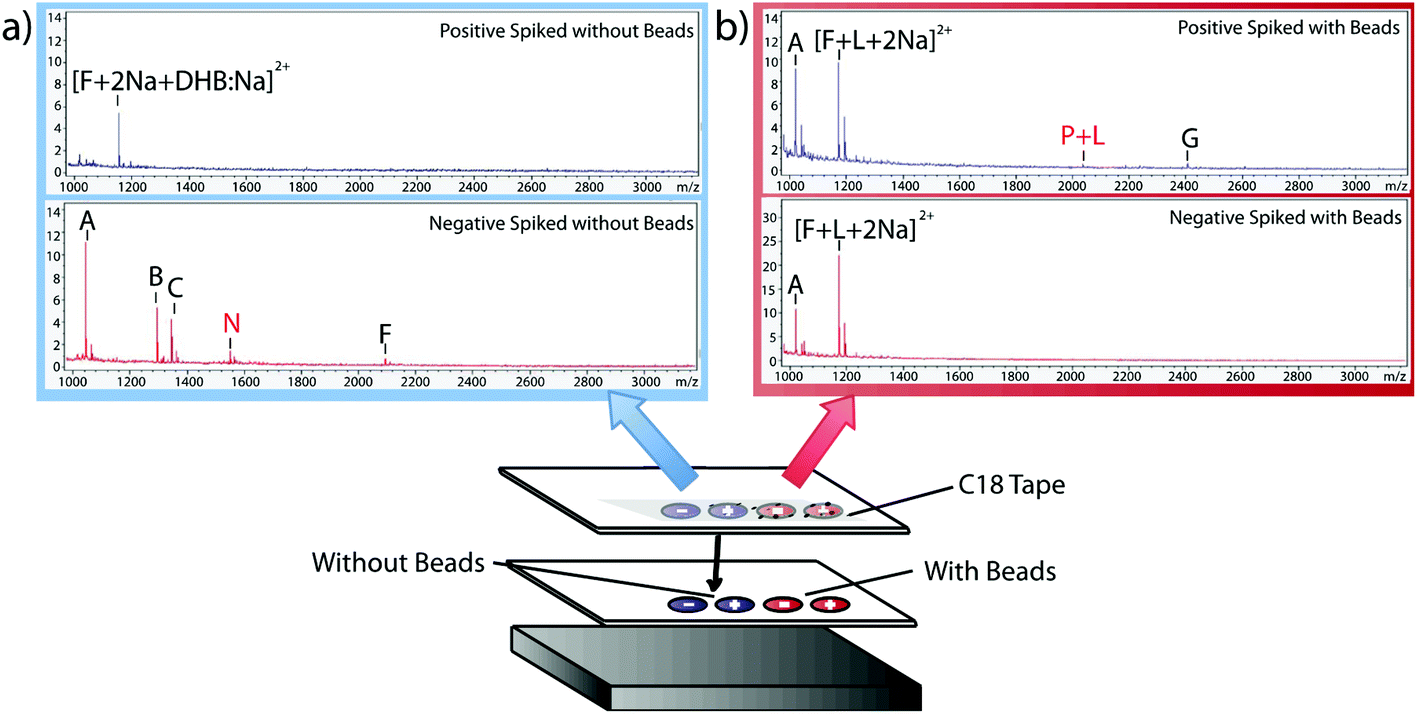
Having verified the effectiveness of this method with respect to selectivity in a complicated matrix, the next goal was to incorporate this bead-based approach into a normal blotting protocol, like those constructed by Caprioli et al.7 First, we tested the reaction chemistry on a solid substrate. Peptide solutions spiked with either the positive or negative control peptides were spotted onto ITO-coated glass slides and reacted with high concentrations of linker and magnetic beads (Fig. 3). These beads were then blotted onto C18 tape using a transfer magnet and analyzed using MALDI-TOF IMS. Peptide solutions that were blotted using the beads were compared to peptide solutions blotted onto a C18 tape collection surface without the presence of linker or beads (Fig. 5). A subset of peptides was blotted more efficiently in the presence of beads; in this case, angiotensin II, ACTH clip 1–17, and the positive control peptides. In addition, it appears that none of the positive control peptides transferred in the absence of beads and none of the negative control peptides transferred in the presence of beads. This is due to the positive control peptide's ability, and the negative control peptide's lack of ability to conjugate to the heterobifunctional crosslinker and thus bind to the magnetic beads. Without the use of the active extraction approach, the positive control peptide was left behind. These data indicate that the magnetic bead-based active extraction method can be used to selectively blot certain subsets of peptides. Although not all of the species in the peptide standard were blotted in the presence of beads, a specific subset of peptides were transferred more efficiently by beads when compared to passive diffusion. In addition, these results verify that the reaction chemistry can be achieved on a solid substrate as well as in-solution.
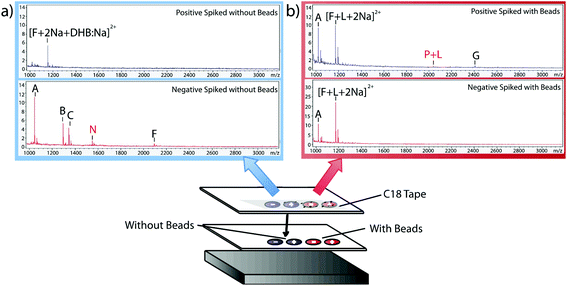 |
| | Fig. 5 MALDI-TOF IMS of peptides on C18 collection surface using passive diffusion (a) versus magnetic bead-based active extraction (b). Positive and negative control peptides are labeled P and N, respectively. The positive control was present only after blotting with the active extraction approach. In addition, it is clear that magnetic bead-based extraction only extracts a certain subset of peptides when compared to passive diffusion. | |
Extraction from mouse brain tissue section
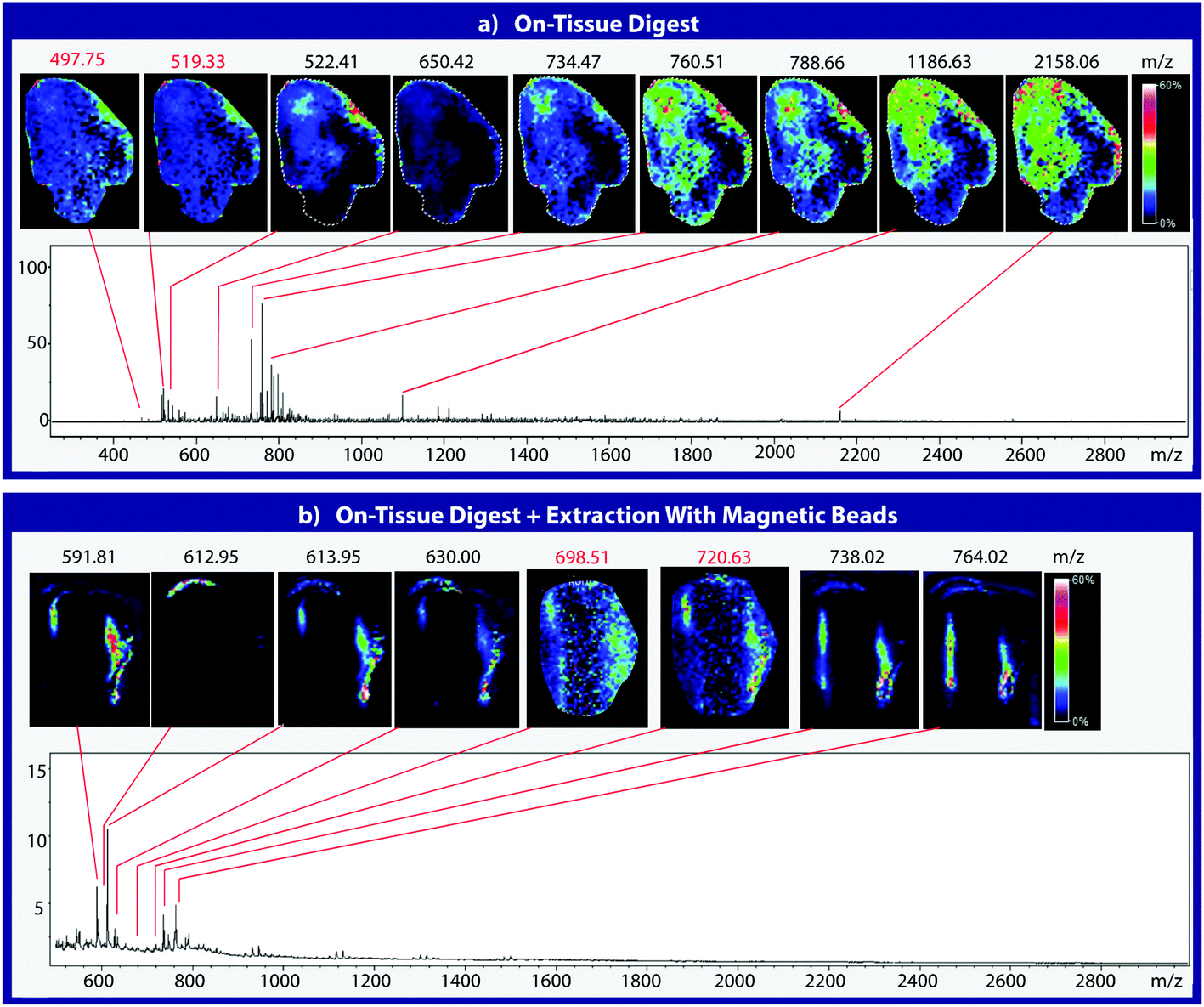
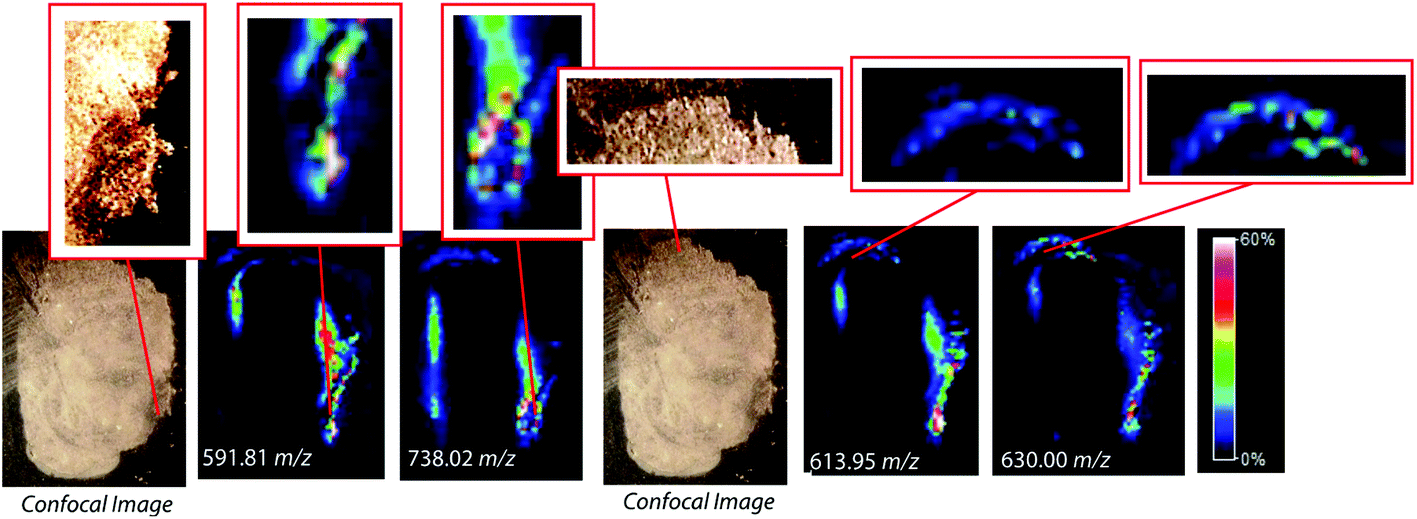
To assess the effectiveness of this method with respect to retaining spatial localization and the potential translational impact of this technique, tryptic peptides from a mouse tissue section were extracted onto a C18 collection surface using the magnetic bead-based extraction approach. The tissue blot, along with an un-extracted tryptically digested tissue section, were imaged using MALDI-TOF IMS (Fig. 6). Comparison of the two tissue sections show several analytes that are detected in both the tissue blot and the tissue section, which are highlighted in red. In the tissue blot, analytes that were successfully extracted have an additional 201 Da due to the presence of the linker, as demonstrated in Fig. 1. These analytes are detected in both samples without significantly compromising their spatial localization. Both analytes appear to be centralized to the right side of the tissue and around the edges. Although heat maps for the two samples are not completely identical, it is expected that the two tissue sections will have slight variations, as they are consecutive slices. In addition, it is possible that the active extraction mechanism was able to extract species that would have otherwise been suppressed in a complex tissue sample, leading to additional localization on the upper left region of the tissue section. Active extraction also yields analytes that were not observed in the digested tissue section (Fig. 6). Analytes at 591 m/z, 738 m/z, 613 m/z and 630 m/z were observed, co-localizing with distinct substructures of the brain after MALDI-TOF IMS analysis (Fig. 7). In addition, conservation of these substructures when compared to a confocal microscopy image of the tissue slice indicates that the method is able to extract these peptides without compromising their spatial localization. It is important to emphasize that these analytes, along with their native m/z counterparts, were not observed when imaging the digested tissue section using MALDI-TOF IMS. These results suggest that the magnetic bead-based extraction approach allows the detection of additional peptides from tissue samples without compromising their spatial localization, suggesting that this method would be a good complementary approach to tryptic digestion or tissue blotting methods for imaging peptides in tissue samples.
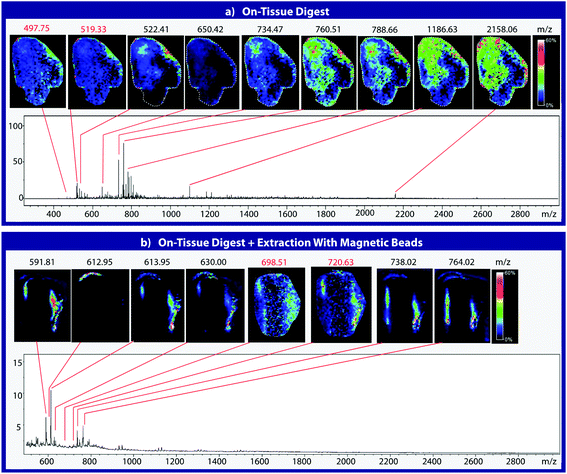 |
| | Fig. 6 MALDI-TOF IMS of mouse brain tissue sections after on-tissue digestion with trypsin (a). Tissue sections were then prepared using the magnetic bead-based active extraction approach and blotted onto a C18 collection surface (b). The presence of linker results in the addition of approximately 201 Da, as indicated by m/z highlighted in red. Species extracted using the linker appear to have similar spatial localization when compared to their native m/z counterparts. | |
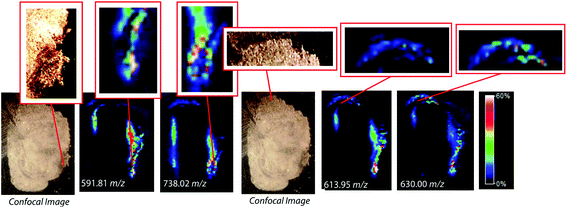 |
| | Fig. 7 Comparison of confocal microscopy of mouse brain tissue section and MALDI-TOF IMS of a mouse brain tissue section after on-tissue tryptic digestion and magnetic bead-based peptide extraction. Several tissue substructures remain distinguishable after blotting with the active extraction method. | |
Conclusions
Throughout this study, we have demonstrated that this magnetic bead-based active extraction approach, previously described by our lab, is not only translatable to a solid-surface peptide extraction, but is an effective method for isolating peptides from tissue samples. Our approach is able to extract analytes that were not observed in tryptically digested tissue samples, allowing a unique subset of detectable peptides to be obtained from a tissue section. In addition this method was successful in doing so, while maintaining spatial localization of the extracted species.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
ABH was supported by the National Institutes of Health (R01GM110406), and the National Science Foundation (CAREER Award, CHE-1351595). The UltrafleXtreme instrument (MALDI-TOF-TOF) was acquired through National Science Foundation award #1625944. We gratefully acknowledge the assistance of the Notre Dame Mass Spectrometry and Proteomics Facility (MSPF) and the generous donation of tissue samples by the M. Sharon Stack laboratory at the University of Notre Dame.
References
- T. C. Rohner, D. Staab and M. Stoeckli, MALDI mass spectrometric imaging of biological tissue sections, Mech. Ageing Dev., 2005, 126(1), 177–185 CrossRef CAS PubMed.
- M. Stoeckli,
et al., Imaging mass spectrometry: a new technology for the analysis of protein expression in mammalian tissues, Nat. Med., 2001, 7(4), 493–496 CrossRef CAS PubMed.
- V. Ntziachristos, C. Bremer and R. Weissleder, Fluorescence imaging with near-infrared light: new technological advances that enable in vivo molecular imaging, Eur. Radiol., 2003, 13(1), 195–208 Search PubMed.
- R. J. A. Goodwin, S. R. Pennington and A. R. Pitt, Protein and peptides in pictures: Imaging with MALDI mass spectrometry, Proteomics, 2008, 8, 3785–3800 CrossRef CAS PubMed.
- E. M. Weaver and A. B. Hummon, Imaging mass spectrometry: From tissue sections to cell cultures, Adv. Drug Delivery Rev., 2013, 65, 1039–1055 CrossRef CAS PubMed.
- B. Spengler, Post-source Decay Analysis in Matrix-assisted Laser Desorption/Ionization Mass Spectrometry of Biomolecules, J. Mass Spectrom., 1997, 32, 1019–1036 CrossRef CAS.
- R. M. Caprioli, T. B. Farmer and J. Gile, Molecular Imaging of Biological Samples: Localization of Peptides and Proteins Using MALDI-TOF MS, Anal. Chem., 1997, 69, 4751–4760 CrossRef CAS PubMed.
- H. Li and A. B. Hummon, Imaging mass spectrometry of three-dimensional cell culture systems, Anal. Chem., 2011, 83(22), 8794–8801 CrossRef CAS PubMed.
- X. Liu and A. B. Hummon, Mass spectrometry imaging of therapeutics from animal models to three-dimensional cell cultures, Anal. Chem., 2015, 87(19), 9508–9519 CrossRef CAS PubMed.
- X. Liu and A. B. Hummon, Chemical imaging of platinum-based drugs and their metabolites, Sci. Rep., 2016, 6, 1–10 CrossRef CAS PubMed.
- G. J. Labonia, S. Y. Lockwood, A. A. Heller and D. M. Spence, Drug penetration and metabolism in 3D cell cultures treated in a 3D printed fluidic device: assessment of irinotecan via MALDI imaging mass spectrometry, Proteomics, 2016, 1814–1821 CrossRef CAS PubMed.
- M. Z. Wang and M. C. Fitzgerald, A solid sample preparation method that reduces signal suppression effects in the MALDI analysis of peptides, Anal. Chem., 2001, 73, 625–631 CrossRef CAS PubMed.
- P. Chaurand, M. Stoeckli and R. M. Caprioli, Direct Profiling of Proteins in Biological Tissue Sections by MALDI Mass Spectrometry, Anal. Chem., 1999, 71, 5263–5270 CrossRef CAS PubMed.
- E. Fournaise and P. Chaurand, Increasing specificity in imaging mass spectrometry: high spatial fidelity transfer of proteins from tissue sections to functionalized surfaces, Anal. Bioanal. Chem., 2015, 407(8), 2159–2166 CrossRef CAS PubMed.
- L. A. Weston,
et al., Selective, bead-based global peptide capture using a bifunctional cross-linker, Anal. Chem., 2013, 85, 10675–10679 CrossRef CAS PubMed.
- K. K.-C. Yeung,
et al., CHCA or DHB? Systematic comparison of the two most commonly used matrices for peptide mass fingerprint analysis with MALDI MS, Spectroscopy, 2010, 25(2), 48–62 Search PubMed.
- M. R. Groseclose,
et al., Identification of proteins directly from tissue: in situ tryptic digestions coupled with imaging mass spectrometry, J. Mass Spectrom., 2007, 42(2), 254–262 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *
*