
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn vitro field potential monitoring on a multi-microelectrode array for the electrophysiological long-term screening of neural stem cell maturation†
Diana
Seidel
a,
Heinz-Georg
Jahnke
 a,
Beate
Englich
a,
Mathilde
Girard
b and
Andrea A.
Robitzki
*a
a,
Beate
Englich
a,
Mathilde
Girard
b and
Andrea A.
Robitzki
*a
aCentre for Biotechnology and Biomedicine (BBZ), University of Leipzig, Division of Molecular Biological-Biochemical Processing Technology, D-04103 Leipzig, Germany. E-mail: andrea.robitzki@bbz.uni-leipzig.de; Fax: +49 341 9731249; Tel: +49 341 9731241
bCECS, I-STEM, AFM, Institute for Stem Cell Therapy and Exploration of Monogenic Diseases, Genopole Campus 1, 5 rue Henri Desbruères, 91030 Evry Cedex, France
First published on 12th April 2017
Abstract
Due to the lack of appropriate cell models as well as automated electrophysiology monitoring technologies, the standardized identification of neurotoxic or protective effects in vitro remains a major problem in today's pharmaceutical ingredient development. Over the past few years, in vivo-like human pluripotent stem cell-derived neuronal networks have turned out to be a promising physiological cell source, if the establishment of robust and time-saving functional maturation strategies based on stable and expandable neural progenitor populations can be achieved. Here, we describe a multi-microelectrode array (MMEA)-based bioelectronics platform that was optimized for long-term electrophysiological activity monitoring of neuronal networks via field potential measurements. Differentiation of small molecule-based neuronal progenitors on MMEAs led to functional neurons within 15 days. More strikingly, these functional neuronal cultures could remain electrophysiologically stable on the MMEAs for more than four weeks. The observed electrophysiological properties correlated with the expression of typical neuron subtype markers and were further validated by specific neurotransmitter applications. With our established monitoring platform, we could show for the first time the long-term stability of the neural stem cell-like progenitor population to differentiate to electrophysiologically active dopaminergic neuronal networks for more than 80 passages. In conclusion, we provide a comprehensive long-term stable field potential monitoring platform based on stem cell-derived human neuronal networks that can be automated and up-scaled for standardized high-content screening applications e.g. in the field of neurotoxic and neuroprotective therapeutics identification.
Introduction
In recent years, human pluripotent stem cells have emerged as a promising tool for the generation of physiological and pathological in vitro cell-based models.1 Especially, a high demand for pluripotent stem cell-derived neuronal tissues exists, since there is no broad access to human primary neurons and immortalized neuronal cell lines are not able to reflect native phenotype concerning metabolism and electrophysiological activity.2In contrast to the well-investigated but susceptible and time-consuming direct neuronal conversion of human pluripotent stem cells,3 neural progenitor cell lines (NPC) with a distinct proliferation capacity can be generated as an intermediate cell population allowing the reliable terminal differentiation of the desired neural tissue (neuron-glia and neuron subtype proportions) in manageable time intervals.4 However, most NPC generation protocols are chemically undefined, contain multiple manual manipulation steps and are not robust in terms of efficiency and NPC homogeneity or stability, which hinders standardization, lab-to-lab reproducibility and scale-up.5 Currently, long-term neuroepithelial-like stem cells (lt-NES) are the only progenitor population that have been described to be expandable over more than 100 passages without losing its proliferation and highly neurogenic differentiation potential.6
Besides a high-end cell model, non-invasive bioanalytical measurement techniques are necessary for non-invasive label-free monitoring over time. In this context, we published an actual work that demonstrates the feasibility of using impedance spectroscopy for monitoring and quantifying the initial differentiation process of neuronal differentiation.7 Nevertheless, for late maturation and functional monitoring of finally differentiated and mature neuronal cultures, the monitoring of neuronal network electrophysiology is crucial, especially in safety assessment to identify the effect of active ingredients on human neuronal cell models. In contrast to traditional technologies such as patch clamp that are invasive and single cell-dependent and hinder scale-up, microelectrode array (MEA)-based field potential recording fulfills the demands of advanced neuronal network electrophysiology characterization.8,9 However, the application of stem cell-derived neurons includes additional requirements such as the support of long-term culture and functional differentiation into spontaneously active neurons in an environment only partially resembling the situation in vivo.
The implementation of neuronal networks directly differentiated from human embryonic stem cells (hES) for MEA-based electrophysiology analysis was reported previously.10 However, the generation of active neuronal networks originating from human induced pluripotent stem cells (hiPS) in vitro was found to be more complicated.11,12 Furthermore, to date no research study has been reported that describes the use of stable, non-commercial, human NPC line-derived neurons for electrophysiology recording on MEA.
Here, we demonstrate for the first time the use of a novel, self-developed long-term stable hiPS-derived neural stem cell-like population in combination with our in-house, neuro-optimized MMEA-based field potential recording technology for the non-invasive, sensitive analysis of spontaneous and modulated long-term neuron network electrophysiology.
Experimental
hiPS lines and culture
The hiPS lines IMR90-4 (hiPS1), IMR90c01 (hiPS2), and 4603c27 (hiPS3) were used for NPC differentiation. hiPS characteristics are stated in ESI Table S1.† The culture procedure is described in the ESI Methods.†NPC generation and differentiation
hiPS were exposed to a NPC generation and cultivation strategy.4Neuronal differentiation of NPC was induced after at least ten passages in neuronal differentiation medium (NDM) or NDM (0.17 ml cm−2) conditioned for four days on a confluent primary rat astrocyte layer. The composition of NDM is given in ESI Table S2.†
MMEA fabrication
MMEAs were produced in our clean room facility by the lift-off technique. Under clean room standard conditions, a glass substrate surface (Borofloat 49/49/1.1 mm, Industriearmaturen Göttgens, Germany) was cleaned in a cascade of ultrapure water, acetone, isopropanol and ultrapure water each for 2 min into a pulsed ultrasonic bath followed by a cascade of piranha etch and ultrapure water to remove organic contaminants. After drying the glass substrate via a spindryer and baking on a 200 °C hotplate for 5 min, spin-coating of the positive resist (AR-P 3510, Allresist, Germany) for 1w54e was done as stated in the manufacturer's manual, creating coatings with a thickness of 3 μm. After baking for 90 s at 95 °C, the MMEA layout was passed on the substrate via a photomask structure (chromium mask, Compugraphics, Germany) using 10 s UV-light (350 nm to 425 nm) exposure (MA6 Mask Aligner, SÜSS MicroTec, Germany). Structures were developed in AR-P 300-35, and the substrate was rinsed and dried. DC sputtering (CREAMET 500, CREAVAC GmbH, Germany) of 50 nm ITO (In2O3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :SnO2, 90/10 wt%, EVOCHEM, Germany) as the adhesion layer followed by 350 nm gold (99.99%) as the electrode material was processed under an argon atmosphere at 4.0 × 10−3 mbar and 1.0 A/0.7 A. The remaining photoresist and metal deposition thereon were removed in acetone, and the sensor array was cleaned in isopropanol and ultrapure water prior to dehydration at 200 °C for at least 30 min. To produce a cell adhesion-supporting surface as well as an insulation of the circuit paths, a passivation layer of negative photoresist NANO™ SU8-2 (Micro Resist Technology, Germany) was used and processed via spin coating, pre-baking (1 min at 65 °C followed by 1 min at 95 °C), UV-light exposure for 4.5 s, post-baking (1 min at 65 °C and an additional 1 min at 95 °C) and development (1 min, MicroChem's mr-Dev 600, Allresist, Germany). The resultant film thickness of 1 μm of SU8 layer was verified using a confocal microscope (Micro Spy Topo, FRT GmbH, Germany). The MMEA was cleaned in ultrapure water, spin-dried and plasma-cleaned at 400 mA for 7 min in the vacuum chamber (CREAMET 500, CREAVAC GmbH) at 2.5 × 10−2 mbar with an argon flow of 0.2 sccm. A culture chamber in 4 wells of 384-well format (Greiner Bio-One, Germany) was bonded with silicon (Sylgard 184 Silicone Elastomer, Dow Corning) onto the sensor array. To ensure consistent MMEA quality, parameters such as the total number of functional electrodes, homogeneity of the passivation layer and cleanness of the electrode surface were controlled by performing a blank value impedance measurement in 0.1% Triton in phosphate-buffered saline.
:SnO2, 90/10 wt%, EVOCHEM, Germany) as the adhesion layer followed by 350 nm gold (99.99%) as the electrode material was processed under an argon atmosphere at 4.0 × 10−3 mbar and 1.0 A/0.7 A. The remaining photoresist and metal deposition thereon were removed in acetone, and the sensor array was cleaned in isopropanol and ultrapure water prior to dehydration at 200 °C for at least 30 min. To produce a cell adhesion-supporting surface as well as an insulation of the circuit paths, a passivation layer of negative photoresist NANO™ SU8-2 (Micro Resist Technology, Germany) was used and processed via spin coating, pre-baking (1 min at 65 °C followed by 1 min at 95 °C), UV-light exposure for 4.5 s, post-baking (1 min at 65 °C and an additional 1 min at 95 °C) and development (1 min, MicroChem's mr-Dev 600, Allresist, Germany). The resultant film thickness of 1 μm of SU8 layer was verified using a confocal microscope (Micro Spy Topo, FRT GmbH, Germany). The MMEA was cleaned in ultrapure water, spin-dried and plasma-cleaned at 400 mA for 7 min in the vacuum chamber (CREAMET 500, CREAVAC GmbH) at 2.5 × 10−2 mbar with an argon flow of 0.2 sccm. A culture chamber in 4 wells of 384-well format (Greiner Bio-One, Germany) was bonded with silicon (Sylgard 184 Silicone Elastomer, Dow Corning) onto the sensor array. To ensure consistent MMEA quality, parameters such as the total number of functional electrodes, homogeneity of the passivation layer and cleanness of the electrode surface were controlled by performing a blank value impedance measurement in 0.1% Triton in phosphate-buffered saline.
Field potential measurements of cellular network electrophysiology
NPC (0.25 m cells per cm2) were seeded on Matrigel-coated (Corning, 1:30 dilution in DMEM/F12) MMEAs in NDM and cultured for ten to 15 days to develop electrophysiological activity. Two-third of the medium was changed at least every third day.
Two-third of the medium was changed 6 h before electrophysiology measurements. MMEAs were placed in a heatable MEA1060 amplifier system (Multi Channel Systems, Germany) and equilibrated for 10 min. Field potential streams were recorded from electrodes with a sampling rate of 40 kHz by using MC-Rack v3.7 software (Multi Channel Systems, Germany). The number of spikes per min and the spike rate were determined at 10 min intervals. To assure statistically analyzable data sets over a long time range, 2–3 independent experiments per differentiated NPC population each comprising five MMEAs were carried out. Despite a number of electrodes available that showed spontaneous but temporary activity, conservatively only electrodes with distinct long-term stable electrophysiological activity (spike amplitude: >5 × SD [∼15 μV], spikes per minute: >60, spike rate: >10 s−1) were analyzed and included in the statistics. The parameter values for selecting the data were reasonably based on the overall activity pattern of hiPS-derived neuron cultures as already described elsewhere.13–15 The number of analyzed electrodes (distributed over one to four MMEAs per condition and time point) ranged from four to twelve and was expressed as the mean ± s.e.m.
To determine the neurotransmitter influence, one constantly active MMEA per NPC population was chosen for experiments. Parameter number of spikes per minute was measured for 5 min, the substance was applied in an effective concentration (ESI Table S5†) and the change in the number of spikes per minute was analyzed over 10 min. The effect of the neurotransmitter on the firing rate was determined as the number of spikes per minute after the application was normalized to the number of spikes per minute before application, described as the mean ± s.e.m.
Molecular biological analysis
Flow cytometry, immunocytochemical staining and quantitative real-time (qRT) PCR analyses are described in the ESI.†Statistical analysis
All statistical analyses were done using GraphPad Prism 5. Values are given as the means ± s.e.m. unless described differentially. Multiple group comparisons were made by 2-way ANOVA and Bonferroni's post hoc test. Comparisons between two groups were made by t-test. Differences between two means were considered to be significant if p < 0.05, very significant if p < 0.01, and highly significant if p < 0.001.Results
Optimization of MMEAs for long-term neuron cultures and electrophysiology monitoring
For the detailed analysis of the electrophysiology of a complex neuronal network, the MEA layout had to be adapted. In general, the electrode size and density are critical factors for monitoring neuronal activity. While a smaller electrode diameter and increased electrode number are aspired for an optimum resolution and maximal signal, increasing the impedance of the electrode and therewith the background noise limits the positive effects of the miniaturization.16 In this context, we analyzed the influence of the electrode size on the noise (Fig. 1a, right). While electrode diameters of 50 μm or more showed comparable, not significantly different maximal noise signals of 6–12 μV. Smaller electrode sizes led to highly significant increases in maximal noise to two- (20 μm) and fourfold (10 μm) the value of 50 μm electrodes. It is noteworthy that the variance of the maximal noise also clearly increased with decreasing the electrode diameter from 0.6 μV (50 μm) to 5.0 μV (25 μm) and 7.6 μV (10 μm). Thus, for the identification of small action potential amplitudes (>15 μV), electrodes with a diameter of 50 μm or higher are favorable. However, to achieve a high spatial resolution and avoid averaging effects, an electrode diameter of 50 μm was chosen for the MMEA layout (Fig. 1b).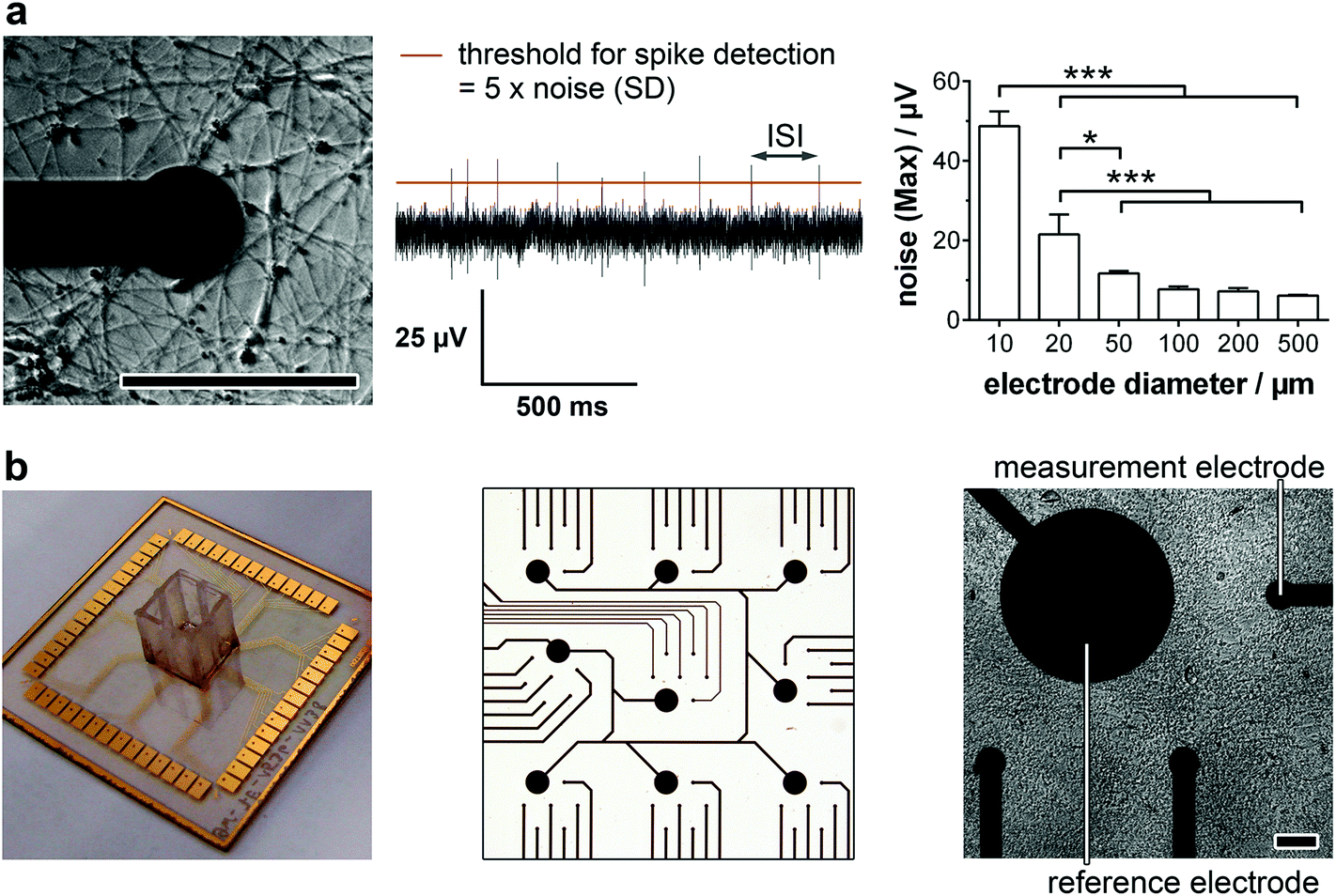 | ||
| Fig. 1 MMEA validation for electrophysiology monitoring of neurons. (a) Micrograph of a neuronal network at one electrode (left) and a corresponding field potential trace (middle). For spike detection, a threshold (red line) is set and the number of spikes per minute and spike rate (1/ISI) were analyzed. Dependence of maximal noise signal on the electrode diameter (right). n = 4 electrodes. (b) 1w54e MMEA (left: 1 well [0.67 cm2] per MEA with 54 measurement electrodes), detailed electrode layout (middle) and MMEA with neuron cell layer (right). ISI = inter-spike interval. Scale bars 100 μm. | ||
To allow for distinct neuronal action potential identification the background noise of the chosen 50 μm electrodes had to be dynamically determined by calculating the standard deviation and setting a detection threshold five times the standard deviation (Fig. 1a, middle). Since the background noise of the microelectrodes (50 μm diameter) was in the range of 3 μV (ESI Fig. S1†), the detection of small action potentials with amplitudes of at least 15 μV was possible. Microelectrodes with a noise higher than 4 μV were discarded from the monitoring.
Besides the electrode diameter, the electrode distance was also adapted to the neuron morphology with small cell bodies and extended processes. A 500 μm distance of six electrodes within a delimited area (1.5 × 1.5 mm) was chosen to assure the spatial resolution of small network activities. The regular arrangement of nine of these electrode fields over an array (6.0 × 6.0 mm) even allows the identification of large interconnected networks (Fig. 1b).
For long-term adherence and maturation of neuronal progenitor cells, the surface of the MMEA had to be functionalized. Therefore, the gold electrodes and SU8 polymer passivation were plasma-treated and coated with high concentrations of Matrigel each time before cell seeding. To achieve a low-density neuronal network after differentiation, which, with respect to electrode size and arrangement, allowed for optimal spatial resolution of the recorded action potentials and prevented average signal detection (Fig. 1a, left), NPC had to be seeded with a density of 150k–250k cells per cm2.
High-quality neuronal network properties of a well-characterized neural stem cell-like progenitor population on MMEA
To guarantee the application of a standardized high-quality neural progenitor population on our MMEA-based electrophysiology screening platform, we relied on a chemically-defined, small molecule-based NPC generation protocol without the need for subjective manual cell selection.4 During the hiPS–NPC transition, cell morphology as well as marker expression were tracked in detail. Within 20 days, an equally primed progenitor population could be established of the three applied hiPS lines that showed expression of typical ectodermal/neural progenitor markers without mesendodermal contaminants or spontaneous neuronal pre-differentiation (ESI Fig. S2–S4a†). The generated NPC could be expanded easily (population doubling time ∼70 h) and retained their primitive anterior mid-/hindbrain identity (ESI Fig. S4b and c†) as well as high neurogenic/low gliogenic potential over more than 30 passages proving high availability, which is an essential criterion for standardized high-throughput in vitro cell systems (ESI Fig. S5†). With these results, we could validate the hES-based protocol of Li and colleagues to be functional for a subset of hiPS lines as well.The establishment of an electrophysiologically active neuronal network is a more stringent criterion for high quality NPC identification than neural marker expression. When progenitor cells were differentiated on MMEAs, the first action potentials could be detected between days 12 and 15. This finding correlated well with molecular biology. After two weeks of differentiation, the dendrite marker MAP-2 and the synapse marker Synapsin I colocalized and showed a significant gene expression increase. Furthermore, the relative amount of Synapsin I in MAP-2+ neurons was found to be similar for all the established progenitor lines, showing their homogeneity (Fig. 2a).
 | ||
| Fig. 2 MMEA-based field potential recording of neuronal network electrophysiology. (a) Immunostaining of the co-localized neuronal marker MAP-2 (red) and the synapse marker Synapsin I (green) of NPC2diff. Gene expression induction of MAP-2 and SYNI; quantified relative to the NPC state (dashed line; 100%). Immunostaining fluorescence intensity of Synapsin I relative to MAP-2. Scale bar 50 μm. n = 3 experiments. (b) Field potential traces of different NPC-derived neuron cultures. (c) Morphological and electrophysiological validation of NPC2diff network activity by similar activity patterns (arrows) of channels 1–4 representing four adjacent electrodes (C1–C4). Scale bar 200 μm. | ||
When we then analyzed neuron activity traces, we could identify intervals of high activity (train spiking) separated by low-activity sequences (no or tonic, single spike activity). Spikes within one sequence were equal in amplitude ranging from 20 μV to 200 μV with most spike trains exhibiting an amplitude of 60–80 μV (Fig. 2b). Based on the background noise of approx. 3 μV (1 × SD; ESI Fig. S1†) and maximum noise peaks in the range of 10 μV (Fig. 1a), the detection of neuronal action potentials in the range of 20 μV and higher was clearly feasible. Therewith, the signal–noise ratio of our MMEA was validated to be superior to commercially available MEAs for field potential applications.14,15,17
Excitingly, clear network activities could be observed in all neuron cultures from day 15 on comparing spike patterns of adjacent electrodes (Fig. 2c). Thus, neuronal cultures were able to generate small interconnected networks of at least 1.5 × 1.5 mm dimension, which were mapped by the optimized MMEA electrode layout. Networks extended to the 9 × 7 electrode arrangement (6.0 × 6.0 mm) could not be detected.
With this finding, we are the first to show a clear network activity of human neurons even after short-time maturation periods.10,14,18
For a deeper understanding of the observed activity traces, we analyzed typical neuron network characteristics such as long-term activity and subtype specification in detail.
Long-term stable dopaminergic neuron network electrophysiology and systemic neurotransmitter modulation on MMEA
To assure interference-free long-term electrophysiology recording, we first analyzed the background noise stability of our microelectrodes (ESI Fig. S1b†). After a slight increase on day 20 (3.3 μV vs. 2.8 μV at the beginning), a very stable background signal could be determined (3.4 μV at day 25/30 and 3.3 μV at day 45).The long-term culture of physiologically functional human neurons is a very complex and versatile process, which is reflected in the overall action potential recording success. Percentage of active MMEAs decreased from 29.2 ± 7.1% at day 15 to 20.0 ± 0.0% at day 45. The proportion of active electrodes per MMEA stayed constant over time (day 15: 6.52 ± 0.66%; day 45: 6.48 ± 0.84%) and was comparable with results determined by others for hiPS-derived neurons.14
To quantify the electrophysiology characteristics of the long-term active human neuronal networks (Fig. 3a), the parameters spike number per minute and spike rate (1/ISI) were determined. The homogeneity of the progenitor population was again verified by neuron cultures exhibiting comparable spike numbers per minute (85–126) and spikes rates (12–18 s−1) (Fig. 3b). These active network characteristics reached the maximum (200–400 spikes per min, spike rate of 20–30 s−1) around day 30 and remained constant for more than two further weeks (Fig. 3c), which is in line with previous studies on mature human neural stem cells12 and primary human neurons.18
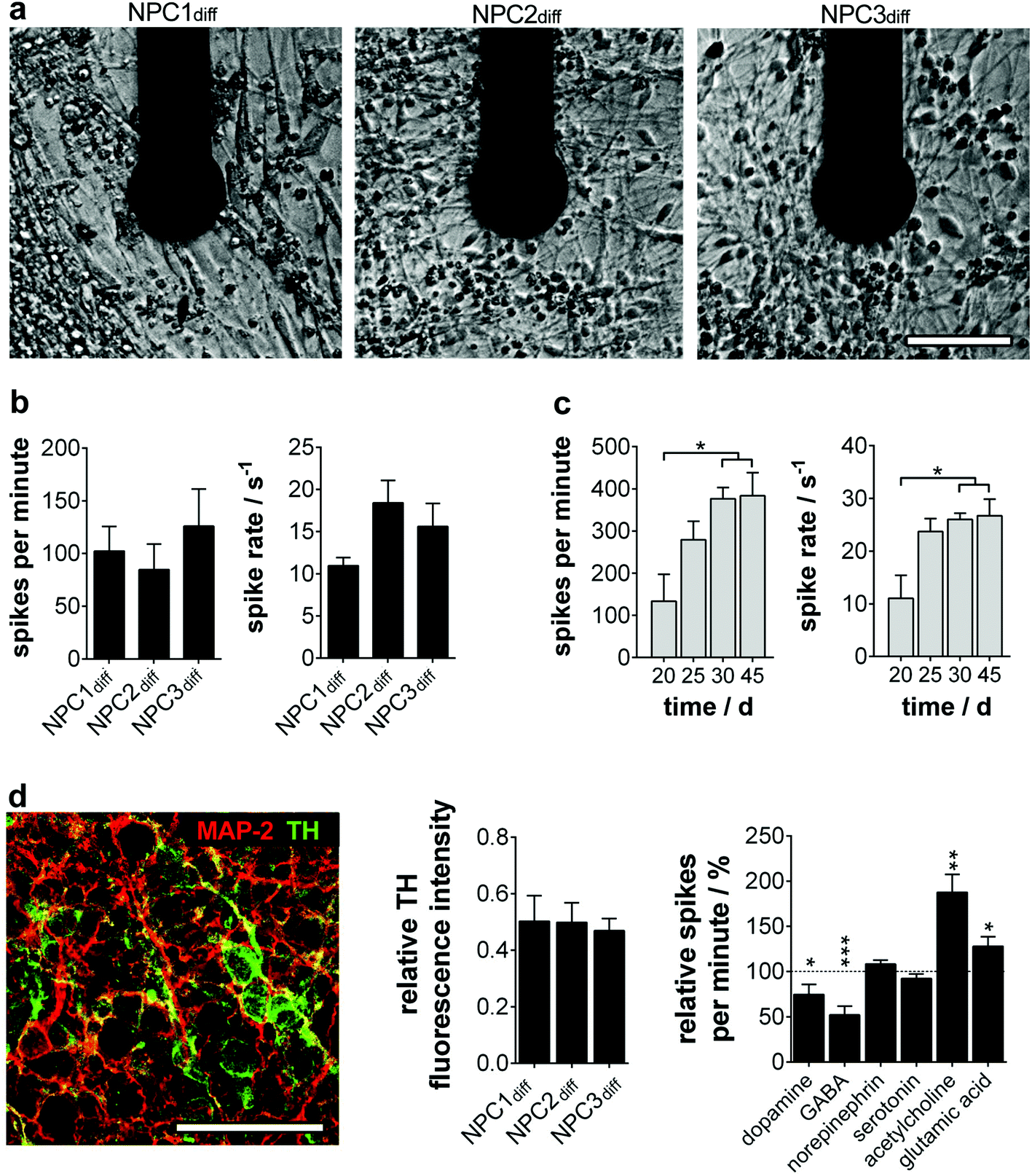 | ||
| Fig. 3 Neurotransmitter-induced changes of long-term stable electrophysiology parameters on MMEA. (a) Neuronal cells after 15 days of differentiation at one exemplary electrode. (b) Quantified activity parameters of neurons at day 15 of differentiation and (c) long-term stability analysis of NPC2diff electrophysiology parameters. n = 4–8 electrodes. (d) Neuronal subtype analysis. NPC2diff immunostaining of MAP-2 and TH (left). TH fluorescence intensity quantification relative to MAP-2. n = 3 experiments. Firing rate change (relative spikes per minute) after neurotransmitter application on NPC1diff cultures. n = 4–10 electrodes; significances relative to untreated cultures (dashed line; 100%). Scale bars 50 μm. | ||
Furthermore, we wondered on the subtype identity of NPC-derived neuronal networks, especially since subtype specification plays a major role in in vitro disease modelling. In accordance with the original data of Li et al., we found a predominant TH+ phenotype with ∼50% of MAP-2+ (Fig. 3d), whereas markers for cholinergic, glutamatergic and GABAergic neurons were only expressed sporadically (ChAT 1/1000; vGlut1 1/10000; GAD65/67 1/10000 MAP-2+ cells). The cell population was subsequently identified as dopaminergic neurons showing significantly decreased electrophysiological activity when dopamine was applied. A TH-expressing adrenergic subtype could be excluded by ineffective norepinephrine treatment (Fig. 3d). Furthermore, neurotransmitters GABA, glutamic acid and acetylcholine induced significant changes in the neuron firing rate. These inhibitory and excitatory inputs are consistent with in vivo and in vitro data on dopaminergic neurons.19,20
The absence of burst firing and the moderate tonic or spike train activity in the neuronal networks is consistent with the identified subtype specification. Previous studies described dopaminergic neurons in vitro to have a pacemaker phenotype resistant to burst firing even in the presence of activating stimuli.20,21
Verification of long-term neural stem cell-like progenitor character for advanced electrophysiology recording on MMEAs
To be applicable in a broad temporal as well as quantitative context in today's pharmaceutical assays, a functional well-characterized and stable cellular phenotype has to be assured.Therefore, we tested if the NPC population showed consistent maintenance and differentiation characteristics in early (<25 passages) and late (>80 passages) culture periods. Comparing gene and protein expression as well as localization, we found no differences in the progenitor state and even a significantly advanced neurogenic potential partially accompanied by a gliogenic phenotype for long-term NPC (ESI Fig. S6a and b†). This glial determination is a typical characteristic of prolonged progenitor maintenance.22,23 NPC ‘maturation’ was also verified by the significant decline of the rhombomeric marker KROX20. The increase of GBX2 points to the stabilization of the anterior mid-/hindbrain identity during prolonged culture (ESI Fig. S6c†).
We then analyzed the ability of long-term progenitors to form active neuronal networks on MMEA. No significant difference in the ability to express and structure Synapsin I in colocalization to MAP-2 was observed (Fig. 4a). As described previously, the spike traces showed the same pacemaker-like tonic and train firing pattern typical of dopaminergic neurons in vitro, which was also reflected in comparable activity parameters at day 15 of differentiation (Fig. 4b and c). The long-term stability of neuronal network activity for more than 45 days on MMEA was as independent of the NPC passage number (Fig. 4d).
 | ||
| Fig. 4 Stable electrophysiology characteristics of early- and late-passage NPC-derived neurons. (a) Cultured (early <25 passages; late >80 passages) and 15-day differentiated NPC2diff were stained for colocalization of the dendritic marker MAP-2 (red) and the synaptic marker Synapsin I (green). Scale bar 50 μm. Fluorescence intensity quantification of marker staining (right). n = 3 experiments. (b) Field potential trains, (c) MMEA-based field potential analysis of the activity parameters the number of spikes per minute and the spike rate and (d) stability over prolonged culture periods (day 15–day 45) for late passage NPC2diff. n = 4–9 electrodes. | ||
When analyzing the subtype specification of old passage NPC, we again found a mainly TH+ phenotype (Fig. 5a). However, quantitative analysis of protein and gene expression showed a significant decrease of TH+/MAP-2+ neurons during long-term culture from 50% to 38%. Furthermore, a significant increase of TPH1, a serotonergic neuron marker gene, could be observed (Fig. 5b, top).
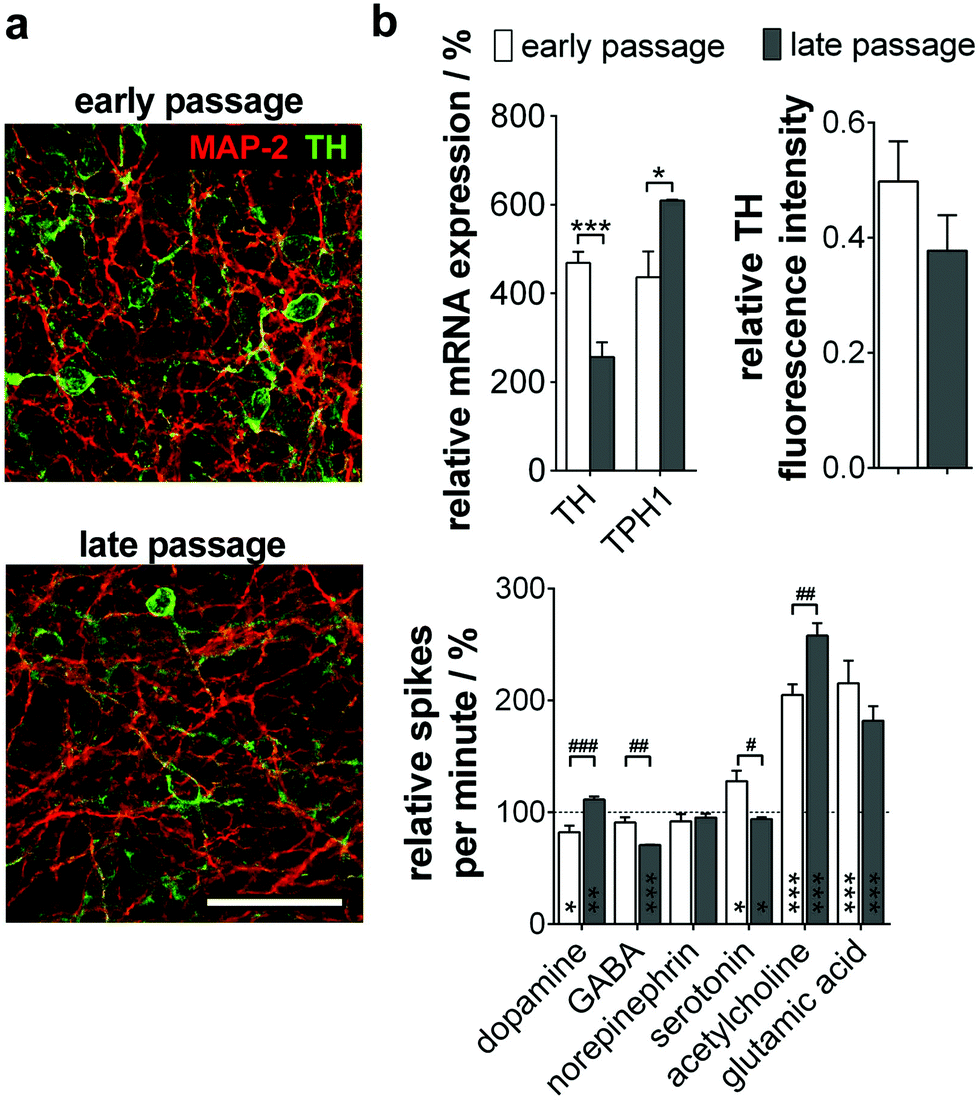 | ||
| Fig. 5 MMEA-based field potential detection of neuronal subtype proportions in neuron cultures of late-passage NPC. (a) Cultured (early <25 passages; late >80 passages) and 15-day differentiated NPC2 were stained for the dendritic marker MAP-2 (red) and the dopaminergic neuron marker TH (green). Scale bar 50 μm. (b) (Top, left) Identification of neuron subtype shifts via gene expression profile. n = 3 experiments. (Top, right) TH fluorescence intensity quantification relative to MAP-2. n = 3 experiments. (Bottom) Validation of subtype proportion-dependent activity changes after the application of typical neurotransmitters. n = 4–7 electrodes. *Significances relative to untreated cultures (dashed line; 100%); #significances between NPCs. | ||
We could prove these results by MMEA-assisted neuron electrophysiology recording under neurotransmitter control. Coherent with the subtype shift, treatment with dopamine did not change firing in neurons beyond passage 80, whereas serotonin inhibited the neuron firing rate significantly (Fig. 5b, bottom). Effects are exerted by inhibitory autoreceptors on the particular neuron subtype controlling neurotransmitter release. Furthermore, long-term passaged and differentiated NPC reacted significantly more sensitively to GABA and acetylcholine application. For both subtypes, functional interactions with serotonergic neurons in the Ralph nucleus and the hippocampus in vivo are described.24,25
Discussion
Based on the demand for functional assays on human neuronal networks in the context of safety assessments as well as pathology modeling and therapeutics development, both components, the monitoring technique as well as the biological part, have to be optimized and adapted with regard to signal feasibility and stability as well as time to assay (cultivation and maturation of neuronal networks) and long-term stability.The use of passive microelectrode array-based field potential recording for neuron network electrophysiology has been intensively described during the last 20 years.9,16 Due to its advantages in patch clamp and voltage-sensing optical measurements, this technology will become the standard for neuron activity recordings of complex electrogenic cell networks, tissues or even organs on a chip within the next few years.26
In this context, we introduce a novel microelectrode array specifically designed for the functional monitoring of neuronal networks. The nine separated fields (1.5 × 1.5 mm; total 6.0 × 6.0 mm) guarantee the optimal mapping of single neurons as well as network morphology. Based on the background noise and signal analysis, we identified unmodified planar gold electrodes with a diameter of 50 μm as optimal for the feasible and long-term stable detection of even small action potentials in the range of 20 μV and higher. This finding is also in line with previous studies on MEA-based field potential applications.16 Concerning the electrode material, in general noble metals (gold and platinum) are the first choice in an electrolyte environment because they are chemically inert. Nevertheless, in our opinion gold is more suitable since it has a higher conductivity allowing longer and thinner circuit paths than platinum and, moreover, there are hints that platinum electrodes can form neurotoxic species under electrochemical oxidative conditions as occurs in neuronal stimulation experiments.27 In contrast to gold, electrodes made of ignoble metals like tungsten generally interfere with neuron function or even cell viability.28
The use of unprocessed planar gold electrodes perfectly matches prerequisites concerning standardized and easy production of such kinds of MEAs.27,28 With our MMEA, complex surface enhancements such as coatings with polymers and/or carbon nanotubes or special production techniques e.g. nanocolumnar titanium nitride coatings are not necessary.29 This is again an advantage with regard to limited long-term stability and biocompatibility of these electrode modifications (degradation/release of polymer/carbon nanotubes).30 With the developed MMEAs coupled to a standardized recording platform, we provide an easy-to-use tool for neuron cell culture, electrophysiology measurements and data processing.
On the side of the biological part, mostly primary neuronal cultures of rodents have been applied for pharmaceutical electrophysiology recording on MEA in recent years. Based on the limited tissue availability and xenogeneity, the demands of today's pharmacological screening in standardization, automation and scale-up are not fulfilled.18
Human neuron cultures derived from pluripotent stem cells can overcome these drawbacks. However, only a few studies have described the direct differentiation of hES to highly active and stable neuronal networks.15,31 Additionally, as a reason for insufficient gene silencing after retroviral reprogramming, the electrophysiological network maturation of advantageous hiPS cells (no ethical concerns, patient-specific disease modeling) was found to be even more complicated.11 Furthermore, the generation of an intermediate progenitor population, which can be maintained and finally mature on demand in much shorter periods, is necessary to accelerate the extensive time course (more than two month) of functional hES/hiPS neuronal differentiation. Thus, with regard to patient-specific applications and availability, the robust and straightforward hiPS-derived NPC maturation protocol validated in this study offers a clear advantage over commercially available hiPS-based neurons, which were recently described to form long-term active neuronal networks on CMOS-based electrode-arrays.14
To achieve a readily available and long-term stable monitoring system, we combined our MMEAs with neural progenitor cell lines that were differentiated on the MMEAs. As a pre-requisite for the successful long-term cultivation of these cells on the MMEA, we identified a plasma surface cleaning before each use. The highly proliferative (population doubling time of 3 days) neural stem cell-like lines were established using a small molecule derivation strategy.4 When differentiated on highly polar plasma-treated MMEAs, evenly distributed low-density active neuron networks were established within two weeks. Typical neuron clustering impeding network electrophysiology recording was prevented.32 A stable neuron network activity over an area of at least 1.5 × 1.5 mm with a spike number of 200–400 spikes per min and a spike rate of 20–30 s−1 with signal amplitudes of 20–200 μV could be assured over one further month. These long-term network activity characteristics of the mature hiPS-based NPC population were found to be superior to monolayer data for direct stem cell-derived neurons with 100 spikes detected per minute after one month on MEA.13,14,33 More strikingly, all three hiPS lines applied in this study, although obtained from different sources, were comparable in their molecular and activity parameters, showing the stringency of the NPC generation protocol. Furthermore, changes in subtype determination of a neuronal network could be identified with high sensitivity by applying the hiPS-derived NPC model on our MMEA-based electrophysiology platform under neurotransmitter control.
To be applicable for standardized pharmacological screening, NPCs have to provide sufficient cell numbers for parallel testing and to show a stable phenotype over high passage numbers. To date, Koch and colleagues have been the only ones to identify long-term neuroepithelial-like stem cells (lt-NES) that meet these criteria.6
Now, we could also prove a small molecule-based protocol to generate a neural stem cell-like population, which can be cultured over more than 80 passages without losing their consistent ability to proliferate and differentiate into high-quality, active networks of dopaminergic/serotonergic neurons.
Conclusion
We could prove, for the first time, functional hiPS-derived neural progenitor network maturation and long-term electrophysiology characterization on a specifically optimized MMEA-based field potential technology platform. Importantly, the MMEA layout can be transferred to a 96 well format allowing an easy scale-up of the array.34 Together with the readily available and scalable, long-term neural stem cell population, the technology platform offers an ideal model for automated and standardized biomedical applications e.g. safety assessment, therapeutics identification and optimization or hiPS transgenic disease modelling.Acknowledgements
This work was funded by the European Community's 7th Framework Program (FP7/2007–2013 and Cosmetics Europe based on Grant Agreement No. 266753 (Scr&Tox)). The image acquisition facility (confocal microscope) was funded by the Free State of Saxony and the European Union (SMWK/EFRE – Grant No. 100185265).References
- V. Tabar and L. Studer, Nat. Rev. Genet., 2014, 15, 82–92 CrossRef CAS PubMed.
- A. D. Ebert and C. N. Svendsen, Nat. Rev. Drug Discovery, 2010, 9, 367–372 CrossRef CAS PubMed.
- J. Sandoe and K. Eggan, Nat. Neurosci., 2013, 16, 780–789 CrossRef CAS PubMed.
- W. Li, W. Sun, Y. Zhang, W. Wei, R. Ambasudhan, P. Xia, M. Talantova, T. Lin, J. Kim, X. Wang, W. R. Kim, S. A. Lipton, K. Zhang and S. Ding, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 8299–8304 CrossRef CAS PubMed.
- A. Morizane, D. Doi, T. Kikuchi, K. Nishimura and J. Takahashi, J. Neurosci. Res., 2011, 89, 117–126 CrossRef CAS PubMed.
- P. Koch, T. Opitz, J. A. Steinbeck, J. Ladewig and O. Brustle, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 3225–3230 CrossRef CAS PubMed.
- D. Seidel, J. Obendorf, B. Englich, H. G. Jahnke, V. Semkova, S. Haupt, M. Girard, M. Peschanski, O. Brustle and A. A. Robitzki, Biosens. Bioelectron., 2016, 86, 277–286 CrossRef CAS PubMed.
- A. Stett, U. Egert, E. Guenther, F. Hofmann, T. Meyer, W. Nisch and H. Haemmerle, Anal. Bioanal. Chem., 2003, 377, 486–495 CrossRef CAS PubMed.
- G. W. Gross, B. K. Rhoades, H. M. Azzazy and M. C. Wu, Biosens. Bioelectron., 1995, 10, 553–567 CrossRef CAS PubMed.
- L. Yla-Outinen, J. Heikkila, H. Skottman, R. Suuronen, R. Aanismaa and S. Narkilahti, Front. Neuroeng., 2010, 3, 111 CAS.
- A. Odawara, Y. Saitoh, A. H. Alhebshi, M. Gotoh and I. Suzuki, Biochem. Biophys. Res. Commun., 2014, 443, 1176–1181 CrossRef CAS PubMed.
- K. Fukushima, Y. Miura, K. Sawada, K. Yamazaki and M. Ito, J. Biomol. Screening, 2016, 21, 54–64 CrossRef CAS PubMed.
- S. Illes, S. Theiss, H. P. Hartung, M. Siebler and M. Dihne, BMC Neurosci., 2009, 10, 93 CrossRef PubMed.
- H. Amin, A. Maccione, F. Marinaro, S. Zordan, T. Nieus and L. Berdondini, Front. Neurosci., 2016, 10, 121 Search PubMed.
- T. J. Heikkila, L. Yla-Outinen, J. M. Tanskanen, R. S. Lappalainen, H. Skottman, R. Suuronen, J. E. Mikkonen, J. A. Hyttinen and S. Narkilahti, Exp. Neurol., 2009, 218, 109–116 CrossRef PubMed.
- M. E. Obien, K. Deligkaris, T. Bullmann, D. J. Bakkum and U. Frey, Front. Neurosci., 2014, 8, 423 Search PubMed.
- S. Illes, M. Jakab, F. Beyer, R. Gelfert, S. Couillard-Despres, A. Schnitzler, M. Ritter and L. Aigner, Stem Cell Rep., 2014, 2, 323–336 CrossRef PubMed.
- A. Napoli and I. Obeid, J. Cell. Biochem., 2016, 117, 559–565 CrossRef CAS PubMed.
- L. Liu, R. Zhao-Shea, J. M. Mcintosh, P. D. Gardner and A. R. Tapper, Mol. Pharmacol., 2012, 81, 541–548 CrossRef CAS PubMed.
- A. A. Grace, S. B. Floresco, Y. Goto and D. J. Lodge, Trends Neurosci., 2007, 30, 220–227 CrossRef CAS PubMed.
- J. N. Guzman, J. Sanchez-Padilla, C. S. Chan and D. J. Surmeier, J. Neurosci., 2009, 29, 11011–11019 CrossRef CAS PubMed.
- T. Xu, M. Zhang, T. Laurent, M. Xie and S. Ding, Stem Cells Transl. Med., 2013, 2, 355–361 CrossRef CAS PubMed.
- D. S. Kim, D. R. Lee, H. S. Kim, J. E. Yoo, S. J. Jung, B. Y. Lim, J. Jang, H. C. Kang, S. You, D. Y. Hwang, J. W. Leem, T. S. Nam, S. R. Cho and D. W. Kim, PLoS One, 2012, 7, e39715 CAS.
- M. Garcia-Alloza, N. Zaldua, M. Diez-Ariza, B. Marcos, B. Lasheras, F. Javier Gil-Bea and M. J. Ramirez, J. Neuropathol. Exp. Neurol., 2006, 65, 1074–1081 CrossRef CAS PubMed.
- D. Gervasoni, C. Peyron, C. Rampon, B. Barbagli, G. Chouvet, N. Urbain, P. Fort and P. H. Luppi, J. Neurosci., 2000, 20, 4217–4225 CAS.
- I. L. Jones, P. Livi, M. K. Lewandowska, M. Fiscella, B. Roscic and A. Hierlemann, Anal. Bioanal. Chem., 2011, 399, 2313–2329 CrossRef CAS PubMed.
- K. M. Kovach, D. W. Kumsa, V. Srivastava, E. M. Hudak, D. F. Untereker, S. C. Kelley, H. A. von Recum and J. R. Capadona, J. Neurosci. Methods, 2016, 273, 1–9 CrossRef CAS PubMed.
- H. Charkhkar, C. Frewin, M. Nezafati, G. L. Knaack, N. Peixoto, S. E. Saddow and J. J. Pancrazio, Biosens. Bioelectron., 2014, 53, 316–323 CrossRef CAS PubMed.
- P. Fattahi, G. Yang, G. Kim and M. R. Abidian, Adv. Mater., 2014, 26, 1846–1885 CrossRef CAS PubMed.
- U. A. Aregueta-Robles, A. J. Woolley, L. A. Poole-Warren, N. H. Lovell and R. A. Green, Front. Neuroeng., 2014, 7, 15 Search PubMed.
- M. Makinen, T. Joki, L. Yla-Outinen, H. Skottman, S. Narkilahti and R. Aanismaa, J. Neurosci. Methods, 2013, 215, 88–96 CrossRef CAS PubMed.
- V. V. Klinshov, J. N. Teramae, V. I. Nekorkin and T. Fukai, PLoS One, 2014, 9, e94292 Search PubMed.
- B. L. Robinette, J. A. Harrill, W. R. Mundy and T. J. Shafer, Front. Neuroeng., 2011, 4, 1 CrossRef PubMed.
- M. Eichler, H. G. Jahnke, D. Krinke, A. Muller, S. Schmidt, R. Azendorf and A. A. Robitzki, Biosens. Bioelectron., 2015, 67, 582–589 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6an02713j |
| This journal is © The Royal Society of Chemistry 2017 |