
A kanamycin sensor based on an electrosynthesized molecularly imprinted poly-o-phenylenediamine film on a single-walled carbon nanohorn modified glassy carbon electrode†
Shuang
Han
a,
Bingqian
Li
a,
Ze
Song
a,
Sihao
Pan
a,
Zhichao
Zhang
a,
Hui
Yao
a,
Shuyun
Zhu
*b and
Guobao
Xu
*c
aCollege of Applied Chemistry, Shenyang University of Chemical Technology, Shenyang 110142, China. E-mail: unihanshuang@163.com
bShandong Provincial Key Laboratory of Life Organic Analysis, College of Chemistry and Chemical Engineering, Qufu Normal University, Qufu, Shandong 273165, China. E-mail: shuyunzhu1981@163.com
cState Key Laboratory of Electroanalytical Chemistry; Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun 130022, China. E-mail: guobaoxu@ciac.ac.cn
First published on 21st November 2016
Abstract
A single-walled carbon nanohorn (SWCNH) has been used to construct a molecularly imprinted electrochemical sensor for the first time. Kanamycin, a widely used aminoglycoside antibiotic, is used as a representative analyte to test the detection strategy. The kanamycin sensor was constructed by the electropolymerization of a molecularly imprinted poly-o-phenylenediamine film on a SWCNH modified glassy carbon electrode. The sensor was investigated in the presence or absence of kanamycin by cyclic voltammetry to verify the changes in the redox peak currents of K3Fe(CN)6. The sensor exhibits a linear range of 0.1–50 μM with a detection limit of 0.1 μM. It also shows high recognition ability, indicating that the SWCNH-based molecularly imprinted sensor is promising.
Introduction
Antibiotics, chemical substances produced by microorganisms, are widely used to kill or inhibit the growth of bacteria. They have been an essential part of human and veterinary medicine for the treatment of infectious diseases caused by bacteria.1–3 The antibiotics can be classified into five groups: penicillins, tetracyclines, macrolides, aminoglycosides, and amphenicols.4,5Kanamycin, produced by fermentation of Streptomyces kanamyceticus, is a member of the aminoglycoside family of antibiotics. It can induce mistranslation and indirectly inhibit translocation during protein synthesis, resulting in the production of abnormal proteins, which are harmful to the bacteria.6,7 Therefore, kanamycin is used to treat a variety of serious infections caused by susceptible strains of E. coli, Proteus species, Enterobacter aerogenes, Klebsiella pneumoniae, Serratia marcescens, and Acinetobacter species, and as a second-line treatment of Mycobacterium tuberculosis.8 Kanamycin exhibits a comparatively narrow safety margin and is potentially ototoxic and nephrotoxic.9 In addition, the abuse of kanamycin results in a large amount of residues in foodstuffs and the environment, leading to serious bacterial resistance, which can endanger the health of humans and animals. Therefore, it is urgent to establish sufficiently sensitive and selective methods for the detection of the kanamycin residue in terms of food security and environmental safety.
Recently, various analytical methods have been developed for the determination of kanamycin, such as HPLC,10–12 capillary electrophoresis,13 immunoassay,14 square-wave cathodic adsorptive stripping voltammetry,15 surface plasmon resonance,16 surface-enhanced Raman scattering,17 colorimetry,18–20 electrochemiluminescence,21,22 and fluorescence spectrometry.23–26 Although these methods show high sensitivity and accuracy, time-consuming analytical procedures and expensive equipment limit their extensive use. Therefore, it is urgent to establish a more facile and rapid assay for the detection of kanamycin. An electrochemical method is attractive for the determination of kanamycin, because it is simple, cost effective and fast. For example, Guo et al. investigated a multi-walled carbon nanotube-ionic liquid-nanoporous PtTi alloy–aptamer composite film electrode for the determination of kanamycin.27 Huang's group reported a sandwich-type electrochemical aptasensor for sensitive detection of kanamycin by assembling graphene–polyaniline and polyamidoamine dendrimer–Au nanoparticle nanocomposites on the surfaces of a glassy carbon electrode.28
Molecularly imprinting polymer (MIP) sensors have advantages such as strong affinity, excellent selectivity and toughness. MIP electrochemical sensors have been rapidly developed to detect different targets by combining various electrochemical techniques with a variety of MIP materials.29 For example, Liu's group has developed a novel strategy for the cyclic voltammetry determination of kanamycin using polypyrrole MIP film electrodes coupled with bioelectrocatalysis.30 In order to enhance the sensitivity and selectivity of the electrochemical sensors, a variety of nanomaterials have been employed to modify electrodes.
A single-walled carbon nanohorn (SWCNH) is a kind of nanocarbon of high purity, since no metal catalyst is involved in its synthesis by laser ablation on highly purified graphite.31,32 They are structures of single-graphene tubules with highly strained conical ends.33 They have been actively studied for electrochemical sensing of analytes because of their unique structure and extraordinary physical properties,34–36 such as large specific surface area, ability to accumulate analytes, high thermal and electrical conductivities, great mechanical strength and outstanding ability to mediate electron transfer for a wide range of electroactive species.37–39
In this study, a poly(o-phenylenediamine) (PPD) film was electropolymerized onto a carboxylic-group-functionalized SWCNH (SWCNH-COOH)-modified glassy carbon electrode (GCE/SWCNH-COOH/PPD) using cyclic voltammetry (CV) (Scheme 1) to make an electrochemical kanamycin MIP sensor. The resulting sensor has been successfully applied to analyze kanamycin with high sensitivity, selectivity, and recovery.
 | ||
| Scheme 1 Schematic representation of the fabrication procedures of the kanamycin MIP sensor. | ||
Experimental
Materials
o-Phenylenediamine (o-PD) was purchased from Sinopharm Chemical Reagent Co., Ltd. Kanamycin was obtained from Xiya Reagent Company. All chemicals were of analytical reagent grade and used as received. Acetate buffer (0.2 M, pH 4.8) was made from CH3COOH and CH3COONa solutions. 0.1 M phosphate buffer was prepared from NaH2PO4 and Na2HPO4. All solutions were prepared with double distilled water.Apparatus
All the electrochemical measurements were carried out on a CHI 660C electrochemical workstation (CH Instruments, Chenhua Co., Shanghai, China) connected to a three-electrode configuration, consisting of a modified GCE (3 mm in diameter) as the working electrode, a potassium chloride-saturated calomel electrode (SCE) as the reference electrode, and a platinum wire as the auxiliary electrode. The Fourier-transform IR (FTIR) spectrum was recorded by using a NEXUS 470 FTIR spectrometer (America). HRTEM measurements were performed on a FEI TECNAI G2 F20 microscope that was operated at an accelerating voltage of 200 kV. All measurements were carried out at ambient temperature.Preparation of SWCNH-COOH modified GCEs
SWCNHs were treated with 30% HNO3 to obtain SWCNH-COOH according to our previous reports.40,41 The SWCNH-COOH suspension was prepared by dispersing 1 mg of SWCNH-COOH in 1 mL double-distilled water by ultrasonic agitation and a relatively stable suspension would be obtained. A GCE was polished with 1.0, 0.3, and 0.05 μm aqueous slurry of alumina on a polishing cloth and then washed in an ultrasonic bath of ethanol and water, respectively. The cleaned GCE was then coated by 10 μL of the black suspension of 1 mg mL−1 SWCNH-COOH and dried under ambient temperature to obtain the SWCNH-COOH film electrode (GCE/SWCNH-COOH).Preparation of MIP and non-molecular imprinted polymer (NMIP) sensors
In this study, electropolymerization was used for PPD deposition. The MIP GCE/SWCNH-COOH/PPD was prepared in acetate buffer (pH 4.8) containing 5.0 mM o-PD and 1.0 mM kanamycin by 30 cycles of cyclic voltammetric measurements between 0 and 0.8 V at a scan rate 0.05 V s−1. After the electropolymerization, the MIP GCE/SWCNH-COOH/PPD was immersed in ethanol for 10 min to remove kanamycin from the MIP film. The NMIP GCE/SWCNH-COOH/PPD was prepared under the same conditions without adding kanamycin.Electroanalytical measurements
The MIP GCE/SWCNH-COOH/PPD was first incubated in different concentrations of kanamycin solutions for 300 s. The electrochemical experiments were performed at a scanning potential of 0.05 V s−1 in 5.0 mM K3Fe(CN)6/0.1 M KCl. After each experimental run, the sensor was washed in ethanol for 10 min to remove kanamycin and was reusable after this cleaning process.Results and discussion
Characterization of SWCNH-COOH
To improve the dispersibility, SWCNHs were treated with 30% HNO3 solution. Fig. 1A shows the HRTEM image of SWCNH-COOH, which maintains a dahlia-like morphology.38 The FTIR spectrum of SWCNH-COOH is shown in Fig. 1B. The peak at about 1577 cm−1 is assigned to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching mode that is associated with sidewall defects in the SWCNH-COOH. The peaks at about 3424 cm−1, 1384 cm−1 and 1123 cm−1 were assigned to the alcohol hydroxyl O–H stretching mode, O–H deformation mode and C–O stretching mode, respectively. The peaks at 1716 cm−1 and 1431 cm−1 were clearly assigned to the CO and O–H stretching mode, indicating the generation of COOH groups.41,42
C stretching mode that is associated with sidewall defects in the SWCNH-COOH. The peaks at about 3424 cm−1, 1384 cm−1 and 1123 cm−1 were assigned to the alcohol hydroxyl O–H stretching mode, O–H deformation mode and C–O stretching mode, respectively. The peaks at 1716 cm−1 and 1431 cm−1 were clearly assigned to the CO and O–H stretching mode, indicating the generation of COOH groups.41,42
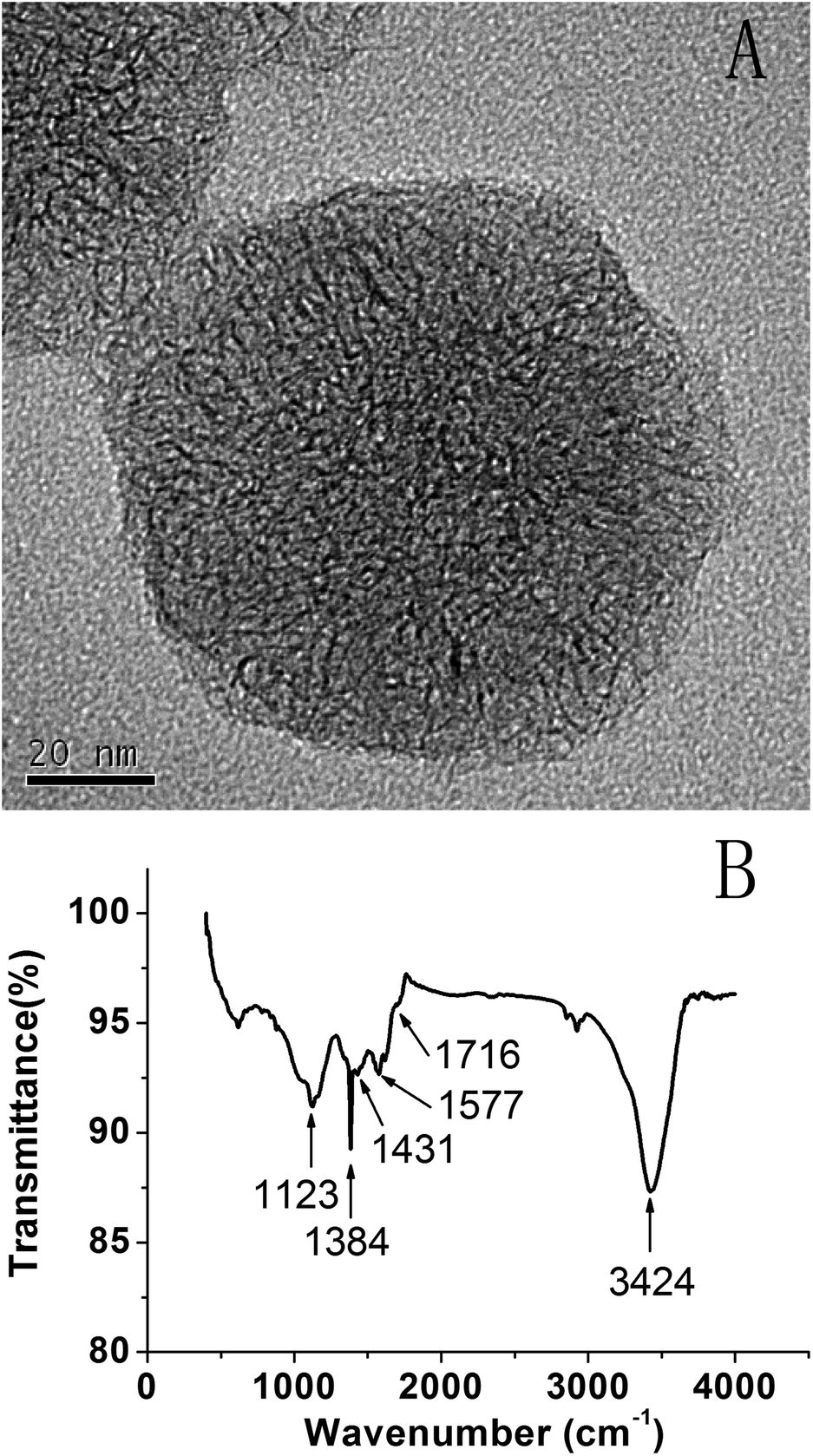 | ||
| Fig. 1 (A) HRTEM image and (B) the FTIR spectrum of SWCNH-COOH. | ||
Electropolymerization of the o-PD MIP film on GCE/SWCNH-COOH
The PPD MIP films were prepared by the electropolymerization of the o-PD monomer in pH 4.8 acetate buffer in the absence and presence of a kanamycin template, respectively (Fig. 2A and B). The oxidation wave of o-PD appears to be completely irreversible. The oxidation peak currents decreased with the increase of CV cycles, indicating the formation of the nonconductive film on the electrode surface. When the number of cycles approached 30, the current of the oxidation peak almost disappeared. By comparison, the CV obtained in the absence of a kanamycin template is similar to that in the presence of a kanamycin template during the electropolymerization.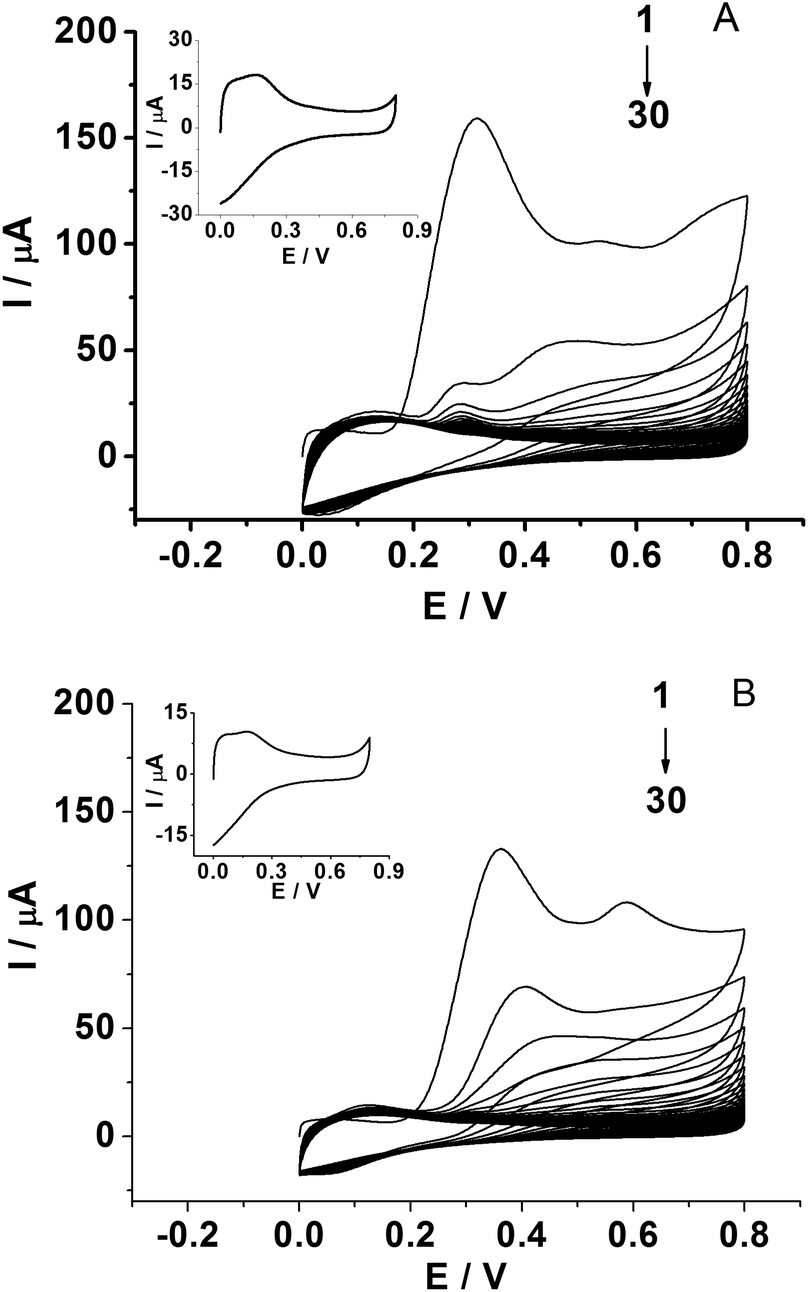 | ||
| Fig. 2 CVs for the electropolymerization of 5.0 mM o-PD on GCE/SWCNH-COOH in the absence (A) and in the presence (B) of 1.0 mM kanamycin in pH 4.8 acetate buffer (scan rate: 0.05 V s−1). Inset: CVs of GCE/SWCNH-COOH/PPD electrodes obtained in pH 4.8 acetate buffer (scan rate: 0.05 V s−1). | ||
Electrochemical characterization of the MIP GCE/SWCNH-COOH/PPD and NMIP GCE/SWCNH-COOH/PPD
Fig. 3 shows CVs of different modified electrodes in 5.0 mM K3Fe(CN)6 containing 0.1 M KCl at a scan rate of 0.05 V s−1. A pair of well-defined reversible redox peaks of K3[Fe(CN)6] was observed at the bare GCE (curve a). After SWCNH-COOH was immobilized on the bare GCE surface, the peak current of K3Fe(CN)6 was slightly increased due to the excellent electrical conductivity and larger special surface area of the SWCNH-COOH (curve b). However, after the electropolymerization of o-PD on the electrode surface, the CV peaks of the probe disappeared completely (curve c), demonstrating the formation of an insulating compact layer of PPD by electropolymerization. This PPD films prevented the diffusion of the probe to reach the electrode surface, and further hindered electron transfer. After the MIP GCE/SWCNH-COOH/PPD was washed in ethanol solution, the current peak increased (curve d). This indicates the removal of the template kanamycin in the MIP films and the formation of porous films, which are beneficial for the diffusion of the probe and subsequent electron transfer with the underlying electrode. The immersion of the kanamycin-free MIP GCE/SWCNH-COOH/PPD into kanamycin solution led to a decrease in the probe current signal (curve e) because the binding of kanamycin hindered the electron transfer to the electrode surface. The interactions, such as hydrogen-bonding interactions, may lead to the binding of kanamycin to the modified electrode surface. As for the NMIP GCE/SWCNH-COOH/PPD (Fig. S1†), the change of current was negligible from curve a to curve b, because no imprinted cavities were obtained after immersing the NMIP GCE/SWCNH-COOH/PPD in ethanol.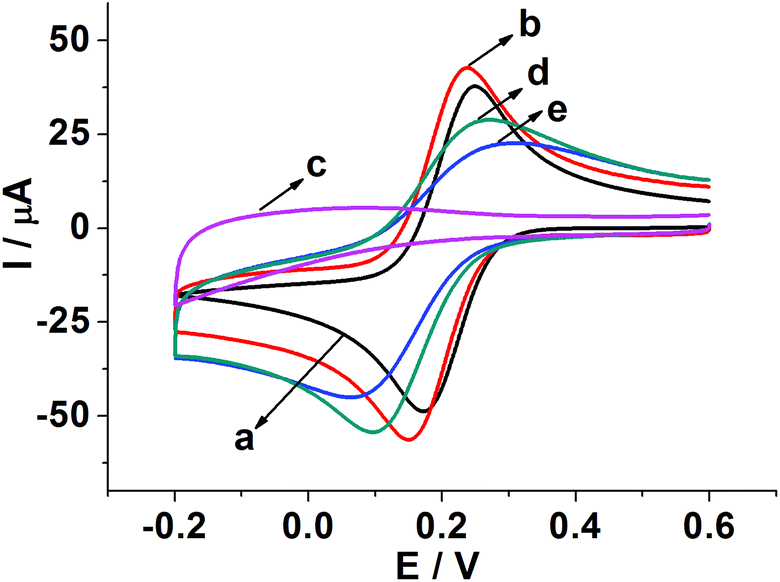 | ||
| Fig. 3 CVs of 5.0 mM K3[Fe(CN)6]/0.1 M KCl on the bare GCE (a); GCE/SWCNH-COOH (b); GCE/SWCNH-COOH after electropolymerization (c); MIP GCE/SWCNH-COOH/PPD after the removal of kanamycin (d); MIP GCE/SWCNH-COOH/PPD after incubation of kanamycin (e). | ||
Meanwhile, the effect of the scan rate on the MIP GCE/SWCNH-COOH/PPD was investigated. As shown in Fig. S2,† the peak currents increase with increasing scan rates from 0.01 to 0.6 V s−1. Moreover, both the anodic peak current (Ipa) and the cathodic peak current (Ipc) are proportional to the square root of the scan rate (ν and the unit is V s−1), suggesting a typical diffusion controlled electrochemical behavior (Fig. 4).
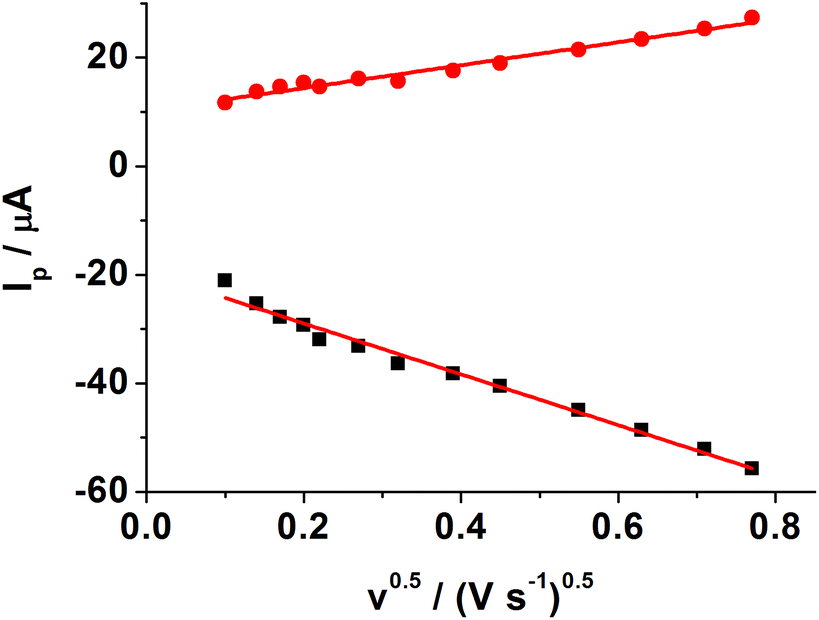 | ||
| Fig. 4 Relationship between the square root of scan rate and anodic and cathodic peak currents. Experimental conditions: MIP GCE/SWCNH-COOH/PPD in 5.0 mM K3[Fe(CN)6]/0.1 M KCl at different scan rates: 0.01, 0.02, 0.03, 0.04, 0.05, 0.075, 0.1, 0.2, 0.3, 0.4, 0.5, 0.6 V s−1. | ||
Optimization of the experimental parameters
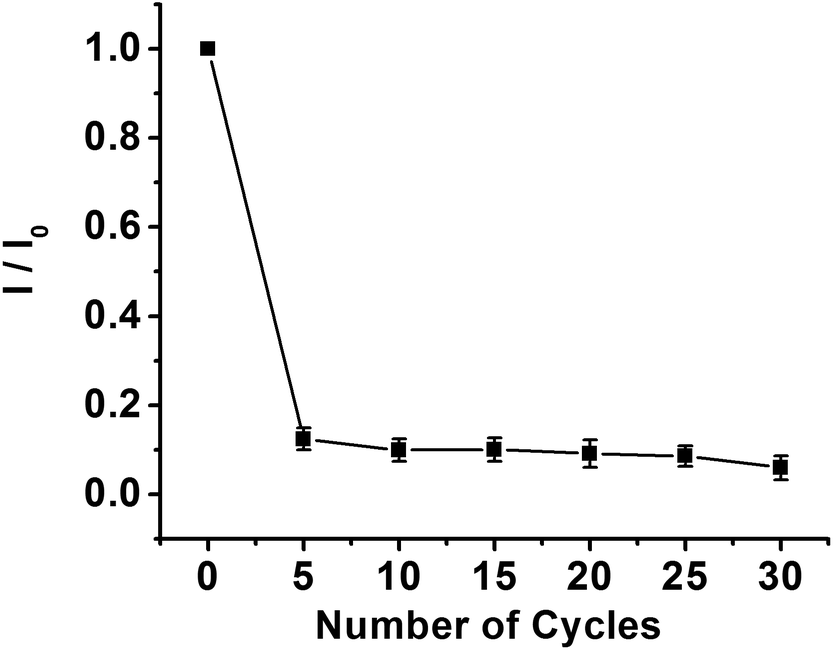 | ||
| Fig. 5 Effect of the electropolymerization cycles on the peak current of the CV response in the presence of 5.0 mM K3[Fe(CN)6] in 0.1 M KCl. I0 is the current of the MIP GCE/SWCNH-COOH/PPD after elution. I is the current of the MIP GCE/SWCNH-COOH/PPD after incubation of kanamycin. | ||
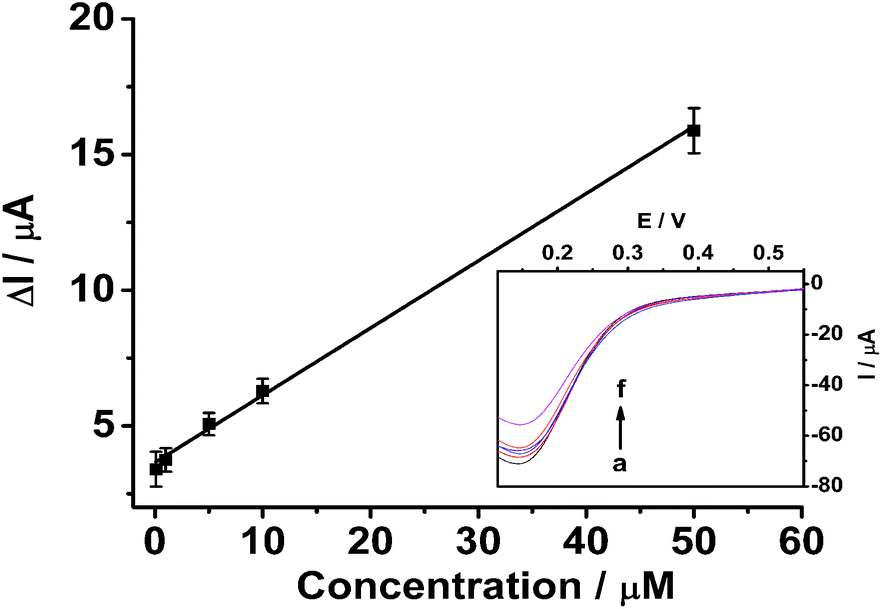 | ||
| Fig. 6 Linear calibration curve of ΔI with kanamycin concentrations. Inset: Linear sweep voltammograms of 5.0 mM K3[Fe(CN)6]/0.1 M KCl at 0.05 V s−1 for kanamycin-free MIP films after rebinding kanamycin at different concentrations (a)–(f): 0, 0.1, 1, 5, 10 and 50 μM. | ||
The selectivity of the imprinted sensor was evaluated by testing the CV response of kanamycin in the presence of ascorbic acid (AA), glucose, and chloramphenicol (CAP). Fig. S6† shows that 25 μM AA, 50 μM glucose, and 25 μM CAP have little effect on the determination of 10 μM kanamycin. To investigate the repeatability, 10 μM kanamycin was determined using the same MIP GCE/SWCNH-COOH/PPD. The calculated RSD was about 3.12% (n = 6). This good reproducibility reveals that the binding of kanamycin is reversible and the MIP GCE/SWCNH-COOH/PPD could be regenerated and reused. After the sensor was exposed to air for 10 days at room temperature, it reserved 90.06% of its original response.
Conclusions
In this work, a sensitive amperometric kanamycin MIP sensor was implemented via an electropolymerizing kanamycin imprinted PPD film on GCE/SWCNH-COOH. The electropolymerization procedure was very simple, rapid, and controllable. The MIP GCE/SWCNH-COOH/PPD has been proved to be selective, repeatable, and stable for the determination of kanamycin.Acknowledgements
This work was supported by the National Natural Science Foundation of China (no. 21344008, 21475123, 21405094, and 21674066), the Ministry of Education of Liaoning Province (no. L2015434, L2016022), the Scientific Public Welfare Research Foundation of Liaoning Province (no. 2015004016), the Natural Science Foundation of Shandong Province (no. ZR2013BQ018), and the Open Funds of the State Key Laboratory of Electroanalytical Chemistry (no. SKLEAC201506). The authors highly appreciate Professor S. Iijima (Solution Oriented Research for Science and Technology in Japan Science and Technology Agency) for the generous offer of SWCNHs.Notes and references
- X. F. Zhang, Y. Zhang, H. Zhao, Y. J. He, X. J. Li and Z. B. Yuan, Anal. Chim. Acta, 2013, 778, 63–69 CrossRef CAS PubMed.
- J. Adrian, S. Pasche, G. Voirin, J. Adrian, D. G. Pinacho, H. Font, F. Sánchez-Baeza, M.-P. Marco, J. M. Diserens and B. Granier, Trends Anal. Chem., 2009, 28, 769–777 CrossRef CAS.
- D. Gendrel, M. Chalumeau, F. Moulin and J. Raymond, Lancet Infect. Dis., 2003, 3, 537–546 CrossRef CAS PubMed.
- D. G. Kennedy, R. J. McCracken, A. Cannavan and S. A. Hewitt, J. Chromatogr., A, 1998, 812, 77–98 CrossRef CAS PubMed.
- F. L. Li, Y. M. Guo, X. Sun and X. Y. Wang, Eur. Food Res. Technol., 2014, 239, 227–236 CrossRef CAS.
- Y. Zhu, P. Chandra, K. M. Song, C. Ban and Y. B. Shim, Biosens. Bioelectron., 2012, 36, 29–34 CrossRef CAS PubMed.
- M. A. Kohanski, D. J. Dwyer, J. Wierzbowski, G. Cottarel and J. J. Collins, Cell, 2008, 135, 679–690 CrossRef CAS PubMed.
- V. Manyanga, R. L. Dhulipalla, J. Hoogmartens and E. Adams, J. Chromatogr., A, 2010, 1217, 3748–3753 CrossRef CAS PubMed.
- R. Oertel, V. Neumeister and W. Kirch, J. Chromatogr., A, 2004, 1058, 197–201 CrossRef CAS PubMed.
- Y. Zhang, H. M. He, J. Zhang, F. J. Liu, C. Li, B. W. Wang and R. Z. Qiao, Biomed. Chromatogr., 2015, 29, 396–401 CrossRef CAS PubMed.
- K. N. Patel, R. S. Limgavkar, H. G. Raval, K. G. Patel and T. R. Gandhi, J. Liq. Chromatogr. Relat. Technol., 2015, 38, 716–721 CrossRef CAS.
- Y. Zhang, H. M. He, J. Zhang, F. J. Liu, C. Li, B. W. Wang and R. Z. Qiao, Biomed. Chromatogr., 2015, 29, 396–401 CrossRef CAS PubMed.
- Y. C. Zhu, H. Y. Zhao, F. G. Ni, X. H. Yang and W. E. Gan, J. Chromatogr., B: Biomed. Appl., 2009, 877, 333–338 CrossRef PubMed.
- J. Peng, Y. W. Wang, L. Q. Liu, H. Kuang, A. K. Li and C. L. Xu, RSC Adv., 2016, 6, 7798–7805 RSC.
- J. L. Yan, Russ. J. Electrochem., 2008, 44, 1334–1338 CrossRef CAS.
- M. Frasconi, R. Tel-Vered, M. Riskin and I. Willner, Anal. Chem., 2010, 82, 2512–2519 CrossRef CAS PubMed.
- A. Zengin, U. Tamerb and T. Caykara, Anal. Chim. Acta, 2014, 817, 33–41 CrossRef CAS PubMed.
- Y. Y. Xu, T. Han, X. Q. Li, L. H. Sun, Y. J. Zhang and Y. S. Zhang, Anal. Chim. Acta, 2015, 891, 298–303 CrossRef CAS PubMed.
- X. F. Zhang, Y. Zhang, H. Zhao, Y. J. He, X. J. Li and Z. B. Yuan, Anal. Chim. Acta, 2013, 778, 63–69 CrossRef CAS PubMed.
- K. M. Song, M. Cho, H. Jo, K. Min, S. H. Jeon, T. Kim, M. S. Han, J. K. Ku and C. Ban, Anal. Biochem., 2011, 415, 175–181 CrossRef CAS PubMed.
- M. Zhao, Y. Zhuo, Y. Q. Chai and R. Yuan, Biomaterials, 2015, 52, 476–483 CrossRef CAS PubMed.
- H. Wang, W. W. Wu, D. Y. Wei, Z. Y. Guo and S. Wang, J. Electroanal. Chem., 2014, 735, 136–141 CrossRef CAS.
- Y. P. Xing, C. Liu, X. H. Zhou and H. C. Shi, Sci. Rep., 2015, 5, 8125 CrossRef CAS PubMed.
- J. Chen, Z. H. Li, J. Ge, R. Yang, L. Zhang, L. B. Qu, H. Q. Wang and L. Zhang, Talanta, 2015, 139, 226–232 CrossRef CAS PubMed.
- H. Li, D. E. Sun, Y. J. Liu and Z. H. Liu, Biosens. Bioelectron., 2014, 55, 149–156 CrossRef CAS PubMed.
- R. Y. Wang, R. Wang, B. Y. Ge, X. L. Jia, Z. G. Li and J. B. Chang, Anal. Methods, 2013, 5, 5302–5308 RSC.
- W. J. Guo, S. Na, X. L. Qin, M. S. Pei and L. Y. Wang, Biosens. Bioelectron., 2015, 74, 691–697 CrossRef CAS PubMed.
- W. Xu, Y. Wang, S. Liu, J. H. Yu, H. Z. Wang and J. D. Huang, New J. Chem., 2014, 38, 4931–4937 RSC.
- L. Ge, S. M. Wang, J. H. Yu, N. Q. Li, S. G. Ge and M. Yan, Adv. Funct. Mater., 2013, 23, 3115–3123 CrossRef CAS.
- W. J. Lian, S. Liu, L. Wang and H. Y. Liu, Biosens. Bioelectron., 2015, 73, 214–220 CrossRef CAS PubMed.
- S. Iijima, M. Yudasaka, R. Yamada, S. Bandow, K. Suenaga, F. Kokai and K. Takahashi, Chem. Phys. Lett., 1999, 309, 165–170 CrossRef CAS.
- R. N. Ma, M. H. Lu, L. Ding, H. X. Ju and Z. W. Cai, Chem. – Eur. J., 2013, 19, 102–108 CrossRef CAS PubMed.
- Z. C. Zhang, S. Han, C. Wang, J. P. Li and G. B. Xu, Nanomaterials, 2015, 5, 1732–1755 CrossRef CAS.
- X. Q. Liu, L. H. Shi, H. J. Li, W. X. Niu and G. B. Xu, Biosens. Bioelectron., 2008, 23, 1887–1890 CrossRef CAS PubMed.
- S. Y. Zhu, J. Zhang, X. E. Zhao, H. Wang, G. B. Xu and J. M. You, Microchim. Acta, 2013, 181, 445–451 CrossRef.
- S. Y. Zhu, X. E. Zhao, G. Chen, H. Wang, G. B. Xu and J. M. You, Ionics, 2015, 21, 1–7 CrossRef.
- M. F. Zhang, M. Yang, C. Bussy, S. Iijima, K. Kostarelos and M. Yudasaka, Nanoscale, 2015, 7, 2834–2840 RSC.
- M. Yang, M. F. Zhang, M. Kunioka, R. Yuge, T. Ichihashi, S. Lijima and M. Yudasaka, Carbon, 2015, 83, 15–20 CrossRef CAS.
- B. Russell, A. D. Migone, J. Petucci, M. M. Calbi, M. Yudasaka and S. Iijima, Phys. Chem. Chem. Phys., 2016, 18, 15436–15446 RSC.
- C. M. Yang, H. Noguchi, K. Murata, M. Yudasaka, A. Hashimoto, S. Iijima and K. Kaneko, Adv. Mater., 2005, 17, 866–870 CrossRef CAS.
- S. Y. Zhu, Z. Y. Liu, L. Z. Hu, Y. L. Yuan and G. B. Xu, Chem. – Eur. J., 2012, 18, 16556–16561 CrossRef CAS PubMed.
- S. Y. Zhu, X. E. Zhao, J. M. You, G. B. Xu and H. Wang, Analyst, 2015, 140, 6398–6403 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6an02338j |
| This journal is © The Royal Society of Chemistry 2017 |