
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Copper(I)-catalyzed sulfonylative Suzuki–Miyaura cross-coupling†
Yiding
Chen
and
Michael C.
Willis
 *
*
Department of Chemistry, University of Oxford, Chemical Research Laboratory, Mansfield Road, Oxford, OX1 3TA, UK. E-mail: michael.willis@chem.ox.ac.uk
First published on 28th February 2017
Abstract
Using a simple copper(I) catalyst has allowed a high yielding sulfonylative-Suzuki–Miyaura cross-coupling reaction to be developed. The process provides a single step route to diaryl sulfones from the direct combination of aryl boronic acids, sulfur dioxide and aryl iodides, and represents the first sulfonylative variant of a classic cross-coupling reaction. Sulfur dioxide is delivered from the surrogate reagent, DABSO. Variation of the reaction conditions allowed interruption of the sulfonylative-Suzuki coupling, resulting in the formation of a presumed Cu–sulfinate intermediate. These sulfinates could be trapped as their sodium salts and treated with electrophiles to allow access to arylalkyl sulfones, β-hydroxyl sulfones, sulfonamides and sulfonyl fluorides.
Introduction
The Suzuki–Miyaura cross-coupling reaction1 is the preeminent method for forming carbon–carbon bonds in the pharmaceutical industry.2 This classic reaction combines aryl boronates and boronic acids with aryl halides under the action of a palladium catalyst, delivering biaryl products in an efficient manner. The popularity of this method stems from the wide availability and low-toxicity of boron-derived substrates, mild reaction conditions, associated good functional group tolerance, and general robustness of the transformations. Carbonylative versions of these reactions have also been developed,3 in which a molecule of carbon monoxide is incorporated in the coupling reaction, diverting the process from biaryl formation and resulting in a ketone product.4 However, despite the electronic similarity between carbon monoxide and sulfur dioxide,5 a sulfonylative variant, which would provide valuable sulfone products, has not been realized (Fig. 1a). When the utility of the sulfone functional group is considered – they feature in numerous biologically active molecules, including many marketed pharmaceuticals and agrochemicals (Fig. 1b), are common motifs in designed materials, and they serve as versatile intermediates for organic synthesis – this absence is striking.6 In this Edge Article, we show that by using a copper(I) catalyst, a high yielding sulfonylative-Suzuki–Miyaura cross-coupling reaction is possible.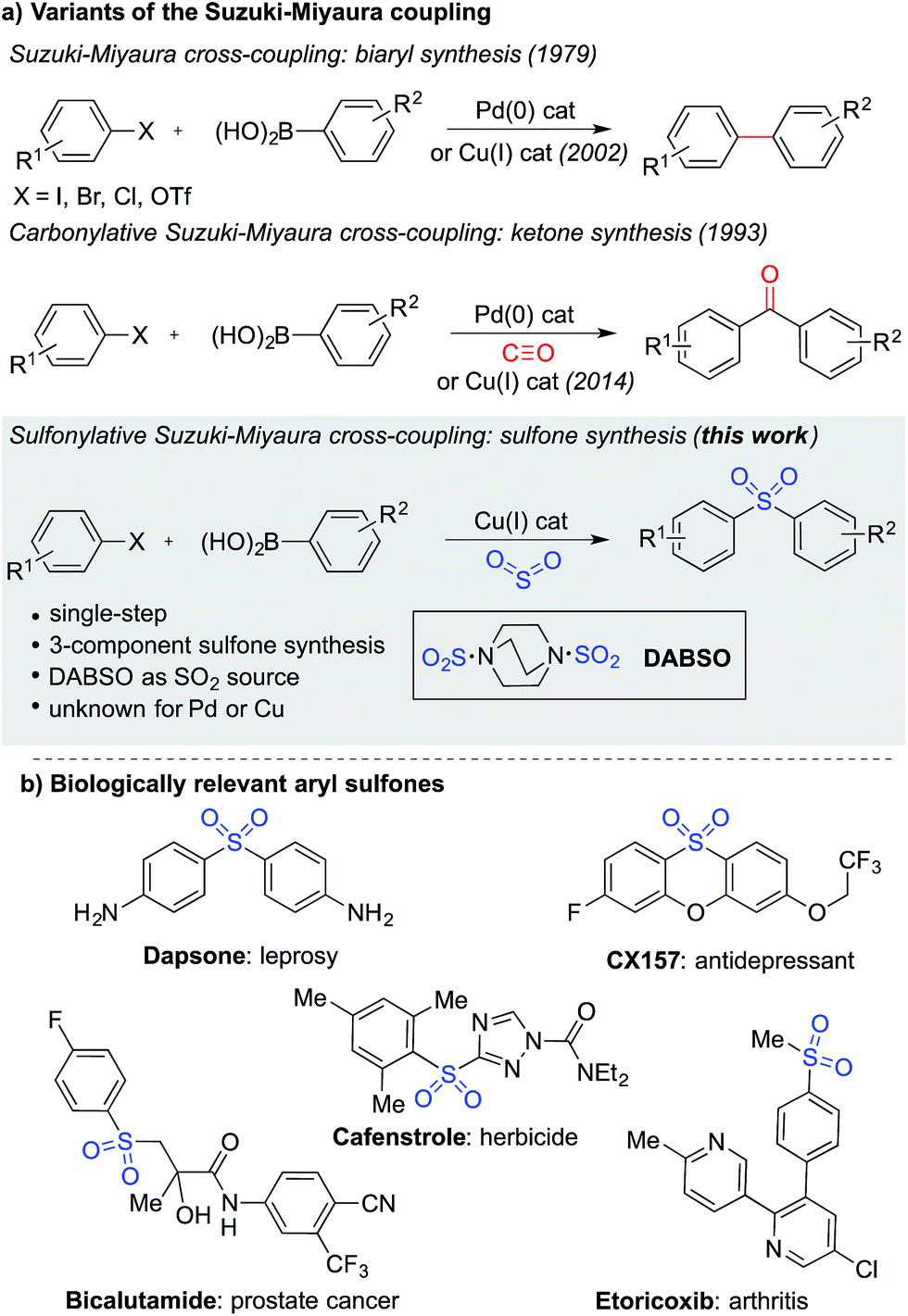 | ||
| Fig. 1 (a) Variants of the Suzuki–Miyaura cross-coupling reaction, and (b) examples of biologically relevant sulfones. | ||
Results and discussion
A variety of methods are available for the synthesis of sulfones,6,7 including a number of transition metal-catalyzed approaches.8 However, many of these classical syntheses are either multi-step, or employ harmful, non-selective reagents, significantly detracting from their utility. Recent years have seen catalytic methods for sulfur dioxide incorporation9 emerge as useful tools for the preparation of sulfonyl-derived functional groups.10 In particular, palladium-catalyzed aryl sulfinate formation has been reported using both aryl halides11 and aryl boronic acids12 as the starting aryl substrates. The sulfinates formed in these reactions can be converted to sulfones, usually by employing a simple electrophilic trap, but this is typically achieved in the second step of the overall reaction.12b Separately, aryl sulfinates have been employed as nucleophiles in a number of palladium-catalyzed coupling reactions with aryl halides.8f,13 We postulated that the integration of these two separate palladium-catalyzed transformations into a single reaction would provide a convenient route for the formation of diarylsulfones, while maintaining many of the desirable features of the classic Suzuki–Miyaura cross-coupling reaction. Despite the encouraging precedents for the separate steps of the proposed coupling, we were never able to achieve the transformation using palladium catalysis. A possible complicating factor with these reactions is the known palladium-catalyzed desulfinative coupling of aryl sulfinates and aryl halides.14Despite this set back, we were still drawn to the advantages that a sulfonylative Suzuki–Miyaura coupling would provide, in particular, the ability to prepare sulfones from the combination of aryl halides and aryl boronic acids – arguably the two coupling partners of choice for synthetic chemists – would provide a conceptually simple, direct route to these valuable products. We turned our attention to the use of alternative catalysts, and were aware that copper(I) catalysts have also been reported to promote the coupling of aryl sulfinates with aryl halides.15,16 Although copper-catalyzed aryl sulfinate formation using sulfur dioxide has also been reported, only the less-available aryl triethoxysilanes can be used as substrates,17 with the use of boronic acids as substrates being unknown.18 Despite this crucial step being unprecedented, we were attracted to the cost-of-goods and sustainability advantages that are potentially available with copper-catalyzed transformations,19 relative to those using palladium, and elected to explore the development of a copper(I)-catalyzed process. Indeed, these advantages have also been recognized in the development of Cu(I)-catalyzed variants of the classic Suzuki–Miyaura cross-coupling reaction,20 as well as a carbonylative version.21
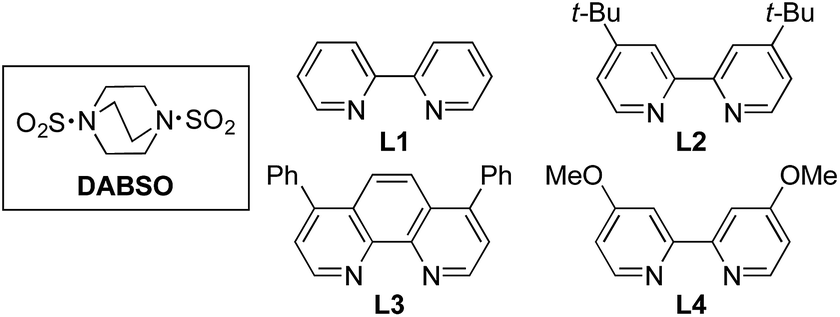
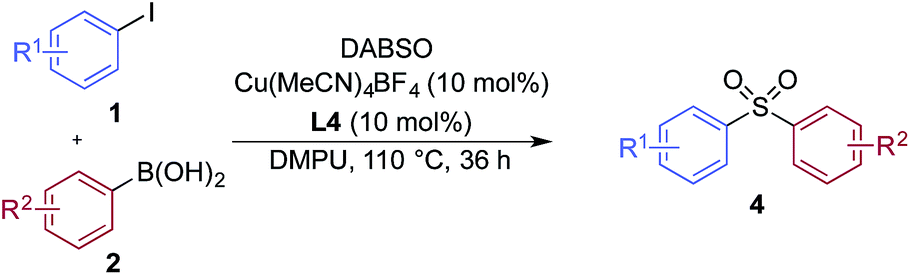
We began our investigation by exploring the coupling of 4-iodotoluene 1a, phenyl boronic acid 2a, and sulfur dioxide. To avoid the direct use of gaseous sulfur dioxide, we employed a surrogate reagent,22 the bis-adduct of sulfur dioxide combined with DABCO, DABSO, which is a bench stable colorless solid and is commercially available.23 Pleasingly, after a variety of copper(I) salts, ligands, solvents, reaction temperatures and additives were explored, we were able to identify a set of optimal conditions, and these are shown in Table 1. The ESI† provides full details, however, the key variations are noted in Table 1. The use of an electron-rich bipyridine ligand, L4,24 in combination with the copper salt Cu(MeCN)4BF4 and the polar aprotic solvent DMPU, allowed an efficient transformation to be achieved.
|
|
||
|---|---|---|
| Entry | Variation from eqn (1) | Yield |
| a Reaction conditions: 1a (0.2 mmol, 1.0 equiv.), DABSO (0.3 mmol, 1.5 equiv.), 2a (0.6 mmol, 3.0 equiv.), solvent (1.0 mL). Yields were measured by HPLC using an internal standard. | ||
| 1 | Cs2CO3 added (2.0 equiv.) | 16% |
| 2 | No Cu, no ligand | 0% |
| 3 | No ligand | 25% |
| 4 | L1 (10%) | 40% |
| 5 | L2 (10%) | 47% |
| 6 | L3 (10%) | 44% |
| 7 | DMF as solvent | 49% |
| 8 | 130 °C | 70% |
| 9 | 24 h | 60% |
| 10 | O2 atmosphere | 3% |
|
||
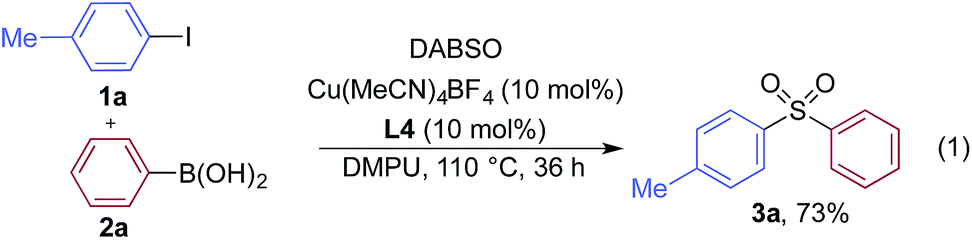
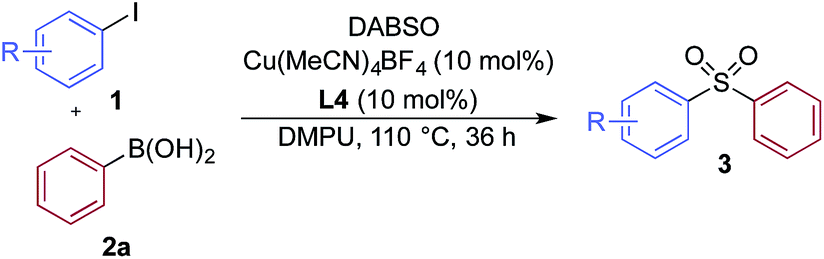
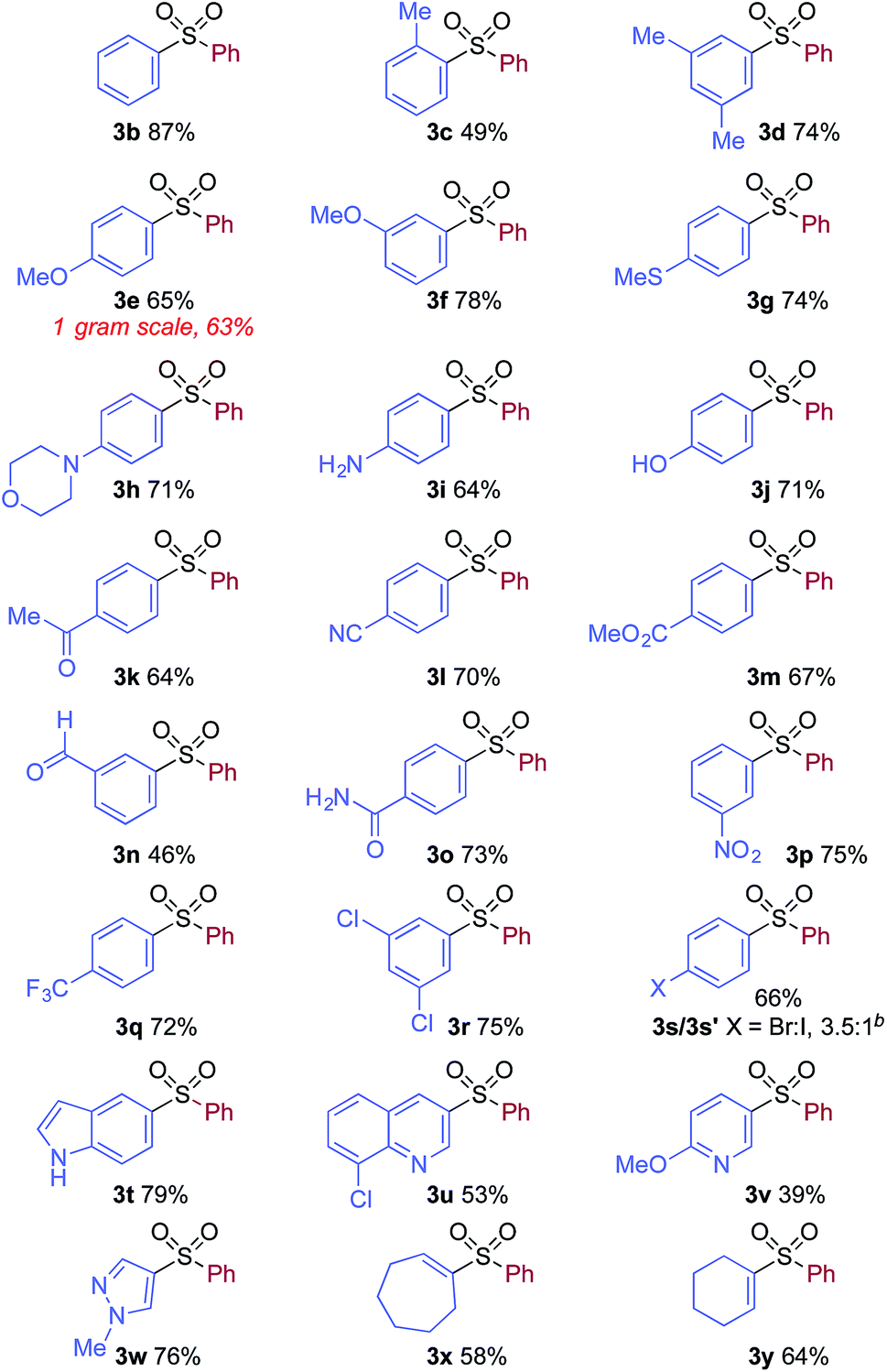
We then used the optimized reaction conditions to explore the scope of the reaction with respect to the variation of the aryl iodide coupling partner, while maintaining phenyl boronic acid as the second aryl component (Table 2). Generally, the process was tolerant of a broad range of both electron-donating and electron-withdrawing functional groups, located at all positions on the aromatic ring. The notable examples include product 3g, which features sulfur atoms with two different oxidation states, an arrangement that would be challenging to prepare using standard sulfone methodology, and sulfone products 3i and 3j which contain free primary amine and phenol groups, respectively. Reactive functional groups such as aldehyde (3n), ketone (3k) and nitrile (3l) were also incorporated without incident. Heterocycle-derived iodides could be transformed to sulfones, with indole (3t), quinoline (3u), pyridine (3v) and pyrazole (3w) derived products being obtained. The products 3x and 3y demonstrate that alkenyl iodide substrates can also be employed. These scoping experiments were routinely performed on a 0.2 mmol scale, however, larger scale reactions were also possible. For example, sulfone 3e was synthesised on a semi-preparative 1 gram scale (5 mmol).
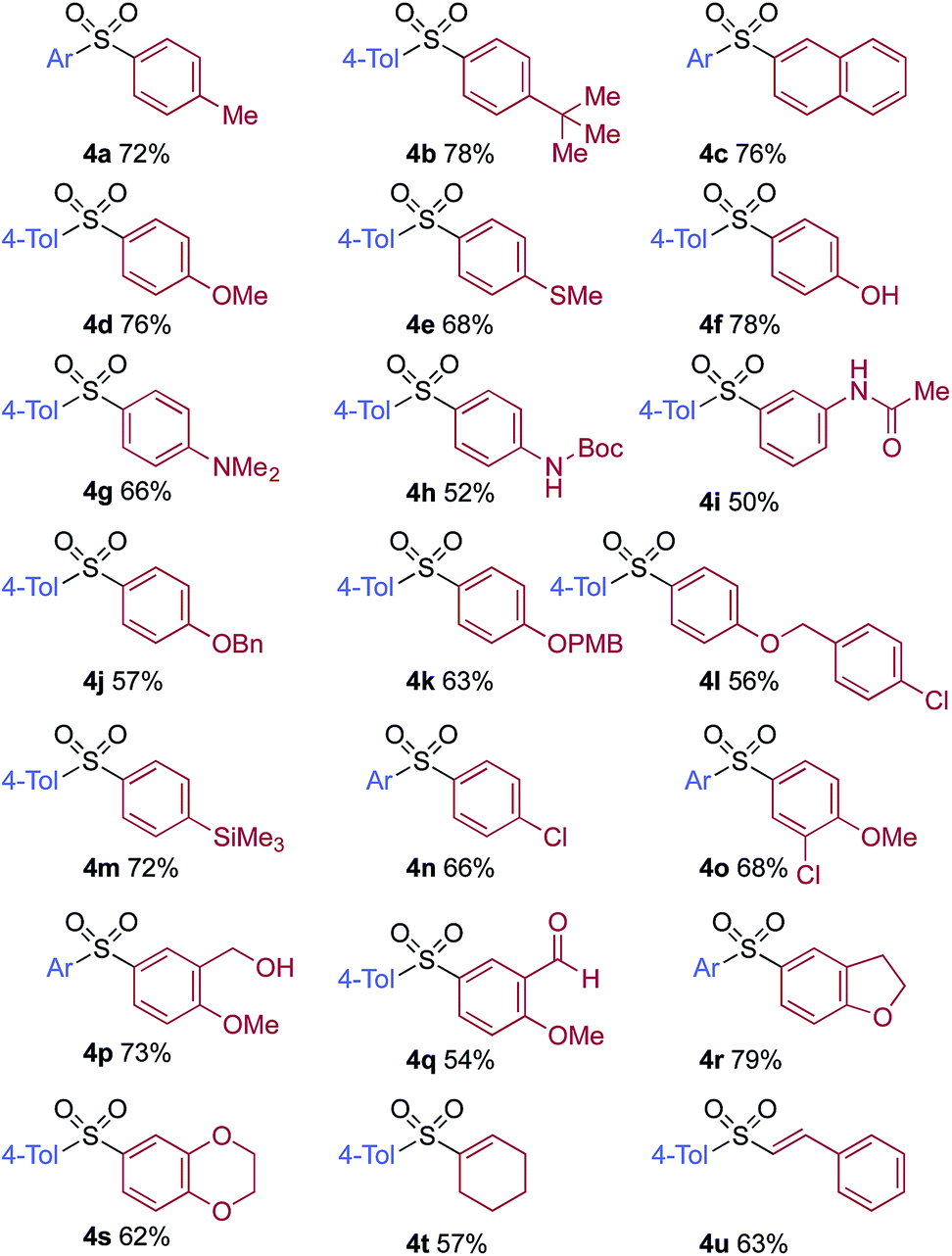
A significant variation of the boronic acid coupling partner was also possible (Table 3). Either 4-iodotoluene or 4-morpholino-iodobenzene was employed as the coupling partner in these transformations, with the choice being dictated by the ease of product purification. A broad range of useful functionalities could be introduced using this chemistry, for example, ether (4d), sulfide (4e), alcohol (4f), amine (4g), carbamate (4h), amide (4i), aryl chloride (4l,n,o), silane (4m), and aldehyde (4q) groups were all tolerated under the reaction conditions. As with the iodide component, heterocyclic variants (4r,s) and alkenyl boronic acids were also useful substrates, with the latter delivering unsaturated sulfone products (4t,u).
|
|
|---|
| a Ar = 4-morpholino–C6H4. Reaction conditions: aryl iodide 1 (0.2 mmol, 1.0 equiv.), DABSO (0.3 mmol, 1.5 equiv.), boronic acid 2 (0.6 mmol, 3.0 equiv.), Cu(MeCN)4BF4 (10 mol%), L4 (10 mol%), DMPU (1.0 mL), 110 °C, 36 h. Isolated yields. |
|
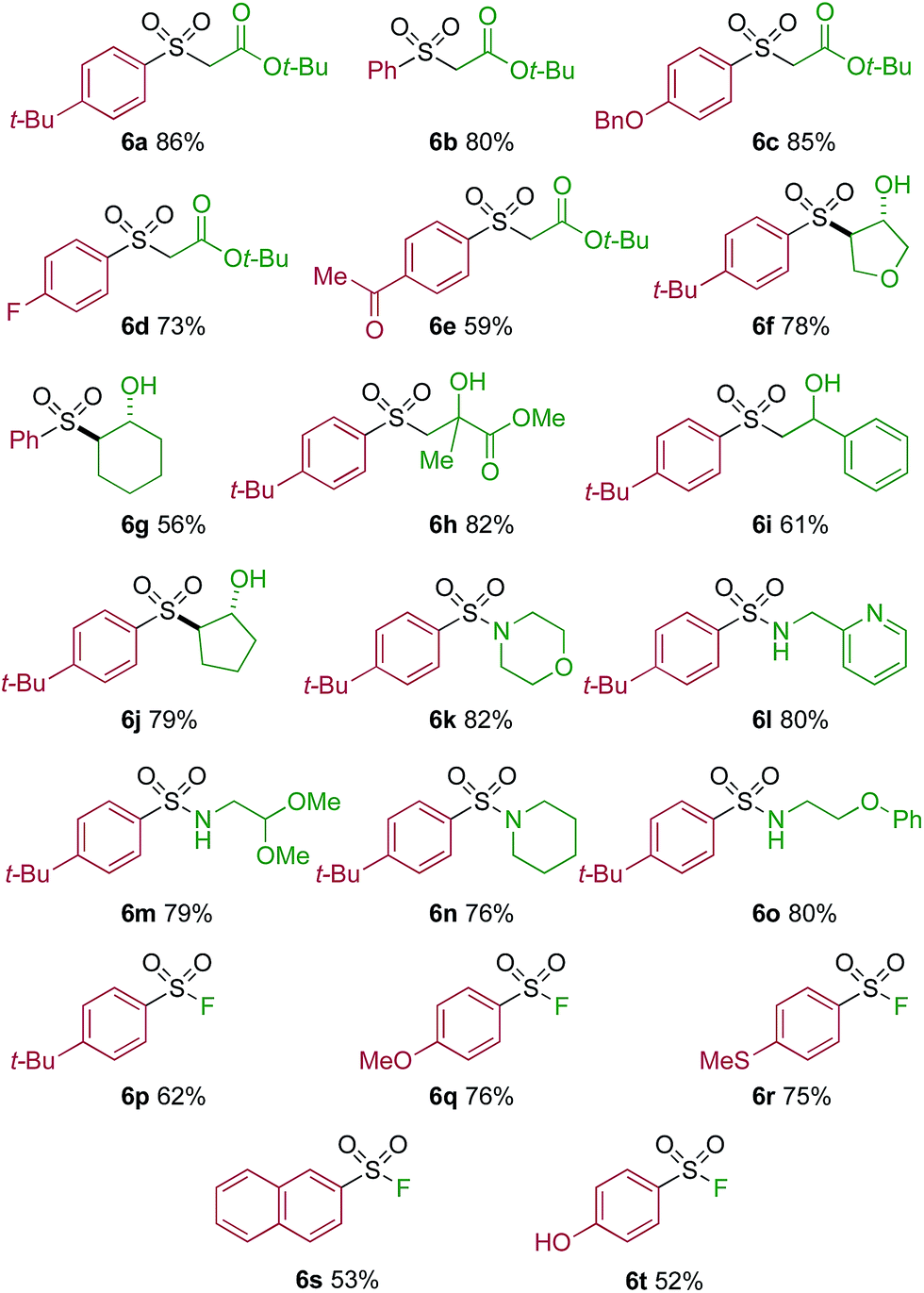
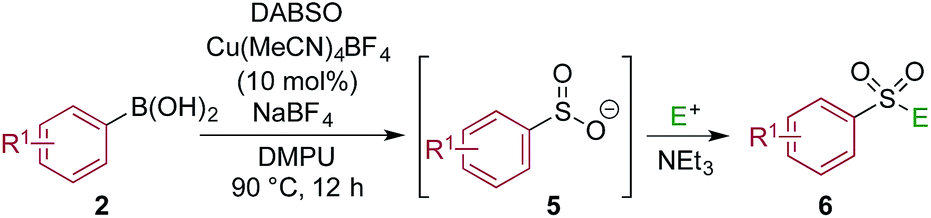
One of the key challenges in developing a direct copper-catalyzed sulfonylative Suzuki–Miyaura reaction was establishing the viability of the copper-catalyzed sulfinate formation from the combination of an aryl boronic acid and sulfur dioxide. Although the broad scope of the targeted reaction, as shown in Tables 2 and 3, goes a significant way towards validating this hypothesis, we wanted to develop reaction conditions that would allow the copper-catalyzed synthesis of discrete sulfinate intermediates. This would then allow these intermediates to be trapped with a variety of electrophiles to deliver structurally diverse products, and would complement the recently described palladium-catalyzed variant of this transformation.12 By employing a stoichiometric amount of NaBF4 in combination with Cu(MeCN)4BF4 as a catalyst, we were able to interrupt the sulfonylative Suzuki coupling and realize an efficient method for the preparation of copper sulfinates from aryl boronic acids (Table 4). Metal sulfinates are not useful products in their own right, and are in fact challenging to isolate, however, they can be converted to a selection of useful sulfonyl-derived functional groups by treatment with appropriate electrophiles. For example, in situ treatment of the sulfinate with an alkyl bromide delivers alkylaryl sulfone products (6a–e).25 The use of epoxides as the electrophiles provides β-hydroxy sulfones (6f–j).25b Treatment with in situ generated chloroamines provides sulfonamides (6k–o),26 and treatment with NFSI, which is a source of electrophilic fluorine, generates the corresponding sulfonyl fluorides (6p–t).27
|
|
|---|
| a Reaction conditions DABSO (0.1 mmol, 0.5 equiv.), boronic acid 2 (0.2 mmol, 1.0 equiv.), NaBF4 (1.0 mmol, 5.0 equiv.), DMPU (1 mL), 90 °C, 12 h; then, for 6a–6e: Et3N (0.3 mmol, 1.5 equiv.), t-butyl-bromoacetate (0.4 mmol, 2.0 equiv.); for 6f–6j: Et3N (0.3 mmol, 1.5 equiv.), epoxide (0.4 mmol, 2.0 equiv.), H2O (2 mL); for 6k–6o: Et3N (0.3 mmol, 1.5 equiv.), amine (0.4 mmol, 2.0 equiv.), NaOCl (0.4 mmol, 2% aq. solution, 2.0 equiv.); for 6p–6t: NFSI (0.3 mmol, 1.5 equiv.), DMPU (0.2 mL). Isolated yields. |
|
Conclusions
In realizing a copper-catalyzed sulfonylative Suzuki–Miyaura cross-coupling, a work-horse reaction of the pharmaceutical industry has been transformed to deliver valuable sulfone products. This process represents the first true sulfonylative-variant of a classic cross-coupling process. A good variation of both coupling partners is possible, delivering sulfone products in high yields. The demonstration that the combination of copper(I) catalysts, aryl boronic acids and a sulfur dioxide surrogate generates aryl sulfinate intermediates is also significant, and bodes well for the development of new reactions which exploit these key intermediates, access to which has been limited when using benign, readily available substrates and sustainable catalysts.Acknowledgements
We thank the EPSRC for the support of this study.Notes and references
- (a) N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS; (b) N. Miyaura, K. Yamada and A. Suzuki, Tetrahedron Lett., 1979, 20, 3437–3440 CrossRef.
- (a) D. G. Brown and J. Bostrom, J. Med. Chem., 2016, 59, 4443–4458 CrossRef CAS PubMed; (b) J. S. Carey, D. Laffan, C. Thomson and M. T. Williams, Org. Biomol. Chem., 2006, 4, 2337–2347 RSC; (c) D. Blakemore, in Synthetic Methods in Drug Discovery: Volume 1, The Royal Society of Chemistry, 2016, vol. 1, pp. 1–69 Search PubMed.
- (a) T. Ishiyama, H. Kizaki, T. Hayashi, A. Suzuki and N. Miyaura, J. Org. Chem., 1998, 63, 4726–4731 CrossRef CAS; (b) T. Ishiyama, H. Kizaki, N. Miyaura and A. Suzuki, Tetrahedron Lett., 1993, 34, 7595–7598 CrossRef CAS.
- A. Brennfuhrer, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2009, 48, 4114–4133 CrossRef PubMed.
- (a) H. S. Klein, Chem. Commun., 1968, 377–378 RSC; (b) G. J. Kubas, Acc. Chem. Res., 1994, 27, 183–190 CrossRef CAS.
- N. S. Simpkins, Sulfones in Organic Synthesis, Pergamon Press, Oxford, 1993 Search PubMed.
- G. Manolikakes, N.-W. Liu and S. Liang, Synthesis, 2016, 48, 1939–1973 CrossRef.
- (a) O. Saidi, J. Marafie, A. E. Ledger, P. M. Liu, M. F. Mahon, G. Kociok-Kohn, M. K. Whittlesey and C. G. Frost, J. Am. Chem. Soc., 2011, 133, 19298–19301 CrossRef CAS PubMed; (b) X. Zhao, E. Dimitrijević and V. M. Dong, J. Am. Chem. Soc., 2009, 131, 3466–3467 CrossRef CAS PubMed; (c) S. Liang, N.-W. Liu and G. Manolikakes, Adv. Synth. Catal., 2016, 358, 159–163 CrossRef CAS; (d) M. Jegelka and B. Plietker, Chem.–Eur. J., 2011, 17, 10417–10430 CrossRef CAS PubMed; (e) M. Ueda and J. F. Hartwig, Org. Lett., 2010, 12, 92–94 CrossRef CAS PubMed; (f) D. C. Reeves, S. Rodriguez, H. Lee, N. Haddad, D. Krishnamurthy and C. H. Senanayake, Tetrahedron Lett., 2009, 50, 2870–2873 CrossRef CAS; (g) W. H. Rao, B. B. Zhan, K. Chen, P. X. Ling, Z. Z. Zhang and B. F. Shi, Org. Lett., 2015, 17, 3552–3555 CrossRef CAS PubMed.
- (a) A. S. Deeming, E. J. Emmett, C. S. Richards-Taylor and M. C. Willis, Synthesis, 2014, 46, 2701–2710 CrossRef; (b) P. Bisseret and N. Blanchard, Org. Biomol. Chem., 2013, 11, 5393–5398 RSC; (c) G. Liu, C. B. Fan and J. Wu, Org. Biomol. Chem., 2015, 13, 1592–1599 RSC.
- (a) B. Nguyen, E. J. Emmett and M. C. Willis, J. Am. Chem. Soc., 2010, 132, 16372–16373 CrossRef CAS PubMed; (b) S. Ye and J. Wu, Chem. Commun., 2012, 48, 10037–10039 RSC; (c) S. Ye and J. Wu, Chem. Commun., 2012, 48, 7753–7755 RSC; (d) E. J. Emmett, C. S. Richards-Taylor, B. Nguyen, A. Garcia-Rubia, B. R. Hayter and M. C. Willis, Org. Biomol. Chem., 2012, 10, 4007–4014 RSC; (e) M. W. Johnson, S. W. Bagley, N. P. Mankad, R. G. Bergman, V. Mascitti and F. D. Toste, Angew. Chem., Int. Ed., 2014, 53, 4404–4407 CrossRef CAS PubMed.
- (a) E. J. Emmett, B. R. Hayter and M. C. Willis, Angew. Chem., Int. Ed., 2014, 53, 10204–10208 CrossRef CAS PubMed; (b) A. Shavnya, S. B. Coffey, A. C. Smith and V. Mascitti, Org. Lett., 2013, 15, 6226–6229 CrossRef CAS PubMed.
- (a) A. S. Deeming, C. J. Russell and M. C. Willis, Angew. Chem., Int. Ed., 2016, 55, 747–750 CrossRef CAS PubMed; (b) A. Shavnya, K. D. Hesp, V. Mascitti and A. C. Smith, Angew. Chem., Int. Ed., 2015, 54, 13571–13575 CrossRef CAS PubMed.
- (a) S. Cacchi, G. Fabrizi, A. Goggiamani, L. M. Parisi and R. Bernini, J. Org. Chem., 2004, 69, 5608–5614 CrossRef CAS PubMed; (b) E. J. Emmett, B. R. Hayter and M. C. Willis, Angew. Chem., Int. Ed., 2013, 52, 12679–12683 CrossRef CAS PubMed.
- (a) D. H. Ortgies, A. Barthelme, S. Aly, B. Desharnais, S. Rioux and P. Forgione, Synthesis, 2013, 45, 694–702 CrossRef CAS; (b) D. H. Ortgies, A. Hassanpour, F. Chen, S. Woo and P. Forgione, Eur. J. Org. Chem., 2016, 2016, 408–425 CrossRef CAS.
- (a) H. Suzuki and H. Abe, Tetrahedron Lett., 1995, 36, 6239–6242 CrossRef CAS; (b) W. Zhu and D. Ma, J. Org. Chem., 2005, 70, 2696–2700 CrossRef CAS PubMed; (c) M. Yang, H. Shen, Y. Li, C. Shen and P. Zhang, RSC Adv., 2014, 4, 26295 RSC; (d) H. Fu, X. Yang and L. Shi, Synlett, 2014, 25, 847–852 CrossRef; (e) B. T. V. Srinivas, V. S. Rawat, K. Konda and B. Sreedhar, Adv. Synth. Catal., 2014, 356, 805–817 CrossRef CAS; (f) Y. Peng, J. Chem. Res., 2014, 38, 447–449 CrossRef CAS.
- C. Beaulieu, D. Guay, Z. Wang and D. A. Evans, Tetrahedron Lett., 2004, 45, 3233–3236 CrossRef CAS.
- (a) X. Wang, L. Xue and Z. Wang, Org. Lett., 2014, 16, 4056–4058 CrossRef CAS PubMed; (b) D. Q. Zheng, R. Y. Mao, Z. M. Li and J. Wu, Org. Chem. Front., 2016, 3, 359–363 RSC.
- R. Mao, D. Zheng, H. Xia and J. Wu, Org. Chem. Front., 2016, 3, 693–696 RSC.
- K. S. Egorova and V. P. Ananikov, Angew. Chem., Int. Ed., 2016, 55, 12150–12162 CrossRef CAS PubMed.
- (a) Y. Zhou, W. You, K. B. Smith and M. K. Brown, Angew. Chem., Int. Ed., 2014, 53, 3475–3479 CrossRef CAS PubMed; (b) S. K. Gurung, S. Thapa, A. Kafle, D. A. Dickie and R. Giri, Org. Lett., 2014, 16, 1264–1267 CrossRef CAS PubMed; (c) J.-H. Li, J.-L. Li, D.-P. Wang, S.-F. Pi, Y.-X. Xie, M.-B. Zhang and X.-C. Hu, J. Org. Chem., 2007, 72, 2053–2057 CrossRef CAS PubMed; (d) M. B. Thathagar, J. Beckers and G. Rothenberg, J. Am. Chem. Soc., 2002, 124, 11858–11859 CrossRef CAS PubMed.
- L. Cheng, Y. Zhong, Z. Ni, H. Du, F. Jin, Q. Rong and W. Han, RSC Adv., 2014, 4, 44312–44316 RSC.
- E. J. Emmett and M. C. Willis, Asian J. Org. Chem., 2015, 4, 602–611 CrossRef CAS.
- H. Woolven, C. Gonzalez-Rodriguez, I. Marco, A. L. Thompson and M. C. Willis, Org. Lett., 2011, 13, 4876–4878 CrossRef CAS PubMed.
- R. A. Altman, E. D. Koval and S. L. Buchwald, J. Org. Chem., 2007, 72, 6190–6199 CrossRef CAS PubMed.
- (a) C. C. Chen and J. Waser, Org. Lett., 2015, 17, 736–739 CrossRef CAS PubMed; (b) A. S. Deeming, C. J. Russell, A. J. Hennessy and M. C. Willis, Org. Lett., 2014, 16, 150–153 CrossRef CAS PubMed; (c) B. N. Rocke, K. B. Bahnck, M. Herr, S. Lavergne, V. Mascitti, C. Perreault, J. Polivkova and A. Shavnya, Org. Lett., 2014, 16, 154–157 CrossRef CAS PubMed.
- (a) A. S. Deeming, C. J. Russell and M. C. Willis, Angew. Chem., Int. Ed., 2015, 54, 1168–1171 CrossRef CAS PubMed; (b) E. F. Flegeau, J. M. Harrison and M. C. Willis, Synlett, 2016, 27, 101–105 CAS.
- (a) J. Dong, L. Krasnova, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2014, 53, 9430–9448 CrossRef CAS PubMed; (b) A. T. Davies, J. M. Curto, S. W. Bagley and M. C. Willis, Chem. Sci., 2017, 8, 1233–1237 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6sc05483h |
| This journal is © The Royal Society of Chemistry 2017 |