
Orientational binding modes of reporters in a viral-nanoparticle lateral flow assay†
Jinsu
Kim
a,
Ryan
Poling-Skutvik
a,
João R. C.
Trabuco
a,
Katerina
Kourentzi
a,
Richard C.
Willson
abc and
Jacinta C.
Conrad
*a
aDepartment of Chemical & Biomolecular Engineering, University of Houston, Houston, TX 77204, USA. E-mail: jcconrad@uh.edu
bDepartment of Biology & Biochemistry, University of Houston, Houston, Texas 77004, USA
cCentro de Biotecnología FEMSA, Tecnológico de Monterrey, Monterrey, Nuevo León, Mexico
First published on 26th September 2016
Abstract
Using microscopy and image analysis, we characterize binding of filamentous viral nanoparticles to a fibrous affinity matrix as models for reporter capture in a lateral flow assay (LFA). M13 bacteriophage (M13) displaying an in vivo-biotinylated peptide (AviTag) genetically fused to the M13 tail protein p3 are functionalized with fluorescent labels. We functionalize glass fiber LFA membranes with antibodies to M13, which primarily capture M13 on the major p8 coat proteins, or with avidin, which captures M13 at the biotin-functionalized tail, and compare orientational modes of reporter capture for the side- versus tip-binding recognition interactions. The number of captured M13 is greater for side-binding than for tip-binding, as expected from the number of recognition groups. Whereas two-thirds of side-bound M13 captured by an anti-M13 antibody bind immediately after colliding with the membrane, tip-bound M13 prominently exhibit three additional orientational modes that require M13 to reorient to enable binding. These results are consistent with the idea that the elongated M13 shape couples with the complex flow field in an open and disordered fibrous LFA membrane to enhance capture.
Introduction
Lateral flow assays (LFAs) are a rapid, inexpensive, and simple format for point-of-care or point-of-use immunoassays.1 Commonly employed for home use as the home pregnancy test (to detect human chorionic gonadotropin), LFAs have been demonstrated to detect antibodies,2 viruses,3–5 bacteria,6,7 parasites,8 nucleic acids,9–11 proteins,12–14 drugs,15,16 and toxins.17,18 In the commonly-employed sandwich lateral flow format, particulate reporters (traditionally gold nanoparticles) are functionalized with antibodies to the analyte of interest. A sample containing analytes is wicked into contact with the reporters in the upstream portion of a membrane by capillary forces. The reporter particles bind to the analyte and are subsequently captured by antibodies at a downstream test line to generate a colorimetric visual signal. Sandwich LFAs require little or no sample preparation and generate a rapid and easily-read result, significant advantages for a point-of-use diagnostic in resource-limited settings.5,7,19 In most current implementations, however, LFAs are semi-quantitative and are insufficiently sensitive to detect analytes at extremely low concentrations – for example, to detect most viruses at clinically-relevant concentrations. Indeed, a wide range of applications requiring point-of-use monitoring, including biopharmaceutical manufacturing, food processing, environmental monitoring, and biodefense, would benefit from new LFA formats with improved sensitivity.One common strategy to increase the sensitivity of sandwich LFAs is to develop new reporters and/or readouts that enable quantitative measures of the analyte concentration. A wide variety of synthetic nanomaterials have been explored as alternatives to colloidal gold, including fluorescent20 or luminescent21 nanoparticles,22 quantum dots,23 iron oxides,24 and phosphors.25,26 Similarly, alternate methods to modulate the flow of reporters through the strip have been proposed, including electrophoresis,27 isotachophoresis,28 and flow delay.29 Finally, proposed ultrasensitive readouts include volumetric magnetization,30,31 near-infrared detection,32 surface-enhanced Raman spectroscopy,33 fluorescence,34,35 chemiluminescence,9,36 phosphorescence,37 and thermal contrast.38 In addition, much recent work has focused on smartphone-compatible readouts39,40 that are suitable for settings lacking sophisticated laboratory infrastructure or instrumentation. Despite these and other significant advances towards quantitative and ultrasensitive LFA formats, there remains a continuing need for flexible, inexpensive approaches that increase LFA sensitivity.
One promising approach to improved LFA sensitivity, by increasing the signal-to-noise ratio at low analyte concentrations, is to employ reporters that exhibit low non-specific binding. Recently, we have employed M13 bacteriophage (M13) decorated with enzymes as viral-nanoparticle reporters in ultrasensitive LFAs.41–44 Filamentous M13 is highly anisotropic, with length of approximately 900 nm (depending on genome size) and width of approximately 6.6 nm. These bacteriophage (“phage”) can be genetically or chemically modified to display a wide range of functional groups for applications in bionanotechnology45,46 and in nanomedicine.47 Here, phage are appealing candidates as LFA reporters because they evolved under Darwinian selection to exhibit low non-specific binding; are readily coupled to recognition elements;48–51 are capturable, in principle, by a single analyte and/or recognition element;52 and are detectable at the single-reporter level. Remarkably, the viral-nanoparticle-based LFAs we have developed to date exhibit sensitivities for model analytes (viruses41–43 and proteins44) that are one-hundred-fold to one-thousand-fold greater than traditional gold-nanoparticle-based LFAs. The mechanisms underlying the ultrasensitivity of viral-nanoparticle-based LFAs, however, remain poorly understood. We speculate that the enhanced sensitivity arises from the binding and capture properties of M13 viral nanoparticles in LFA membranes. Systematic investigations of phage transport and binding at the pore scale are hence expected to generate insight into the origins of the improved sensitivity of viral-nanoparticle-based LFAs.
Here, we characterize the transport and binding properties of functionalized M13 bacteriophage as reporters in a model LFA designed to enable pore-scale imaging. We functionalize glass fiber LFA membranes with either antibodies to M13 phage, which are able to capture the phage by the (approximately 2700) copies of the major p8 coat protein, or with NeutrAvidin, which captures the phage via biotin conjugated to one or more of the five p3 minor coat proteins on the phage tip. By manipulating fluid properties to slow the translocation of phage and match the index of refraction of the glass fibers, we visualize the binding of individual phage over time as they transport through the LFA membrane. We identify four orientational modes by which phage are captured onto the functionalized membranes. For phage that laterally (side-)bind through the anti-M13 antibody, most binding occurs immediately after phage collide with a fiber within the membrane. Three additional modes contribute significantly for phage that tip-bind (via NeutrAvidin) onto fibers, all of which involve the local flow re-orienting phage to align parallel to the fiber prior to binding. These experiments confirm that individual phage can be captured by a single recognition element (at the phage tip), and are consistent with the idea that the anisotropic phage shape coupled to the complex flow promotes reorientation to facilitate binding.
Materials and methods
Culture and titration of M13 phage
AviTag-displaying M13 phage (AviTag-M13) were a gift from Prof. Brian Kay at the University of Illinois at Chicago. The culturing and titering of M13 phage are performed as described previously.53 Briefly, for phage growth, 10 μl of 106 pfu ml−1 AviTag-M13 is pre-mixed with 100 μl of a fresh overnight Escherichia coli TG1 culture. After phage are allowed to adsorb to TG1 for 2 h at 37 °C, the pre-mixture is transferred to 2× yeast extract and tryptone (2 × YT) broth medium in a 2.8 l flask and growth continues overnight at 37 °C. Bacteria are removed from the lysate by centrifugation followed by filtration through a 0.45 μm filter (Corning, #430512), and the purified phage are concentrated in 1 ml of phosphate-buffered saline (PBS) using poly(ethylene glycol) (PEG) precipitation. For phage titration, 300 μl of a mid-log TG1 culture is mixed with 10 μl of the serially-diluted phage stock solutions. After adsorption for 15 min, a solution containing 40 μl of 40 mg ml−1 X-Gal (5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside), 10 μl of 500 mM IPTG (Isopropyl β-D-1-thiogalactopyranoside), and 5 ml of 0.7% top agar is added and spread on a pre-warmed Luria–Bertani (LB) plate. After overnight growth, the blue plaques due to X-Gal and β-galactosidase encoded by the lacZ contained in the AviTag-M13 gene are counted to calculate the concentration of the phage stock solution.Biotinylation of AviTag-M13
AviTag-M13 are partly biotinylated on the p3 tail protein during assembly in E. coli, then further biotinylated in vitro using E. coli biotin ligase (birA) either purchased or prepared by recombinant expression in-house, following the manufacturer's instructions (Avidity, LLC). After incubation for 1 h at room temperature, excess biotinylation reagents are removed from the biotinylated phage solution via PEG precipitation and a 7 K molecular-weight cutoff (MWCO) Zeba spin desalting column. The degree of biotinylation is evaluated using an enzyme-linked immunosorbent assay (ELISA) on NeutrAvidin-coated 96-well microplates (additional details are given in Fig. S1 in the ESI†).AlexaFluor 555 labeling of biotinylated-M13
Biotinylated M13 are modified with AlexaFluor 555 Carboxylic Acid (Succinimidyl Ester, Life Technologies #A-20009) as previously described43 and illustrated schematically in Fig. 1(a). Representative fluorescence micrographs of the AlexaFluor 555 labeled phage are shown in the ESI (Fig. S2†). Phage in these experiments are captured by either one of two recognition elements: by anti-M13 antibodies, which can capture phage on any of the 2700 p8 coat proteins, or by NeutrAvidin, which can capture phage only through the biotin attached to one or more of the five p3 tail proteins (Fig. 1(b)).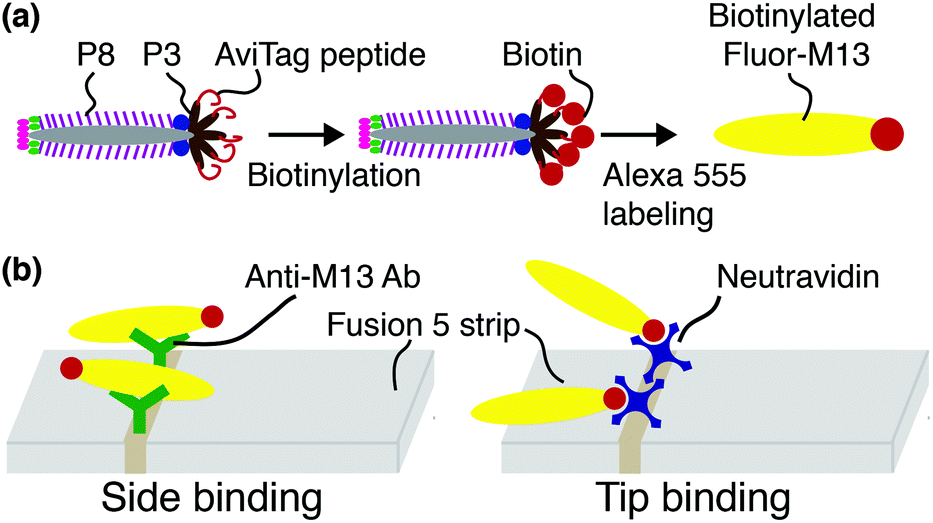 | ||
| Fig. 1 Schematic of phage modification and capture. (a) AviTag M13 are biotinylated on one or more of the five p3 tail proteins located on the phage tip, and then fluorescently labeled with Alexa 555 dye. (b) In side-binding, biotinylated Fluor-M13 are captured by anti-M13 antibodies almost exclusively directed against the p8 major coat proteins. In tip-binding, biotinylated Fluor-M13 are captured by a biotin–NeutrAvidin interaction. | ||
Flow experiments and image analysis
Fusion 5 LFA matrix strips (3 mm × 50 mm × 0.35 mm, GE Healthcare & Life Sciences #8151–9915, pore diameter 11 μm [reported by manufacturer], average fiber diameter D = 4.35 ± 1.90 μm [measured]) are modified with 3 μl of 1 mg ml−1 NeutrAvidin (Thermo Scientific, #3100) or 0.43 mg ml−1 polyclonal rabbit anti-M13 antibody (Novus Biologicals, #NB100-1633) in 50 mM acetate buffer at pH 3.6 by hand spotting at a fixed distance of 1 cm from the downstream end of the strip. After drying in air for 2 h at room temperature, each functionalized strip is placed on a large coverslip (48 × 65 mm, Gold Seal), covered with a second smaller coverslip (22 × 22 mm, Fisher Brand), and moved onto the stage of an epifluorescence microscope (Leica DMI 3000B) equipped with a 63× oil immersion objective lens (NA 1.4, depth of field ∼1 μm and depth of focus ∼0.6 mm), as shown in Fig. S3 in the ESI.† For the model LFA experiments, biotinylated Fluor-M13 are suspended in a background solution containing 0.2% w/v hydrolyzed polyacrylamide (HPAM, FLOPAAM 3330, SNF, weight-averaged molecular weight of 8 MDa) and 30% v/v glycerol in PBS. Subsequently, 25 μL of 107 pfu ml−1 biotinylated Fluor-M13 is dropped on the upstream end of the strip.Biotinylated Fluor-M13 are imaged in capillary-driven transport through the membrane using a sCMOS camera (pco.edge 4.2, 30 frame per s, 20 ms exposure time, 208.3 μm × 213 μm image field of view, pixel resolution 0.42 μm). Recording begins immediately after liquid breakthrough in the field of view and ceases when the phage particles exhibit only Brownian (rather than advective) motion, typically nine minutes. Four movies are analyzed for anti-M13 functionalized membranes; fourteen movies are analyzed for the NeutrAvidin-functionalized membranes. From the concentration of phage in solution, the average fluid velocity, and the focal area, we estimate that ∼1000 phage per movie pass through the field of view. The average number of bound Fluor-M13 phage per movie for anti-M13 antibody, NeutrAvidin, and unfunctionalized (bare) strips is 89.0 ± 19.8, 7.2 ± 1.9, and 0.2 ± 0.4, respectively.
The centroids of individual biotinylated Fluor-M13 are located in two dimensions with resolution of 0.25 pixel (0.1 μm) using the Hough transform algorithm.54 Phage centroid positions are linked into trajectories and tracked over time using multiple hypothesis tracking (MHT) software downloaded from http://www.multiplehypothesis.com/. Analysis of the relative angles between the tracked phage and the fibers on which they are captured is performed manually.
Fluid flow simulation
Fluid flow in a very simplified model membrane is simulated using a 3D laminar flow model in COMSOL Multiphysics 4.1. The simulation box has dimensions of 100 μm × 100 μm × 100 μm with two fibers of radius 2.5 μm spanning the size of the box. The inlet is specified to have a uniform velocity distribution normal to the yz plane at x = 1 with the outlet on the opposite face of the box at x = 0. One fiber is oriented along the (1, 1, 1) vector and makes a 45° angle to inlet flow when projected onto the xy and xz planes. The other fiber is oriented along the (1, −1, 1/3) vector and makes a 45° angle to the flow when projected onto the xy plane and approximately a 20° angle when projected onto the xz plane. The remaining external boundaries are open. The fiber surfaces are specified to have a no-slip boundary condition. The non-Newtonian fluid is modeled as a shear-thinning, power law fluid with a viscosity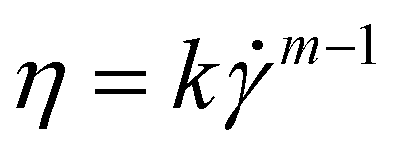 with k = 0.62, m = 0.36, and a zero-shear viscosity of 2.7 Pa s. The parameters are chosen to be consistent with the previously reported rheology of a 0.1 wt% HPAM solution55 and are representative of the fluid used in the phage-tracking experiments.
with k = 0.62, m = 0.36, and a zero-shear viscosity of 2.7 Pa s. The parameters are chosen to be consistent with the previously reported rheology of a 0.1 wt% HPAM solution55 and are representative of the fluid used in the phage-tracking experiments.
Results and discussion
Protocols for a binding-imaging assay in LFA membranes
In viral-nanoparticle LFAs, an aqueous PBS buffer solution rapidly travels through a membrane strip of 3 mm width and 5 cm length in several seconds, driven by capillary force; from the position of the liquid interface as a function of time, we estimate that the average velocity at which phage in PBS flow through the Fusion 5 membrane is 1.8 mm s−1. Although in principle phages moving at these velocities can be imaged using a fast camera, there remains a second limitation for imaging of binding at the pore scale: the refractive index mismatch between the PBS (index of refraction n = 1.33) and the glass Fusion5 membrane (n = 1.51) significantly increases scattering and hence limits the distance into which membranes can be imaged. Therefore, for single-phage imaging experiments we formulate a model viscous solvent that nearly matches the index of refraction of the Fusion 5 membrane to allow us to image up to 20 μm into the membrane from below, using an inverted microscope. We mix glycerol at 30% by volume with 0.2% by weight of partially-hydrolyzed poly(acrylamide) (HPAM) in PBS, and use this as the background solution for phage binding experiments. The viscosity of this solution, measured at a fixed shear rate of 42 s−1 using an Ubbelohde viscometer, is μ = 17.2 mPa s; its density is ρ = 1.08 g ml−1; and its index of refraction is n = 1.39. Because the membrane is disordered, the resistance to flow can be described by an isotropic term on large length scales (i.e. we do not expect that the viscoelastic fluid will generate anisotropic transport).56 We confirm via a control enzyme-linked immunosorbent assay (ELISA) that the binding of the phage is minimally affected by the presence of the glycerol and the HPAM (Fig. S4 in the ESI†) and hence use this solution for imaging experiments.The position of the solution interface x in the LFA membrane scales as the square root of time t, as expected from Washburn's equation57 (x2 = γdt/4η, where γ is the surface tension of the liquid, d is a characteristic pore diameter, and η is the dynamic viscosity) and shown in Fig. 2(a). This result indicates that capillary pressure is sufficient to drive flow of the fluid through the membrane, as in a traditional LFA. Subsequently we image phage as they flow through the membrane at a fixed position (x = 1 cm, y = 10 μm, z = 1.5 mm) downstream from the imbibition pad and initially measure their local maximum velocity as a function of time. The local maximum velocity is highest at the start of the experiment (immediately after fluid breakthrough) and first rapidly then slowly decreases with time, as shown in Fig. 2(b). Although the maximum velocity steadily decreases with time, the local flow profile can exhibit significant spatiotemporal variations (inset to Fig. 2(b)). The Fusion 5 membranes used in our model LFA have an open pore structure, and the local velocity of flow depends on the proximity and position of the fibers. Given the open pore network, we estimate the Reynolds number as Re = ρDV/μ, where ρ is the background solution density, V is the phage velocity, and μ is the solution viscosity; for fibrous membranes, the characteristic length scale D is the fiber diameter, similar to fluid flow around a cylinder.58 The maximum velocity of phages is 252 μm s−1 and hence the maximum Re is approximately 6.80 × 10−5. Although the Reynolds number is low the complex fiber geometry generates curved streamlines, on which phage flow around the fibers (see Movie S1 in the ESI†). The maximum Péclet number of the flow, estimated from Pe = VD/Dp, where Dp = 0.23 μm2 s−1 is the measured translational diffusivity of the biotinylated Fluor-M13 (Fig. S5 in the ESI†), is 4.8 × 103; advective transport dominates until the latest stages of imbibition.
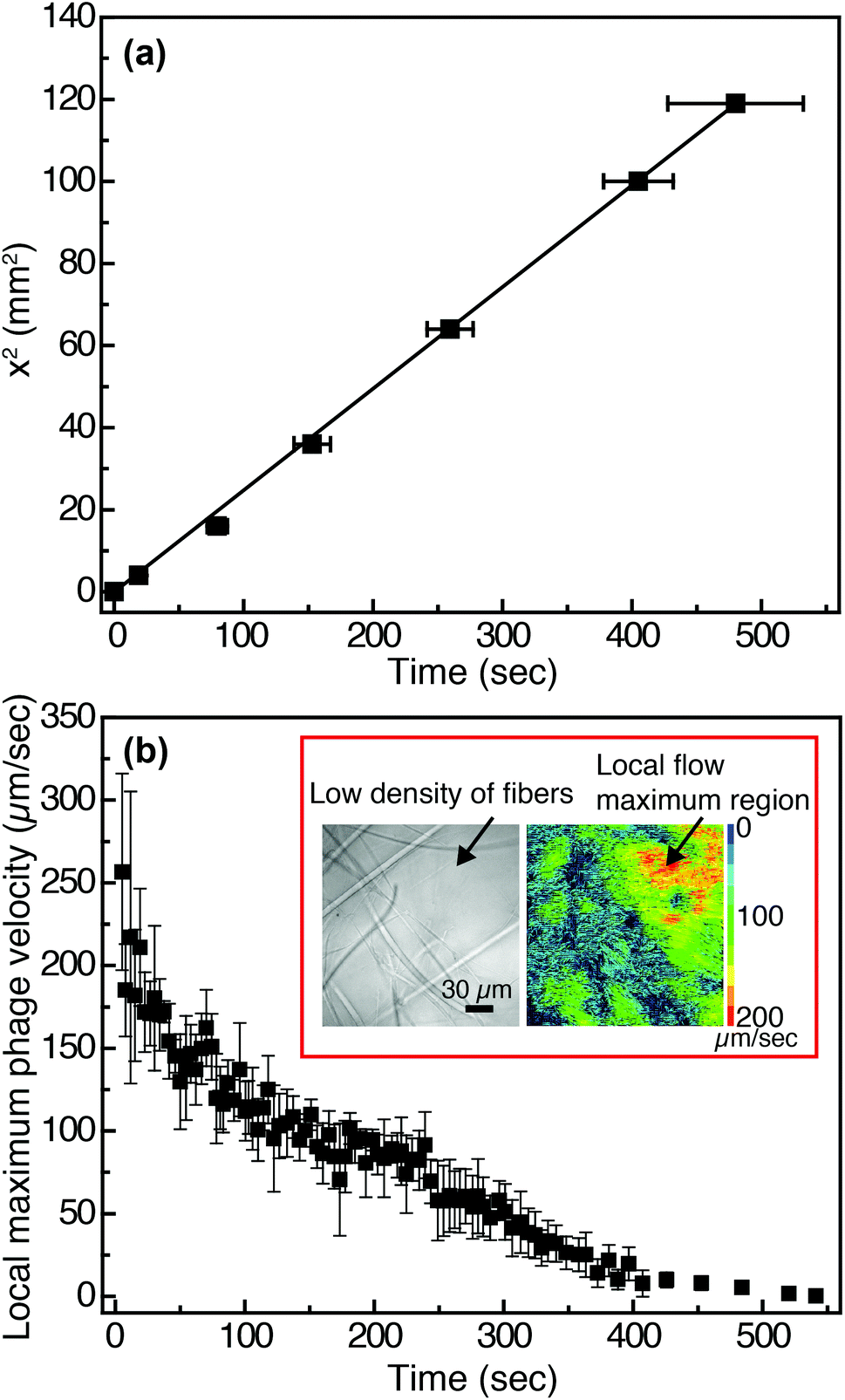 | ||
| Fig. 2 Flow characteristics of biotinylated Fluor-M13 solutions in Fusion 5 membranes. (a) Position of the liquid interface as a function of time for the solution of glycerol and HPAM flowing by capillary action in Fusion 5 membranes. Error bars indicate the standard deviation measured from three independent Fusion 5 membranes. (b) Local maximum velocity of individual biotinylated Fluor-M13 as a function of time in Fusion 5 membranes, measured at a fixed distance of 1 cm down the strip. Error bars indicate the standard deviation of the maximum velocity averaged across all phage in three independent movies. Inset: (Left) The disordered fiber structure of Fusion 5. (Right) Particle image velocimetry (PIV) reveals significant spatial variations in velocity that give rise to a complex local flow profile; the local maximum phage velocity is estimated from the region showing phage of highest velocity in three independent movies. | ||
Classification of orientational phage binding modes
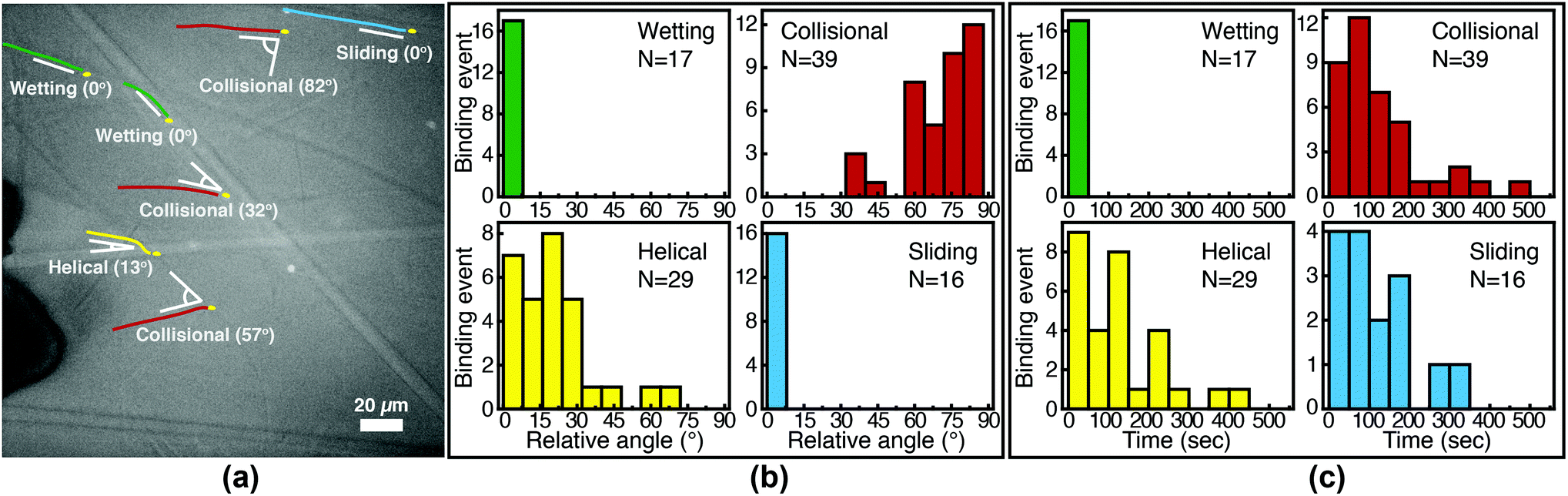
We image phage as they bind to the functionalized Fusion 5 membranes and use the motion of phage relative to the membrane prior to binding to classify each binding event into one of four orientational modes: wetting, collisional, helical, or sliding (Fig. 3). First, immediately after fluid breakthrough a thin film of fluid wets each fiber; phage entrained in this thin liquid layer align preferentially along the fiber and eventually (but not typically immediately) are captured by the recognition agents thereon. This mode, which we label “wetting”, occurs only when the solution/air interface is still visible, i.e. only at short times in the early stage of imbibition. After the strip is fully wetted, we identify three subsequent modes by which phage bind to the membrane. Second, some phage immediately bind to a membrane fiber within a single time step (within 0.04 s) of encountering the fiber and do not travel along it; we term this mode “collisional”. In the remaining two modes, phage orient along the fiber surface and move a finite distance along the fiber prior to irreversibly binding. Here, they move along the fiber for N = 3 to 30 time steps (0.1–1 s), depending on the flow speed; we classify binding as one of two distinct modes based on the shape of the path traced out by the phage along the fiber. Phage binding using the third “helical” mode follow curved trajectories with respect to the fiber axis. Phage binding through the fourth “sliding” mode follow straight trajectories. Quantitatively, it is convenient to use the angle between the phage trajectory and the fiber axis to distinguish these modes: less than 1° for sliding, greater than 1° for helical. | ||
| Fig. 3 Observed FluorM13 orientational modes. Biotinylated FluorM13 bind to functionalized fibers in one of four different orientational modes. (1) “Wetting” binding occurs only during the initial imbibition of fluid into the membrane, during which thin liquid films form along the fibers. Only phage trapped between the fiber and the liquid/air interface are deemed to be using this mode. (2) Phage that bind immediately after encountering the fiber are deemed to use the “collisional” mode. Alternately, phage that do not immediately bind are transported along the fiber prior to binding by the “helical” or “sliding” mode. (3) In the “helical” mode, phage move in curved trajectories over the fiber interface prior to binding. (4) Conversely, in “sliding” mode phage move in nearly linear trajectories along the fiber prior to binding and are oriented parallel to the fiber axis. A decision chart for classifying orientational modes is given in the ESI (Fig. S6†). | ||
Phage captured by anti-M13 antibodies (predominantly side-bound) and by NeutrAvidin (tip-bound) exhibit differences in the distribution of the orientational modes by which they bind to functionalized LFA membranes. Over seventy percent of side-bound phage (total number of binding events N = 356) bind immediately after colliding with a membrane fiber (Fig. 4(a)). In this case the antibodies on the fiber are able to capture phages that are oriented in all directions with respect to the fiber axis, and so the majority of phage do not have to precisely align with the fiber to bind. Nonetheless, one-third of the phage bind after transport along the fiber; filamentous phage have a larger effective capture surface area (relative to a sphere of the same volume) and hence are more likely to be captured before advecting away.
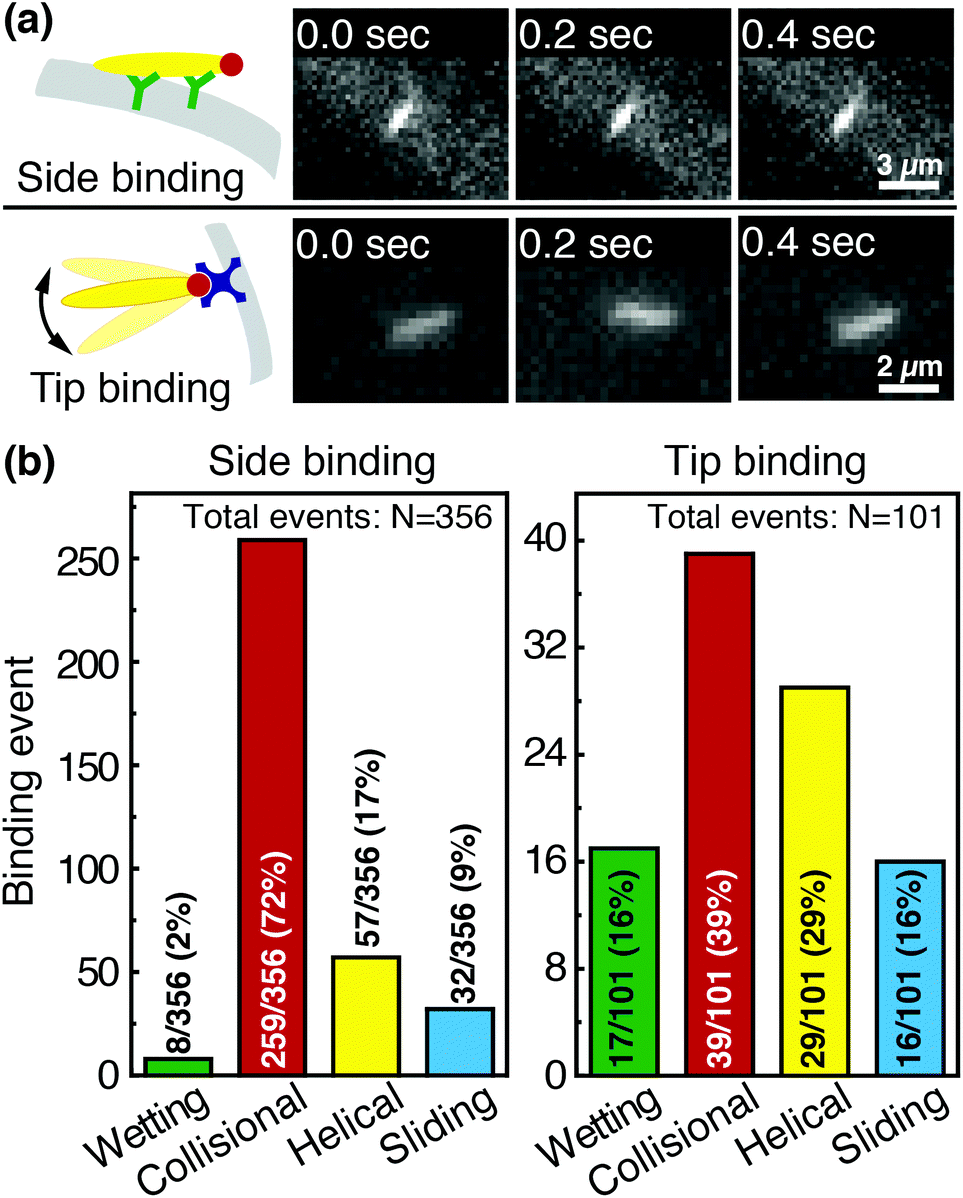 | ||
| Fig. 4 Comparison of side-binding and tip-binding orientational modes. (a) Side-bound phage are securely bound to fibers by anti-M13 antibodies, and the positions of their centers of mass do not fluctuate in time. Tip-bound phage are captured through a biotin–NeutrAvidin interaction, and can still fluctuate in flow. (b) Distribution of orientational modes for side-binding (left) and tip-binding (right) phage. The collisional mode dominates side-binding, as a phage can side-bind to an anti-M13 antibody regardless of its orientation with respect to the fiber. Tip binding, however, requires that phage properly orient with respect to the fibers for binding. Because fewer phage are properly oriented to bind upon first encountering the fiber, binding occurs more frequently through helical and sliding modes that enable phage to reorient along the fiber. | ||
Tip-bound phage, by contrast, exhibit a more uniform distribution of orientational modes. Only N = 101 phage in total are observed to bind across all movies, consistent with a lower binding efficiency of tip-binding, and only forty percent of tip-bound phage use the collisional mode (Fig. 4(b)). We note that the affinity constant of the biotin-NeutrAvidin bond (1015 M−1)59 and the association rate constant (kon = 7.5 × 107 M−1 s−1) are much greater than those for a typical antibody–antigen pair (108 M−1, kon ∼ 104–106 M−1 s−1). If the five biotin sites were distributed over the surface of the phage rather than localized at the tip, we would expect that the number of bound phage would be comparable to that for the antibody–antigen interaction due to the higher affinity and association constant of the biotin–NeutrAvidin bond.60 Moreover, a change in the number of binding sites should not result in changes to orientational modes. The lower frequency of the collisional mode for tip-binding (39%) than for side-binding (72%) suggests that the biotin sites localized at the phage tip impose a geometric constraint on phage binding. This constraint reduces the percentage of phage that collisionally bind and forces the relative angle of collisional binding events to be nearly orthogonal to the fiber surface. Tip-binding phage are unlikely to be properly oriented for binding during flow; even if phage are transported along a streamline and collide with the fiber surface, only approximately half will have the biotin-functionalized tail correctly oriented to bind upon collision.61 Instead, wetting (early in imbibition), helical, and sliding modes are more common for tip-binding than for side-binding. In these modes phage align along the fiber prior to binding, increasing their residence time near the fiber; both reorientation and the long residence time increase the likelihood that the biotin recognition element successfully encounters a NeutrAvidin on the fiber before the phage is advected away from the fiber surface.
We first characterize each orientational mode by the relative (2-D projected) angle at which the phage body is oriented with respect to the fiber axis as it attaches. The positions of phage and of fibers are determined from fluorescence and brightfield images, respectively, that are acquired at the same focal position. For side-binding (capture by anti-M13 antibodies), phage binding via collision preferentially orient at high angles with respect to the fiber axis but exhibit a broad distribution across all angles (Fig. 5(b)). Phage binding via the helical mode preferentially bind at low angles, but with a broad distribution. Wetting and sliding entrain the phage along the fiber and hence phage binding in these modes are oriented along the fiber axis.
 | ||
| Fig. 5 Relative angle and time for side binding. (a) Micrograph with bound phage (yellow ovals) and the orientational mode of binding indicated (by color: red for collisional, green for wetting, blue for sliding, yellow for helical) for a representative side-binding movie. (b) Distributions of relative angle (in degrees) between the phage body and the fiber axis for the N = 356 side-binding events. Phage that bind immediately after colliding with a fiber bind at most angles, consistent with omnidirectional capture ability of the anti-M13 antibody; phage that first reorient to transport along the fiber preferentially but not exclusively bind at low angles with respect to the fiber axis. (c) Distributions of the time of side-binding events after fluid breakthrough. The number of binding events decreases over time. | ||
The timing of binding events after the arrival of the fluid meniscus also depends on orientational mode. All wetting binding events occur right after breakthrough, when the liquid–air interface is still present (Fig. 5(c)). In the other modes, most phage also bind early (i.e. within 100 seconds of breakthrough), when the flux of phage is highest per the velocity measurements in Fig. 2(b). Indeed the binding time distribution is correlated with the velocity, as shown in Fig. S7 in the ESI.† The number of unbound phage is nearly constant across at all times, as shown in Fig. S8 in the ESI,† and hence higher velocity likely increases the rate at which phage encounter fibers. For example, collisional and helical binding events preferentially occur at shorter times after breakthrough; nonetheless, collisional, helical, and sliding binding events occasionally occur even at the longest accessible time scales, for which the advection velocity is nearly zero. Both the high likelihood of binding through collision and the wide distribution of binding angles are consistent with the omnidirectional capture of phage by anti-M13 antibodies on the membrane fibers.
Phage binding to a NeutrAvidin-functionalized membrane, by contrast, must properly orient the biotin group to be captured by one of the NeutrAvidin proteins on the fiber surface. This requirement is a more stringent condition on the orientation of the phage with respect to the fiber axis. As a result, fewer phage are able to immediately bind upon reaching the fiber surface. Indeed, phage that bind in collision are all oriented at a high angle (Fig. 6(b)) so that the biotin functionality encounters the NeutrAvidin binding sites. Phage that are not properly oriented are less likely to bind but can nonetheless bind after reorientation. Notably, phage that arrive at the surface oriented at a low angle (<30°) only bind if they are able to reorient and align along the fiber axis (i.e. in helical or sliding modes) or if they are forced to reorient by the presence of the fluid interface (i.e. in wetting, early after breakthrough). Phage that do not reorient instead slide over the fibers and are transported further into the membrane. Most tip-binding events, as in side-binding, occur early during the fluid flow (Fig. 6(c)), consistent with the higher phage flux during the transient flow on those time scales. All four orientational modes (for both tip- and side-binding phage) appear in fluids of different HPAM concentrations and varying viscosities (Fig. S9–S11†), suggesting that phage orientational modes are independent of fluid rheology.
 | ||
| Fig. 6 Relative angle and time for tip binding events. (a) Micrograph with bound phage (yellow ovals) indicated, along with the orientational mode of binding (by color: red for collisional, green for wetting, blue for sliding, yellow for helical), for a representative tip-binding movie. (b) Distributions of relative angle (in degrees) between the phage body and the fiber axis for the N = 101 tip binding events observed. (c) Distributions of the time of tip-binding events after fluid breakthrough. Most binding occurs in the beginning stage of flow. | ||
Role of tumbling in binding
Coupling between the flow field within the porous membrane and the phage shape promotes reorientation for binding through other mechanisms not described by the four modes above. Fig. 7 illustrates one example, in which a phage tumbles end-over-end prior to capture on the fiber. The physics underlying tumbling is straightforward: M13 phage are elongated. These phage are anisotropic with high aspect ratios; hence different ends of the phage can experience different local fluid environments. Close to the surface of a fiber, no-slip boundary conditions hold; far from the fiber surface, fluid flows rapidly through the open pore network. This geometry generates a gradient of increasing flow speed farther from the surface of the fiber, which can result in a net torque on the elongated phage. A phage encountering a fiber at a nonzero angle hence is rotated by the vorticity field to align parallel to the fiber; for biotin-conjugated phage whose binding group is located far from the fiber surface, this rotation would bring the binding group closer to the fiber surface. Hence we expect near-fiber tumbling (at low Re) to enhance the binding of anisotropic particles by increasing the effective cross-section for capture, relative to that of a sphere of the same volume. | ||
| Fig. 7 Reorientation of biotinylated Fluor-M13 by tumbling for tip binding. A phage approached a NeutrAvidin modified fiber and collided with the fiber at 1.0 s (second image). At later times (after the dashed line), phage tumbled while traveling along the fiber prior to binding in the helical mode. Inset: Illustration of phage orientation during tumbling and binding. | ||
A single phage of length L and radius r has the same volume as a sphere of radius R = (3r2L)1/3 = 27 nm. The anisotropic shape of the phage suggests several ways to define an effective radius that may control capture cross-section. For example, phage have the same surface area as a sphere with  , a hydrodynamic radius RH = kBT/(6πμD) = 55 nm, a radius of gyration
, a hydrodynamic radius RH = kBT/(6πμD) = 55 nm, a radius of gyration 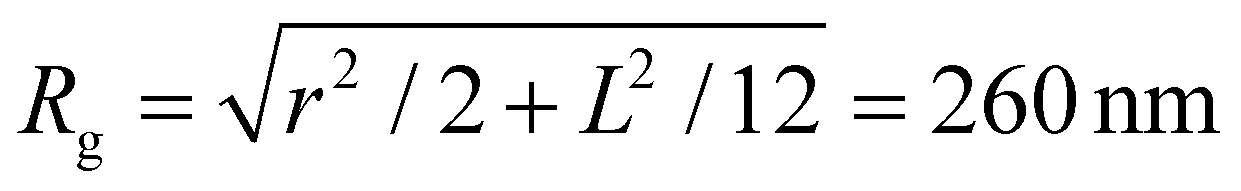 , and the same pervaded volume as a sphere with RV = L/2 = 450 nm, where kB is the Boltzmann constant, T is temperature, μ = 17.2 mPa s is the solution viscosity, and D = 0.236 μm2 s−1 is the translation diffusion. All of these effective radii are larger than the radius of a sphere with the same volume, with relative increases of
, and the same pervaded volume as a sphere with RV = L/2 = 450 nm, where kB is the Boltzmann constant, T is temperature, μ = 17.2 mPa s is the solution viscosity, and D = 0.236 μm2 s−1 is the translation diffusion. All of these effective radii are larger than the radius of a sphere with the same volume, with relative increases of 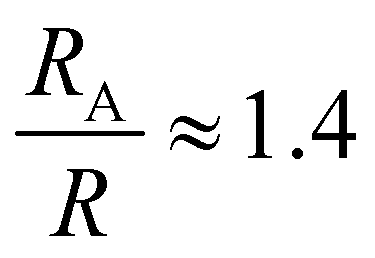 ,
, 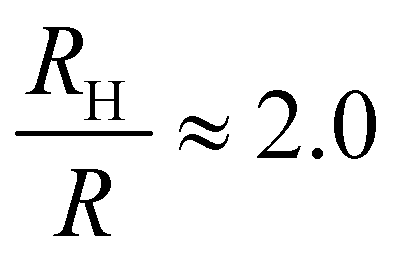 ,
, 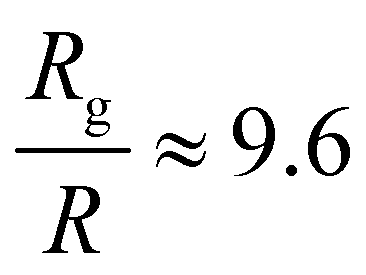 , and
, and 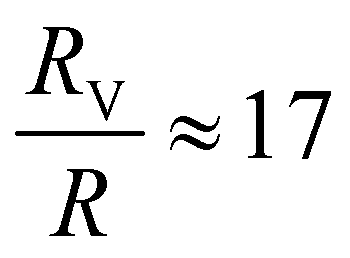 , respectively, but are smaller than the characteristic pore size. Similar arguments regarding capture cross-section have been used to explain the increase of non-specific binding of low-aspect-ratio ellipsoidal nanoparticles in dense packed beds.62
, respectively, but are smaller than the characteristic pore size. Similar arguments regarding capture cross-section have been used to explain the increase of non-specific binding of low-aspect-ratio ellipsoidal nanoparticles in dense packed beds.62
To illustrate the role of local flow fields on particle reorientation, we use a simplified COMSOL model of a shear-thinning non-Newtonian fluid flowing around two fibers. As in the experimental membrane, the local flow velocity depends on proximity and position of the fibers (Fig. 8) and streamlines diverge around the fiber surface. Across a range of flow rates, the vorticity is highest near the fiber surface (Fig. 8(a) and (b)); moreover, because the streamlines typically encounter the fibers at an angle they distort to flow along the fibers. The bending of streamlines along the fibers is similar to the bending of phage trajectories near the fiber observed in the experiments (cf. Fig. 3, 4(a) and 5(a)). These flow features are expected to promote reorientation of anisotropic phage along the fiber axis. We note that this reorientation does not depend on the non-Newtonian character of the solutions; similar flow fields are obtained for a COMSOL simulation of Newtonian water (Fig. 8(c) and (d)). Although this model neglects most of the geometric complexity of the experimental system, it nonetheless suggests that the local flow can promote reorientation of filamentous M13 bacteriophage along the fiber axis, providing one route to increase binding and capture in the open porous Fusion 5 membrane. In a highly porous membrane such as Fusion 5, the diverging streamlines around the fibers reorient phage to promote attachment; in contrast, in dense porous media the converging streamlines in a pore instead transport anisotropic particles towards the pore center and hence have been reported to reduce (non-specific) binding.63
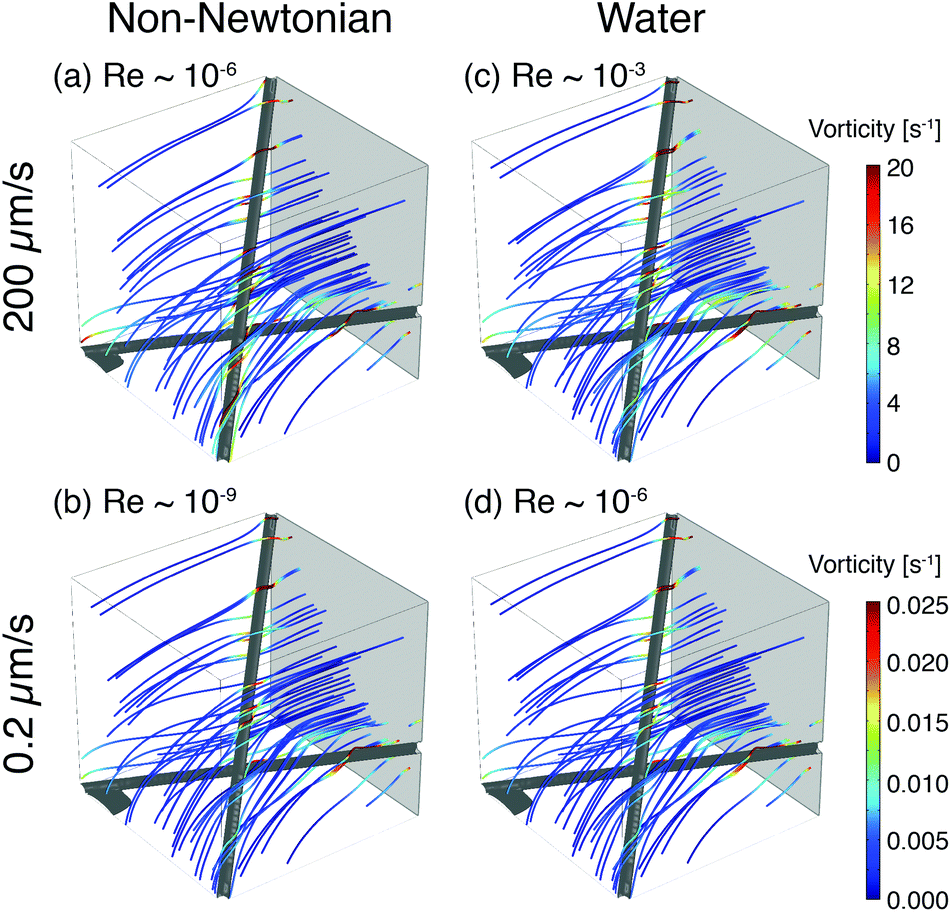 | ||
| Fig. 8 Example streamlines from COMSOL simulations of fluid flow around two fibers for (a, b) a non-Newtonian shear-thinning fluid and (c, d) water at different inlet velocities. Inlet flow is uniform and normal to the gray plane, into the simulation box. Streamlines are colored according to the magnitude of local vorticity. | ||
Conclusions
We characterized the binding and orientational modes of elongated viral nanoparticles to functionalized open-porous membranes at the pore scale using a model LFA. M13 bacteriophage captured by anti-M13 antibodies on the membrane preferentially bind immediately after colliding with a fiber, and do so regardless of their orientation with respect to it. By contrast, a large fraction of the phage captured by a biotin–NeutrAvidin interaction at their tip bind only after reorienting and transporting along the fiber axis. Our results are consistent with (though not proof positive of) the idea that the anisotropic phage shape may promote binding of phage used as reporters in lateral flow immunoassays by two mechanisms: by increasing the capture cross-section and by reorienting in flow to increase capture likelihood. Although in these experiments we fix the phage size and hence cannot differentiate between different effective radii of the phage, all of the effective radii are nonetheless larger than the radius of a sphere of the same volume. Hence anisotropic phage have a larger capture cross section compared to spherical particles. This idea may explain part of the increase in sensitivity of the viral-nanoparticle LFAs compared to traditional gold-nanoparticle-based LFAs, which employ spherical or nearly-spherical reporters that cannot reorient to increase the likelihood of capture.Beyond the specific application to phage-reporter LFAs explored here, we expect that the coupling between reporter shape and binding in a random open porous membrane may inspire new routes towards the design of improved or sensitive LFAs. First, the idea that anisotropic reporters promote binding can be extended to inorganic/abiotic nanoparticles. The excellent control afforded by modern synthetic methods (e.g. for gold nanoparticles, which can be synthesized in a variety of anisotropic morphologies64) would enable systematic studies of the role of particle shape on transport and capture towards identification of design rules for reporters. Second, our results suggest that the orientation of fibers relative to the local flow direction modifies the local vorticity, which in turn generates tumbling near the fiber and increases the likelihood that phage reorient along the fiber. It is thus possible that modifying (in, for example, paper membranes65) either the strip architecture66 or the fiber orientation may increase capture of anisotropic particles and hence also improve LFA sensitivity. These ideas arise in the context of the relatively open LFA membranes studied here. As the characteristic pore size of the membrane is decreased, additional coupling between flow and particle shape may lead to other nanoparticle orientations67 that, in turn, could promote reporter capture and hence increase LFA sensitivity. More broadly, the methods established here to probe the effects of particle anisotropy on capture and binding at the pore-scale may prove useful towards optimizing particle capture in a variety of microscale open porous media, with relevance for applications in on-chip chromatography, virus purification, membrane chromatography, and in microfluidic nanocomposite fabrication.
Acknowledgements
We thank Peter G. Vekilov for use of the pco.edge sCMOS camera, Mohammad S. Safari for assistance with the index of refraction measurement, and Gavin Garvey for the scanning electron micrograph of the Fusion 5 membrane in the ESI.† We acknowledge support from NIAID (1R21AI111120-01A1, to RCW, JCC, and KK), from NSF (CBET-1511789, to JCC and RCW), and from the Welch Foundation (E-1869, to JCC).References
- G. A. Posthuma-Trumpie, J. Korf and A. van Amerongen, Anal. Bioanal. Chem., 2009, 393, 569–582 CrossRef PubMed.
- T. Chinnasamy, L. I. Segerink, M. Nystrand, J. Gantelius and H. A. Svahn, Analyst, 2014, 139, 2348–2354 RSC.
- X. Li, D. Lu, Z. Sheng, K. Chen, X. Guo, M. Jin and H. Han, Talanta, 2012, 100, 1–6 CrossRef PubMed.
- B. A. Rohrman, V. Leautaud, E. Molyneux and R. R. Richards-Kortum, PLoS One, 2012, 7, e45611 Search PubMed.
- S. Vanithamani, S. Shanmughapriya, R. Narayanan, V. Raja, M. Kanagavel, K. Sivasankari and K. Natarajaseenivasan, PLoS One, 2015, 10, e0137130 Search PubMed.
- I.-H. Cho and J. Irudayaraj, Anal. Bioanal. Chem., 2013, 405, 3313–3319 CrossRef PubMed.
- J. Singh, S. Sharma and S. Nara, Anal. Methods, 2015, 7, 9281–9288 RSC.
- S. Y. Ongagna-Yhombi, P. Corstjens, E. Geva, W. R. Abrams, C. A. Barber, D. Malamud and S. Mharakurwa, Malar. J., 2013, 12, 74 CrossRef PubMed.
- Y. Wang, C. Fill and S. R. Nugen, Biosensors, 2012, 2, 32–42 CrossRef PubMed.
- Z. Fang, W. Wu, X. Lu and L. Zeng, Biosens. Bioelectron., 2014, 56, 192–197 CrossRef PubMed.
- Y. Terao, K. Takeshita, Y. Nishiyama, N. Morishita, T. Matsumoto and F. Morimatsu, J. Food Prot., 2015, 78, 1560–1568 CrossRef PubMed.
- P. L. Corstjens, C. J. de Dood, J. J. van der Ploeg-van Schip, K. C. Wiesmeijer, T. Riuttamäki, K. E. van Meijgaarden, J. S. Spencer, H. J. Tanke, T. H. Ottenhoff and A. Geluk, Clin. Biochem., 2011, 44, 1241–1246 CrossRef PubMed.
- F. Mashayekhi, A. M. Le, P. M. Nafisi, B. M. Wu and D. T. Kamei, Anal. Bioanal. Chem., 2012, 404, 2057–2066 CrossRef PubMed.
- Y. K. Oh, H.-A. Joung, H. S. Han, H.-J. Suk and M.-G. Kim, Biosens. Bioelectron., 2014, 61, 285–289 CrossRef PubMed.
- C. Liu, W. Ma, Z. Gao, J. Huang, Y. Hou, C. Xu, W. Yang and M. Gao, J. Mater. Chem. C, 2014, 2, 9637–9642 RSC.
- T. Peng, F. S. Zhang, W. C. Yang, D. X. Li, Y. Chen, Y. H. Xiong, H. Wei and W. H. Lai, J. Food Prot., 2014, 77, 1824–1829 CrossRef PubMed.
- L. Anfossi, C. Baggiani, C. Giovannoli, F. Biagioli, G. D'Arco and G. Giraudi, Anal. Chim. Acta, 2013, 772, 75–80 CrossRef PubMed.
- L. Anfossi, F. Di Nardo, C. Giovannoli, C. Passini and C. Baggiani, Anal. Bioanal. Chem., 2013, 405, 9859–9867 CrossRef PubMed.
- R. Kwizera, J. Nguna, A. Kiragga, J. Nakavuma, R. Rajasingham, D. R. Boulware and D. B. Meya, PLoS One, 2014, 9, e103156 Search PubMed.
- S. Bamrungsap, C. Apiwat, W. Chantima, T. Dharakul and N. Wiriyachaiporn, Microchim. Acta, 2014, 181, 223–230 CrossRef.
- A. S. Paterson, B. Raja, G. Garvey, A. Kolhatkar, A. E. Hagström, K. Kourentzi, T. R. Lee and R. C. Willson, Anal. Chem., 2014, 86, 9481–9488 CrossRef PubMed.
- D. Quesada-González and A. Merkoçi, Biosens. Bioelectron., 2015, 73, 47–63 CrossRef PubMed.
- J. G. Bruno, Pathogens, 2014, 3, 341–355 CrossRef PubMed.
- C. Liu, Q. J. Jia, C. Yang, R. Qiao, L. Jing, L. Wang, C. Xu and M. Gao, Anal. Chem., 2011, 83, 6778–6784 CrossRef PubMed.
- P. Corstjens, M. Zuiderwijk, A. Brink, S. Li, H. Feindt, R. S. Neidbala and H. Tanke, Clin. Chem., 2001, 47, 1885–1893 Search PubMed.
- P. L. Corstjens, M. Zuiderwijk, M. Nilsson, H. Feindt, R. Sam Niedbala and H. J. Tanke, Anal. Biochem., 2003, 312, 191–200 CrossRef PubMed.
- R. Lin, A. Skandarajah, R. E. Gerver, H. D. Neira, D. A. Fletcher and A. E. Herr, Lab Chip, 2015, 15, 1488–1496 RSC.
- B. Y. Moghadam, K. T. Connelly and J. D. Posner, Anal. Chem., 2015, 87, 1009–1017 CrossRef PubMed.
- L. Rivas, M. Medina-Sánchez, A. de la Escosura-Muñiz and A. Merkoçi, Lab Chip, 2014, 14, 4406–4414 RSC.
- J. M. Barnett, P. Wraith, J. Kiely, R. Persad, K. Hurley, P. Hawkins and R. Luxton, Biosensors, 2014, 4, 204–220 CrossRef PubMed.
- A. V. Orlov, V. A. Bragina, M. P. Nikitin and P. I. Nikitin, Biosens. Bioelectron., 2016, 79, 423–429 CrossRef PubMed.
- C. Swanson and A. D'Andrea, Clin. Chem., 2013, 59, 641–648 Search PubMed.
- J. Hwang, S. Lee and J. Choo, Nanoscale, 2016, 8, 11418–11425 RSC.
- G. P. Leonardi, A. M. Wilson and A. R. Zuretti, J. Virol. Methods, 2013, 189, 379–382 CrossRef PubMed.
- L. G. Lee, E. S. Nordman, M. D. Johnson and M. F. Oldham, Biosensors, 2013, 3, 360–373 CrossRef PubMed.
- M. Mirasoli, A. Buragina, L. S. Dolci, M. Guardigli, P. Simoni, A. Montoya, E. Maiolini, S. Girotti and A. Roda, Anal. Chim. Acta, 2012, 721, 167–172 CrossRef PubMed.
- E. M. Tjon Kon Fat, W. R. Abrams, R. S. Niedbala and P. L. A. M. Corstjens, Methods Cell Biol., 2012, 112, 203–234 Search PubMed.
- Z. Qin, W. C. Chan, D. R. Boulware, T. Akkin, E. K. Butler and J. C. Bischof, Angew. Chem., Int. Ed., 2012, 51, 4358–4361 CrossRef PubMed.
- D. J. You, T. S. Park and J. Y. Yoon, Biosens. Bioelectron., 2013, 40, 180–185 CrossRef PubMed.
- D. Erickson, D. O'Dell, L. Jiang, V. Oncescu, A. Gumus, S. Lee, M. Mancuso and S. Mehta, Lab Chip, 2014, 14, 3159–3164 RSC.
- M. Adhikari, S. Dhamane, A. E. V. Hagström, G. Garvey, W.-H. Chen, K. Kourentzi, U. Strych and R. C. Willson, Analyst, 2013, 138, 5584–5587 RSC.
- A. E. V. Hagström, G. Garvey, A. S. Paterson, S. Dhamane, M. Adhikari, M. K. Estes, U. Strych, K. Kourentzi, R. L. Atmar and R. C. Willson, PLoS One, 2015, 10, e0126571 Search PubMed.
- J. Kim, M. Adhikari, S. Dhamane, A. E. V. Hagström, K. Kourentzi, U. Strych, R. C. Willson and J. C. Conrad, ACS Appl. Mater. Interfaces, 2015, 7, 2891–2898 CAS.
- M. Adhikari, U. Strych, J. Kim, H. Goux, S. Dhamane, M.-V. Poongavanam, A. E. V. Hagström, K. Kourentzi, J. C. Conrad and R. C. Willson, Anal. Chem., 2015, 87, 11660–11665 CrossRef CAS PubMed.
- C. M. Soto and B. R. Ratna, Curr. Opin. Biotechnol., 2010, 21, 426–438 CrossRef CAS PubMed.
- S. H. Yang, W.-J. Chung, S. McFarland and S.-W. Lee, Chem. Rec., 2013, 13, 43–59 CrossRef CAS PubMed.
- W.-J. Chung, D.-Y. Lee and S. Y. Yoo, Int. J. Nanomed., 2014, 9, 5825–5836 Search PubMed.
- T. Douglas and M. Young, Science, 2006, 312, 873–875 CrossRef CAS PubMed.
- H. Zhang, Y. Xu, Q. Huang, C. Yi, T. Xiao and Q. Li, Chem. Commun., 2013, 49, 3778–3780 RSC.
- R. J. Citorik, M. Mimee and T. K. Lu, Curr. Opin. Microbiol., 2014, 19, 59–69 CrossRef CAS PubMed.
- J. Litvinov, A. E. V. Hagström, Y. Lopez, M. Adhikari, K. Kourentzi, U. Strych, F. A. Monzon, W. Foster, P. T. Cagle and R. C. Willson, Biotechnol. Lett., 2014, 36, 1863–1868 CrossRef CAS PubMed.
- H. B. Lowman and J. A. Wells, Methods, 1991, 3, 205–216 CrossRef CAS.
- H. J. Kim, M. A. Rossotti, K. C. Ahn, G. G. González-Sapienza, S. J. Gee, R. Musker and B. D. Hammock, Anal. Biochem., 2010, 401, 38–46 CrossRef CAS PubMed.
- T. Peng, A. Balijepalli, S. K. Gupta and T. LeBrun, J. Comput. Inf. Sci. Eng., 2007, 7, 330–338 CrossRef.
- F. Babaye Khorasani, R. Poling-Skutvik, R. Krishnamoorti and J. C. Conrad, Macromolecules, 2014, 47, 5328–5333 CrossRef.
- J. C. Slattery, AIChE J., 1967, 13, 1066–1071 CrossRef.
- E. W. Washburn, Phys. Rev., 1921, 17, 273–283 CrossRef.
- L. Spielman and S. L. Goren, Environ. Sci. Technol., 1968, 2, 279–287 CrossRef.
- N. M. Green, Adv. Protein Chem., 1975, 29, 85–133 CrossRef CAS PubMed.
- K. C. Chang and D. A. Hammer, Biophys. J., 1999, 76, 1280–1292 CrossRef CAS PubMed.
- K. Knez, W. Noppe, N. Geukens, K. P. F. Janssen, D. Spasic, J. Heyligen, K. Vriens, K. Thevissen, B. P. A. Cammue, V. Petrenko, C. Ulens, H. Deckmyn and J. Lammertyn, Anal. Chem., 2013, 85, 10075–10082 CrossRef CAS PubMed.
- T. H. Weiss, A. L. Mills, G. M. Hornberger and J. S. Herman, Environ. Sci. Technol., 1995, 29, 1737–1740 CrossRef CAS PubMed.
- S. Xu, Q. Liao and J. E. Saiers, Environ. Sci. Technol., 2008, 42, 771–778 CrossRef CAS PubMed.
- C. Bao, J. Conde, E. Polo, P. del Pino, M. Moros, P. V. Baptista, V. Grazu, D. Cui and J. M. de la Fuente, Nanomedicine, 2014, 9, 2353–2370 CrossRef CAS PubMed.
- A. K. Yetisen, M. S. Akram and C. R. Lowe, Lab Chip, 2013, 13, 2210–2251 RSC.
- C. Parolo, M. Medina-Sánchez, A. de la Escosura-Muñiz and A. Merkoçi, Lab Chip, 2013, 13, 386–390 RSC.
- M. Trebbin, D. Steinhauser, J. Perlich, A. Buffet, S. V. Roth, W. Zimmermann, J. Thiele and S. Förster, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 6706–6711 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: (1) Movie S1, a representative pore-scale movie of capture of biotinylated Fluor-M13 phage by anti-M13 antibodies on a Fusion 5 membrane; (2) Fig. S1, a comparison of the degree of biotinylation of the Fluor-M13 phage; (3) Fig. S2, representative micrographs of biotinylated Fluor-M13 phage; (4) Fig. S3, schematic of the experimental setup; (5) Fig. S4, degree of binding on biotinylated Fluor-M13 in the presence of glycerol and HPAM; (6) Fig. S5, translational diffusivity of biotinylated Fluor-M13; (7) Fig. S6, a decision tree for classifying orientational modes; (8) Fig. S7, comparison of the binding time distributions and the local maximum velocity; (9) Fig. S8, number of phage in the field of view and the residence time; (10) Fig. S9–S11, comparison of side-binding and tip-binding modes in solutions of varying HPAM concentration and hence different viscosities. See DOI: 10.1039/c6an00567e |
| This journal is © The Royal Society of Chemistry 2017 |