
A paper-based platform for detection of viral RNA†
 ab
Sapna K.
Deo
*a
ab
Sapna K.
Deo
*a
Abstract
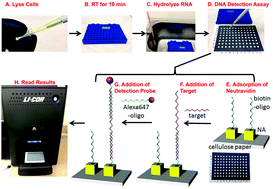
Viral detection presents a host of challenges for even the most sensitive analytical techniques, and the complexity of common detection platforms typically preclude portability. With these considerations in mind, we designed a paper microzone plate-based virus detection system for the detection of viral genetic material that can be performed with simple instruments. The sensing system can detect viral cDNA reverse-transcribed from total RNA extraction by utilizing a biotinylated capture probe and an Alexa Fluor® 647-labeled reporter probe. The biotinylated capture probe was linked to the paper surface via NeutrAvidin® that was physically adsorbed on the paper. After addition of reverse-transcribed sample and reporter probe in sequence, the reverse-transcribed target captured the reporter probe and tethered it to the capture probe in a bridged format. Fluorescence intensity was imaged using a Western blot imaging system, and higher target concentration was visible by the increased emission intensity from Alexa Fluor® 647. By utilizing paper, this detection setup could also serve as a sample concentration method via evaporation, which could remarkably lower the detection limit if needed. This detection platform used Epstein-Barr virus (EBV) RNA as a proof-of-concept by sensing cDNA resulting from reverse transcription and can be further expanded as a general method for other pathogens. EBV is a well-known human tumor virus, which has also recently been linked to the development of cervical cancer. The assay was accomplished within two hours including the room-temperature RNA extraction and reverse transcription steps. Also, this paper microzone plate-based platform can potentially be applicable for the development of point-of-care (POC) detection kits or devices due to its robust design, convenient interface, and easy portability. The experiment could be stopped after each step, and continued at a later time. The shelf-life of the modified paper plate setup was at least 3 months without a discernible change in signal, and the result from day 1 could be read at 3 months – both of which are important criteria for POC analytical testing tools, especially in resource-poor settings. All of the required assay steps could potentially be performed without any significant equipment using inexpensive paper microzone plates, which will be ideal for further development of POC testing devices. Although, this platform is not at the stage where it can be directly used in a point-of-care setting, it does have fundamental characteristics such as a stable platform, a simple detection method, and relatively common reagents that align closely with a POC system.
- This article is part of the themed collection: Coronavirus articles - free to access collection
 Please wait while we load your content...
Please wait while we load your content...

