
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCopper carbenes alkylate guanine chemoselectively through a substrate directed reaction†
Stefanie N.
Geigle
a,
Laura A.
Wyss
b,
Shana J.
Sturla
b and
Dennis G.
Gillingham
a
aDepartment of Chemistry, University of Basel, St. Johanns-Ring 19, CH-4056, Basel, Switzerland. E-mail: dennis.gillingham@unibas.ch
bDepartment of Health Sciences and Technology, ETH Zurich, Schmelzbergstrasse 9, CH-8092 Zurich, Switzerland
First published on 7th September 2016
Abstract
Cu(I) carbenes derived from α-diazocarbonyl compounds lead to selective alkylation of the O6 position in guanine (O6-G) in mono- and oligonucleotides. Only purine-type lactam oxygens are targeted – other types of amides or lactams are poorly reactive under conditions that give smooth alkylation of guanine. Mechanistic studies point to N7G as a directing group that controls selectivity. Given the importance of O6-G adducts in biology and biotechnology we expect that Cu(I)-catalyzed O6-G alkylation will be a broadly used synthetic tool. While the propensity for transition metals to increase redox damage is well-appreciated, our results suggest that transition metals might also increase the vulnerability of nucleic acids to alkylation damage.
Introduction
Catalytic reactions with metal carbenes are broadly applied in synthetic chemistry.1,2 Although metal carbenes are reactive intermediates, their chemoselectivity can be steered through the right combination of metal, ligand, and carbene precursor.3 Even in water metal carbenes can still perform an impressive array of transformations.4–7 In our studies on the post-synthetic tailoring of large nucleic acids through metal catalysis we have found that donor–acceptor substituted carbenes8 derived from Rh(II) and Cu(I) give primarily N–H insertion of exocyclic amine groups in unpaired regions of DNAs and RNAs.9–11 Here we show that unstabilized Cu(I) carbenes derived from α-diazo acetate and amide derivatives have a completely different chemoselectivity: they quickly and cleanly alkylate the O6 position in guanine and inosine (see Fig. 1B for a representative reaction with GMP and ethyl α-diazoacetate (EDA)). Cu(I) is known to bind amides (especially amidates),12 but there is no precedence which would have predicted such a pronounced selectivity for the lactam-like oxygen of guanine. While most instances of copper13 direction by heterocycles in synthetic chemistry14–16 and biology17,18 employ histidine or pyridine (see Fig. 1A), our results show that the purine ring system of guanine is also a powerful directing group in copper catalysed alkylation (see Fig. 1B) reactions.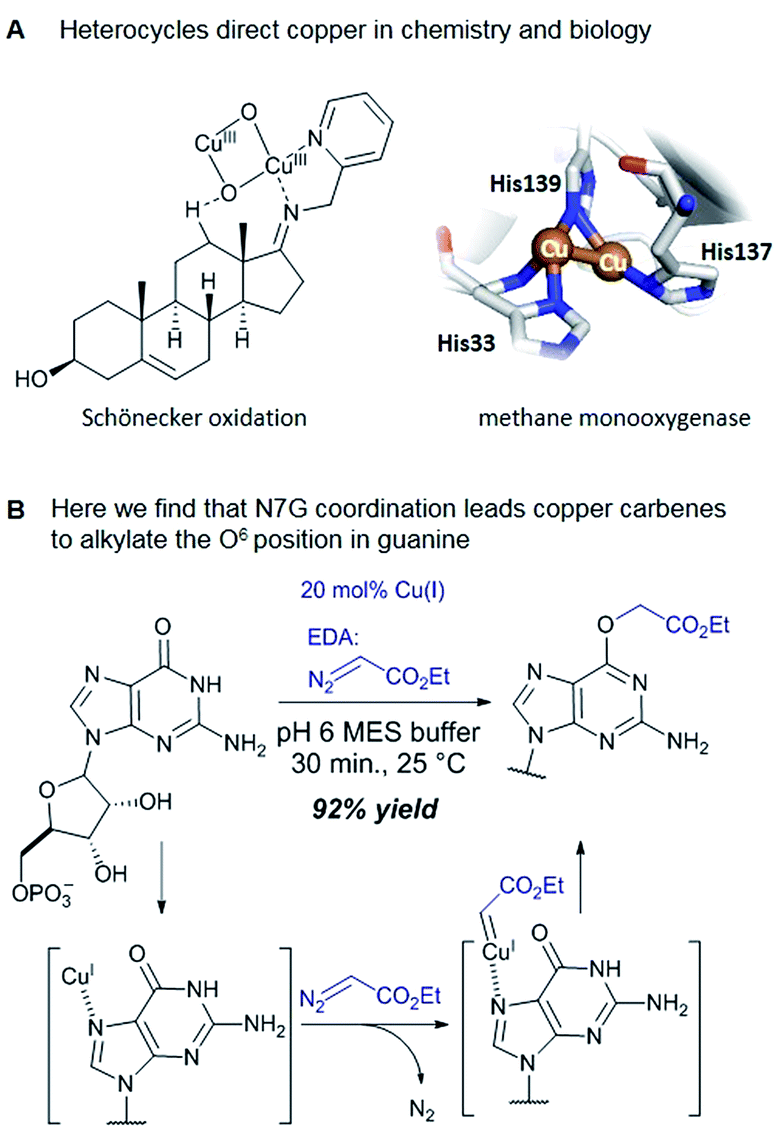 | ||
| Fig. 1 (A) In both synthetic chemistry and biology there are numerous examples of copper chemistry being controlled by coordination to nitrogen heterocycles (PDB ID: 1YEW) (B) N7 of guanines and inosines can direct copper catalysed reactions to the O6 position. | ||
DNA is constantly exposed to electrophiles, both natural and man-made.19 In response, all organisms have evolved DNA surveillance and repair pathways to maintain genomic integrity.20 Alkylation at the O6 of guanine21 is particularly dangerous because polymerases tend to misincorporate thymidines in response to O6-(alkyl)-G during replication, leading to potentially carcinogenic G-to-A transition mutations.22–25 The colon cells of people on high red meat diets have increased carboxymethyl damage at O6-G.26,27 Epidemiological studies have shown that the high-red meat diet demographic in Western cultures suffer as well from a higher prevalence of gastrointestinal cancers.28,29 Whether there is a causative connection between these two occurrences is currently unclear. Studying the biology of O6-(carboxymethyl)-G damage (O6-cmG), however, is hampered by synthetic challenges. Multi-step procedures, protecting group manipulations,30 and the need for strong electrophilic phosphorous reagents,31 combine to make both the synthesis and purification of O6-(alkyl)-G derivatives laborious. With copper catalysed alkylation the carboxymethyl motif is introduced in a single step from naturally occurring precursors.
Results and discussion
Reaction discovery and substrate scope
Cu(I) carbenes derived from α-diazocarbonyl compounds are highly selective for O6 alkylation of unpaired guanines and inosines in ssDNA, monophosphates (MP) and triphosphates (TP) (see Fig. 1B and Table 1). The surprising selectivity prompted us to synthesize an O6-alkyl-inosine adduct by traditional routes to check the site-selectivity: identical characterization data confirm the proposed structures (see the ESI,† page S5-6). The method is the most concise and selective approach ever reported to the O6-(alkyl)-G motif and it is post-synthetic, obviating the need for solid phase DNA synthesis with modified phosphoramidites. The convergent assembly opens the door to a range of O6-(alkyl)-G derivatives from simple starting materials.|
|
|||||
|---|---|---|---|---|---|
| Entry | Substrate | Time (min) | Cosolvent | Conv. (%) | Yield. (%) |
| a Complete conditions: 5 mM substrate, 1 mM CuSO4, 5 mM ascorbate, 50 mM EDA (ethyl diazoacetate), 100 mM MES (2-(N-morpholino)ethanesulfonic acid) pH 6, H2O, 25 °C. b Same conditions as a but with 20% (v/v) of cosolvent. c No ascorbate added. d No CuSO4 or ascorbate added. e Alkylation site was assigned on the basis of tandem MS fragmentation ions. f Six minor mono-alkylation products were detected by ESI, but none in an amount that could be isolated. | |||||
| 1a | dGMP | 30 | None | 98e | 86 |
| 2a | GMP | 30 | None | 96e | 92 |
| 3b | GMP | 30 | DMSO | 93e | — |
| 4c | GMP | 30 | None | 87 (ref. 32) | — |
| 5d | GMP | 150 | None | 0 | — |
| 6a | IMP | 30 | None | 92e | 81 |
| 7a | AMP | 30 | None | 40 | — |
| 8a | CMP | 30 | None | 38 | — |
| 9a | UMP | 30 | None | 21 | — |
| 10a | TMP | 30 | None | 13 | — |
| 11b | GTP | 30 | tBuOH | 98e | 73 |
| 12b | dGTP | 30 | tBuOH | 97e | 65 |
| 13a | d(TGT) | 15/45 | None | 62/81e | — |
| 14a | d(TAT) | 120 | None | 35f | — |
| 15b | d(ATGC) | 150 | tBuOH | 38e | — |
| 16b | d(ATGC) | 150 | Dioxane | 64e | — |
| 17b | d(TTTTGTTTT) | 30 | DMSO | 82e | — |
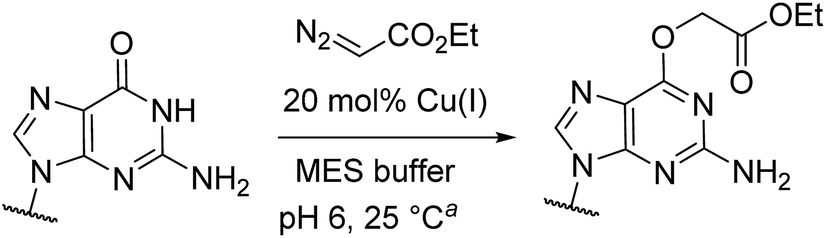
We explored the efficiency and selectivity of O6-G targeting by characterizing reactions with nucleotide monophosphates. Treatment of dGMP or GMP with 20 mol% Cu(II)SO4 (in situ reduced by ascorbate) and ethyl α-diazoacetate (EDA) in aqueous buffer resulted in high conversion and isolated yields of ethylcarboxymethyl product (see Table 1, entry 1 and 2) within 30 min. Under the same conditions, the structural analogues IMP, GTP, and dGTP were efficiently alkylated at their O6 positions (see Table 1, entry 6, 11 and 12). In contrast, reactions without catalyst (entry 5) or with different MPs like UMP, AMP, CMP, or TMP (entries 7–10) resulted in little or no detectable alkylation. Rather, in cases where substrates lacked a guanine, the starting material was partially converted through unselective pathways that we could not completely characterize – in each case some alkylation was usually observed in the MS but these did not give substantial HPLC peaks that we could isolate. The same was also seen in short DNA fragments such as d(TAT) (see entry 14); here again starting material was consumed over time, but no single alkylation product could be isolated. In contrast, when guanine was present post-synthetic modifications of a DNA 3-mer d(TGT) and 9-mer d(TTTTGTTTT) with the EDA-derived Cu(I) carbene gave a single large alkylation product (only O6-G alkylation observed, see Table 1 entries 13 and 17). In the substrates that lack guanine we cannot account for the mass balance: we believe in these cases that oxidation is a side-reaction that consumes starting material when no guanine is available for the carbene-based reaction.
Cosolvent, oligonucleotide size, and diazo substrate all modulate reactivity
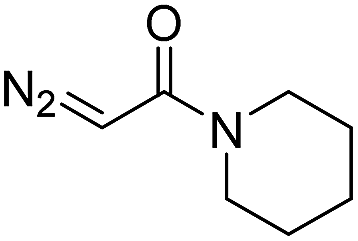
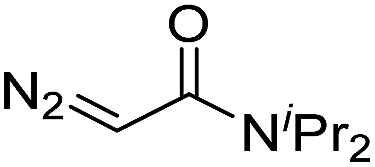
While oligonucleotides also reacted selectively at O6-G, the reactions were slower. For example, d(ATGC) was alkylated at O6-G (determined by MS-MS sequencing, see ESI page S18†) but required longer reaction times and conversion was lower than for the mononucleotides (cf.Table 1 entries 11 & 15). This reduced reactivity can likely be attributed to the unproductive sequestration of copper to other binding sites (phosphate backbone, other nucleobases) in the nucleic acid. The lower reactivity with longer oligonucleotides was mitigated by using 1,4-dioxane as a cosolvent. For example, changing the stock solution of EDA from 10% tBuOH in water to dioxane nearly doubled the conversion of d(ATGC) (cf.Table 1 entries 15 & 16). Nevertheless, these observations point to a limitation of the method: the larger the oligonucleotide the slower it is likely to react.Diazo substrate variation is another approach to improve the reaction with challenging nucleic acid substrates. Diazoacetamides33 (DAAs) led to higher conversions than EDA and high O6 selectivity was maintained (see Table 2). For example, the alkylation of d(ATGC) with DAAs led to conversions of starting material above 60% (see entries 3 & 5, Table 2), whereas the same substrate with EDA led to only 38% conversion (see entry 15, Table 1). While DAAs are better substrates in nucleic acid alkylation, they are also less stable; it was, for example, impossible to prepare a stock solution in 10% tBuOH and water due to rapid decomposition. DMSO as co-solvent stabilizes the DAAs, with the drawback that their reactivity in nucleic acid alkylation is reduced. Changing to a less coordinating co-solvent like dioxane seems to be the best compromise between these competing effects. For example comparing entries 3 & 4 in Table 2 we see that in 20% dioxane higher conversion was obtained in a fifth of the time (62% after 150 minutes versus 74% after 30 minutes). With the more reactive DAAs, even longer DNAs such as 16-mers were alkylated efficiently (see entries 8 & 9, Table 2). The amide substrates offer the additional advantages of stability and modularity. Customization of substrates for pull-downs or with fluorescent tags would simply require the addition of the appropriate amine in the DAA synthesis.33 As an example, modification of guanine at the O6 position of d(ATGC) with a terminal alkyne-bearing diazo substrate (see entry 10, Table 2) followed by a click reaction with azide-conjugated biotin (see the ESI page S31†), installed a complex pull-down tag in only two steps.
|
|
||||
|---|---|---|---|---|
| Entry | Substrate | Diazo compound | Time (min) | Conv. (%) |
| a Complete conditions: 5 mM substrate, 1 mM CuSO4, 5 mM ascorbate, 50 mM diazo compound, 100 mM MES pH 6, DMSO 20% (v/v), H2O, 25 °C. b G-alkylation was assigned on the basis of tandem MS fragmentation ions. c Same conditions as a but with 20% (v/v) dioxane instead of DMSO. | ||||
| 1 | d(TGT) |

|
15 | 78b |
| 2 | d(TGT) |

|
15 | 78b |
| 3 | d(ATGC) |

|
150 | 62b |
| 4 | d(ATGC)c |
|
30 | 74b |
| 5 | d(ATGC) |
|
150 | 61b |
| 6 | d(TTTTGTTTT) |

|
30 | 92b |
| 7 | d(TTTTGTTTT) |

|
30 | 81b |
| 8 | d(AACAGTCATATCCTTA) |

|
180 | 48 |
| 9 | d(AACAGTCATATCCTTA) |

|
180 | 40 |
| 10 | d(ATGC)c |

|
150 | 66b |

Mechanistic hypothesis
The selectivity observed with copper carbenes does not reflect the intrinsic nucleophilicity of DNA nucleophiles.34 In studies with powerful alkylating agents, it has been established that N7G is the most potent nucleophile in both dsDNA and ssDNA. O6-G alkylation is found five to tenfold less, while the remaining heteroatoms account for approximately five percent of alkylation products.21 Since unstabilized Cu(I) carbenes should be highly reactive with water, we expected that the selectivity might stem from pre-association of copper with the guanine nucleobase. Guanine is the most electron rich of the nucleobases and readily chelates with divalent metals, including copper(II).35,36 DFT calculations of molecular dipoles in DNA bases indicate that guanine has the largest molecular dipole (6.9 D) and its negative pole is located between O6 and N7. On the other hand, little is known about the interaction of Cu(I) with DNA,37 likely because the Cu(I) oxidation state has limited stability in aqueous buffer at neutral pH (it tends to oxidize or disproportionate)38 and is therefore difficult to study. Nevertheless, based on analogy to Cu(II), which readily binds guanine N7s, we hypothesize that the selectivity derives from carbene formation with an N7 bound copper atom. Crystal structure data for Cu(II)39 and other divalent metals36 show a strong preference for N7G coordination, with a consequent positioning of the metal approximately 3.5 Å away from O6. The bound carbene would be thus poised to form a metal ylide with the O6 position (see lower mechanistic scheme in Fig. 2), and an irreversible protonation would complete the alkylation.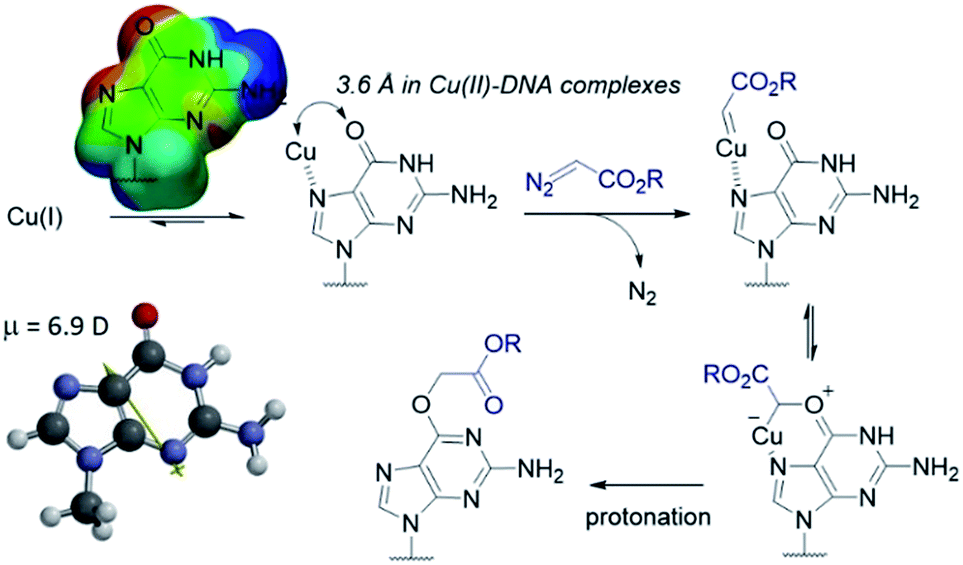 | ||
| Fig. 2 Mechanistic proposal accounting for the high O6-G chemoselectivity. Electrostatic potential map and the molecular dipole moment (6.9 D) were calculated in Spartan′14 with DFT calculations at the EDF2 6-31G* level of theory using 9-methylguanine. | ||
The selectivity that we observe mirrors the preference in sites of Cu-catalysed oxidative (peroxide or superoxide) damage in DNA, which show increased oxidation at and around guanine residues.40 The research on copper-mediated redox damage supports two main pathways. In the first an unbound copper(I) ion releases reactive oxygen species upon oxygen reduction or hydrogen peroxide cleavage, the resulting hydroxyl radical diffuses to the nearest site on DNA and typically leads to DNA fragmentation. This mechanism is unselective and presents a constant danger to DNA and RNA – it is, however, inefficient. In the second mechanism a guanine-bound copper(I) forms a copper(II) oxo complex by reacting with peroxide, leading to oxidative damage of the guanine itself as well as neighbouring bases. These reports on oxidative damage, however, provided no insight on why guanine was the target or how the copper bound. We propose that both the specificity of oxidative damage observed in earlier reports and the alkylation specificity we observe here is a result of the same thing: guanine's superior ability to coordinate copper.
The hypothesis that guanine pre-coordination controls chemoselectivity would predict the following:
(1) Other amide or lactone substrates that lack the strong coordination ability of guanine should be poorly reactive.
(2) Strongly chelating multidentate ligands should be deleterious to reactivity, since the combination of the guanine and the multidentate ligand would not leave space in copper's coordination environment for carbene formation.
Over the next two sections we will examine experimental data for each of these scenarios and demonstrate that the hypothesis holds.
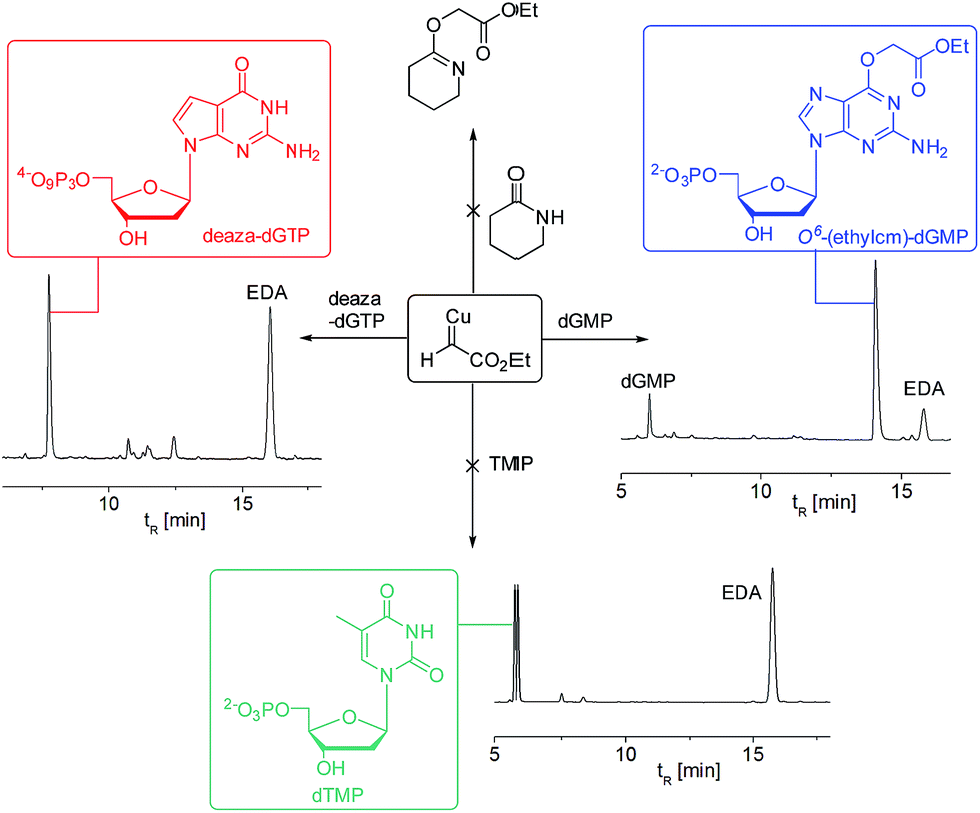 | ||
| Fig. 3 Substrate-based mechanistic probes point to the role of the N7 of guanine in directing the chemoselectivity. Only the dGMP starting material leads to efficient reaction. HPLC traces were recorded after 30 min reaction time. Reaction conditions: 5 mM substrate (dGMP or deaza-dGTP or TMP or α-valerolactone), 1 mM CuSO4, 5 mM ascorbate, 50 mM EDA, 100 mM MES pH 6, dioxane 20% (v/v), H2O, 25 °C. | ||
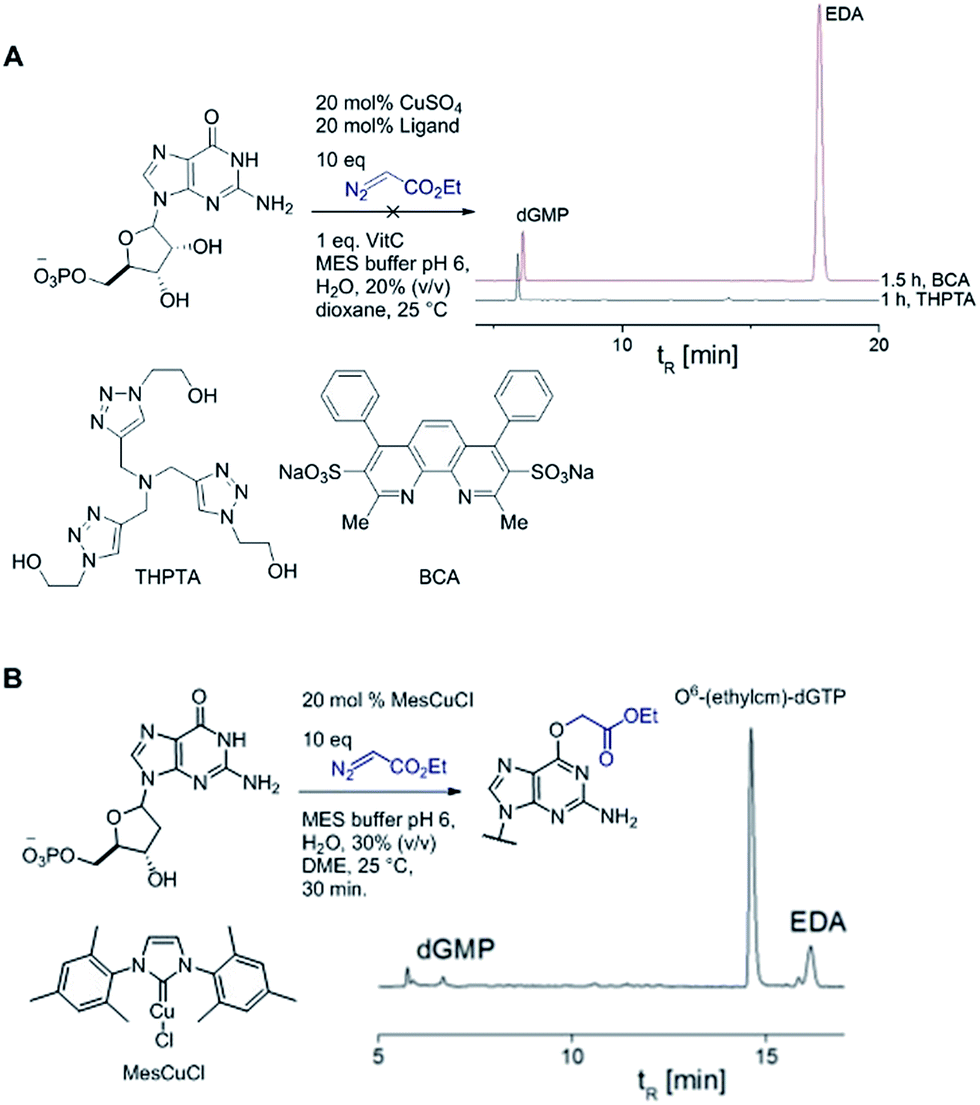 | ||
| Fig. 4 (A) Multidentate ligands like THPTA and bathocuproine disulfonic acid disodium salt (BCA) suppress dGMP alkylation. (B) dGMP alkylation catalysed by MesCuCl is highly efficient. HPLC trace was recorded after 30 minutes reaction time. | ||
A copper catalyst bearing an N-heterocyclic carbene (NHC) ligand behaved differently: mesitylimidazolinium copper chloride (MesCuCl) gave the same alkylation profile as the standard ‘ligandless’ conditions, with O6-G alkylation being the dominant product (see Fig. 4B). It is revealing that the NHC ligand, which is not bidentate and hence would still allow coordination with N7G, is the only type tested that maintained alkylation activity. The NHC ligands have other favourable properties for the alkylation reaction (such as reducing oxidative damage) and we are currently exploring the full potential of NHC ligands in DNA and RNA alkylation.
Biochemical studies with O6-(alkyl)-G adducts
With ready access to O6-G derivatives we next used the products to study two open questions in the biochemistry of O6-G adducts: first, their repair by alkylguanine transferases (AGTs) and second, their incorporation during DNA replication.Human AGT repairs peptide-like carboxamide adducts
AGTs are found in all domains of life and are responsible for repairing O6-G adducts in the genome. Human AGT (hAGT) repairs O6-(alkyl)-G adducts from single stranded and duplex DNAs.44–47 The repair pathway is important since polymerases often mis-incorporate T when bypassing this lesion during replication.48,49 Evolution has found no effective catalytic solution to the direct reversal of O6-G damage; instead AGT acts as a suicide protein. It bears a nucleophilic cysteine that attacks the alkyl group of the adduct and restores the native guanine.44 Previous reports conflict as to whether carboxymethyl damage is repaired by hAGT.50,51 In addition, since diazotized peptides are the likely source of these alkylation adducts,26,50 it is important to understand whether adducts bearing amides are also effectively repaired. Bacteria also produce diazo peptide natural products that may alkylate DNA,52,53 adding additional impetus to understand the repair of amide adducts. Thus, we tested whether a DNA 9-mer modified with a cyclohexyl carboxymethylamide (see Table 2, entry 6), was a substrate for hAGT (see Fig. 5). Although removal of the adduct was slow (reduced rates are expected for ssDNAs),54 analysis of both the deoxyoligonucleotide (see panel B in Fig. 5) and the protein (see panel C in Fig. 5) indicate repair occurred. These results support the most recent claim that carboxymethyl adducts are indeed repaired,51 and it adds the carboxymethyl amide motif to the list of deoxyoligonucleotides repaired by hAGT. | ||
| Fig. 5 (A) De-alkylation of O6-G modified ssDNA 9 mer by hAGT at 37 °C. (B) HPLC analysis of the reaction mixture after 0 (green), 0.5 (purple) and 4 hours (red). (C) High resolution ESI-MS from hAGT after 4 h at 37 °C (the second set of peaks derive from a post-translational modification (likely methylation) of hAGT that we cannot identify, but which has no effect on its function since both masses shift after the dealkylation). | ||
O6-G alkylated triphosphates can be misincorporated during DNA synthesis
To test the impact of O6-alkylation on nucleotide incorporation during DNA synthesis, we characterized O6-(alkyl)-G triphosphates as polymerase substrates. Thus, we synthesized the modified deoxynucleotidetriphosphates O6-(ethyl carboxymethyl)-dGTP and (O6-(ethylcm)-dGTP) and O6-(carboxymethyl)-dGTP (O6-(cm)-dGTP) with our Cu(I) carbene methodology. Selective carboxymethylation of commercial dGTP at the O6 position with EDA as carbene precursor directly delivers O6-(ethylcm)-dGTP and a subsequent ester hydrolysis yields O6-(cm)-dGTP; hence both molecules are obtained in a total of two chemical operations (see Fig. 6A). The direct modification approach offers unprecedented efficiency in the formation of O6-G adducts (in this case from a commercial triphosphate without any protecting group or phosphorylation steps). Previous syntheses of O6-G modified nucleosides and triphosphates involved four31 to seven55–58 steps including triphosphate introduction using strong electrophilic phosphorous reagents.59 | ||
| Fig. 6 (A) Synthesis of O6-(alkyl)-dGTPs with a Cu(I) carbene derived from EDA. (B) Single nucleotide primer extension with O6-(alkyl)-dGTPs opposite DNA templates G, A, T, and C catalyzed by KTqM747K. Reactions were performed at 55 °C for 10 min and contained 50 μM dNTPs. (C) PAGE analysis of the primer extension experiment. M, blank corresponding to 23 nt primer. | ||
To investigate the incorporation of O6-(cm)-dGTP and O6-(ethylcm)-dGTP during DNA synthesis, we performed single nucleotide primer extension experiments with KTqM747K, a mutant of KlenTaq polymerase that bypasses several DNA modifications.60–62 Unmodified 28-mer templates (5′-ATT ATG CTG AGT GAT ATC CCT CTX CTC A) in which X = G, A, T or C were annealed to a 5′-end [γ-32P] radiolabeled 23-mer primer (5′-AGA GGG ATA TCA CTC AGC ATA AT) and then subjected to primer extension conditions (see Fig. 6B). Reaction products were separated by denaturing polyacrylamide gel electrophoresis and visualized with phosphorimaging. Nucleotides O6-(cm)-dGTP as well as O6-(ethylcm)-dGTP were incorporated opposite both C and T, but C was especially well tolerated. Furthermore, whereas with T incorporation the polymerase stalls after the first extension, the O6-(alkyl)-G:C mismatch seems to pose no problem for the polymerase's procession since the second extension also occurs quickly (see the C lane in Fig. 6C). Consistent with steric matching as a key component in polymerase fidelity,63–66 purine mismatches were poorly tolerated (see G and A bands in the gel in Fig. 6C). These experiments serve as the proof-of-concept that O6-(cm)-G damaged nucleotides can be incorporated during DNA replication. In light of these results a full study of how different polymerases (particularly human) deal with O6-(cm)-dGTP in the dNTP pool during replication is warranted.
Conclusions
We have discovered a new way to create O6-G adducts post-synthetically in nucleic acids of varying size, from nucleotide mono- or triphosphates to long oligonucleotides. The ability to obtain high yields of precisely defined O6-G adducts eliminates a major roadblock in studying their biochemistry, a point we have illustrated through the first synthesis of O6-(cm)-dGTP and the first demonstration of its incorporation during DNA replication.Copper-catalysed O6-G alkylation is a dump-and-stir procedure that employs readily available materials. α-Diazo ester substrates are stable enough that several are sold commercially and a simple33 two-step (one-pot) protocol for α-diazo ester synthesis makes nearly any diazo compound in this class synthetically accessible. Research on O6-G adducts in biology67 and biotechnology68 demands ready synthetic access to these adducts and the method we have described is the simplest to date.
Nucleic acids are found in diverse contexts and can fold in myriad ways, complicating attempts at broad generalizations of reactivity. Nevertheless, the weight of evidence suggests N7G is the most powerful nucleophile34 and ligand36 in DNA and RNA. It is a primary target of organic electrophiles,34 as well as organometallic anticancer drugs.69 To the list of N7G's functions we add the role of directing group in copper catalysed reactions. We have focussed on alkylation reactions, but the same effect is likely responsible for the well-known propensity of Fenton-type oxidative damage to be localized around guanines.40
Acknowledgements
D.G.G and S.N.G acknowledges the Swiss National Science Foundation (SNF) for financial support (grant number 200021_149805). L.A.W and S.J.S acknowledge the European Research Council (260341), the SNF (156280), and the ETH research commission (ETH-43 14-1) for financial support. Prof. Nathan Leudtke and Mr Aaron Johnson are gratefully acknowledged for helpful discussions.Notes and references
- M. A. M. M. P. Doyle and T. Ye, Modern Catalytic Methods for Organic Synthesis with Diazo Compounds, Wiley, New York, 1998 Search PubMed.
- A. Ford, H. Miel, A. Ring, C. N. Slattery, A. R. Maguire and M. A. McKervey, Chem. Rev., 2015, 115, 9981–10080 CrossRef CAS PubMed.
- D. Gillingham and N. Fei, Chem. Soc. Rev., 2013, 42, 4918–4931 RSC.
- I. Nicolas, P. Le Maux and G. Simonneaux, Coord. Chem. Rev., 2008, 252, 727–735 CrossRef CAS.
- J. M. Antos and M. B. Francis, J. Am. Chem. Soc., 2004, 126, 10256–10257 CrossRef CAS PubMed.
- B. V. Popp and Z. T. Ball, Chem. Sci., 2011, 2, 690–695 RSC.
- F. Vohidov, J. M. Coughlin and Z. T. Ball, Angew. Chem., Int. Ed. Engl., 2015, 54, 4587–4591 CrossRef CAS PubMed.
- H. M. L. Davies and D. Morton, Chem. Soc. Rev., 2011, 40, 1857–1869 RSC.
- K. Tishinov, K. Schmidt, D. Haussinger and D. G. Gillingham, Angew. Chem., Int. Ed., 2012, 51, 12000–12004 CrossRef CAS PubMed.
- K. Tishinov, N. Fei and D. Gillingham, Chem. Sci., 2013, 4, 4401–4406 RSC.
- N. Fei, D. Haussinger, S. Blumli, B.-J. Laventie, L. D. Bizzini, K. Zimmermann, U. Jenal and D. Gillingham, Chem. Commun., 2014, 50, 8499–8502 RSC.
- G. Rousseau and B. Breit, Angew. Chem., Int. Ed., 2011, 50, 2450–2494 CrossRef CAS PubMed.
- E. I. Solomon, D. E. Heppner, E. M. Johnston, J. W. Ginsbach, J. Cirera, M. Qayyum, M. T. Kieber-Emmons, C. H. Kjaergaard, R. G. Hadt and L. Tian, Chem. Rev., 2014, 114, 3659–3853 CrossRef CAS PubMed.
- B. Schönecker, T. Zheldakova, Y. Liu, M. Kötteritzsch, W. Günther and H. Görls, Angew. Chem., Int. Ed., 2003, 42, 3240–3244 CrossRef PubMed.
- Y. Y. See, A. T. Herrmann, Y. Aihara and P. S. Baran, J. Am. Chem. Soc., 2015, 137, 13776–13779 CrossRef CAS PubMed.
- J. Ohata, M. B. Minus, M. E. Abernathy and Z. T. Ball, J. Am. Chem. Soc., 2016, 138, 7472–7475 CrossRef CAS PubMed.
- K. A. Magnus, B. Hazes, H. Ton-That, C. Bonaventura, J. Bonaventura and W. G. Hol, Proteins, 1994, 19, 302–309 CrossRef CAS PubMed.
- R. Balasubramanian, S. M. Smith, S. Rawat, L. A. Yatsunyk, T. L. Stemmler and A. C. Rosenzweig, Nature, 2010, 465, 115–119 CrossRef CAS PubMed.
- F. Drabløs, E. Feyzi, P. A. Aas, C. B. Vaagbø, B. Kavli, M. S. Bratlie, J. Peña-Diaz, M. Otterlei, G. Slupphaug and H. E. Krokan, DNA Repair, 2004, 3, 1389–1407 CrossRef PubMed.
- Nobel Media AB 2014, http://www.nobelprize.org/nobel_prizes/chemistry/laureates/2015/press.html, 2015.
- We use the convention that exocyclic heteroatoms are labelled with a superscript and endocyclic ones with normal font: E. C. Friedberg, G. C. Walker, W. Siede and R. D. Wood, DNA repair and mutagenesis, American Society for Microbiology Press, 2005 Search PubMed.
- C. Coulondre and J. H. Miller, J. Mol. Biol., 1977, 117, 577–606 CrossRef CAS PubMed.
- M. Sekiguchi and K. Sakumi, Jpn. J. Hum. Genet., 1997, 42, 389–399 CrossRef CAS PubMed.
- G. P. Margison, M. F. Santibáñez Koref and A. C. Povey, Mutagenesis, 2002, 17, 483–487 CrossRef CAS PubMed.
- F. Zhang, M. Tsunoda, K. Suzuki, Y. Kikuchi, O. Wilkinson, C. L. Millington, G. P. Margison, D. M. Williams, C. E. Morishita and A. Takenaka, Nucleic Acids Res., 2013, 41, 5524–5532 CrossRef CAS PubMed.
- B. C. Cupid, Z. Zeng, R. Singh and D. E. G. Shuker, Chem. Res. Toxicol., 2004, 17, 294–300 CrossRef CAS PubMed.
- M. H. Lewin, N. Bailey, T. Bandaletova, R. Bowman, A. J. Cross, J. Pollock, D. E. Shuker and S. A. Bingham, Cancer Res., 2006, 66, 1859–1865 CrossRef CAS PubMed.
- H. zur Hausen, Int. J. Cancer, 2012, 130, 2475–2483 CrossRef CAS PubMed.
- V. Bouvard, D. Loomis, K. Z. Guyton, Y. Grosse, F. E. Ghissassi, L. Benbrahim-Tallaa, N. Guha, H. Mattock and K. Straif, Lancet Oncol., 2015, 16, 1599–1600 CrossRef PubMed.
- Y.-Z. Xu, Tetrahedron, 2000, 56, 6075–6081 CrossRef CAS.
- J. von Watzdorf, K. Leitner and A. Marx, Angew. Chem., Int. Ed., 2016, 55, 3229–3232 CrossRef CAS PubMed.
- R. G. Salomon and J. K. Kochi, J. Am. Chem. Soc., 1973, 95, 3300–3310 CrossRef CAS.
- T. Toma, J. Shimokawa and T. Fukuyama, Org. Lett., 2007, 9, 3195–3197 CrossRef CAS PubMed.
- D. Gillingham, S. Geigle and O. A. von Lilienfeld, Chem. Soc. Rev., 2016, 45, 2637–2655 RSC.
- S. L. Labiuk, L. T. J. Delbaere and J. S. Lee, J. Biol. Inorg. Chem., 2003, 8, 715–720 CrossRef CAS PubMed.
- Y.-G. Gao, M. Sriram and A. H.-J. Wang, Nucleic Acids Res., 1993, 21, 4093–4101 CrossRef CAS PubMed.
- W. A. Prütz, J. Butler and E. J. Land, Int. J. Radiat. Biol., 1990, 58, 215–234 CrossRef.
- D. K. Johnson, M. J. Stevenson, Z. A. Almadidy, S. E. Jenkins, D. E. Wilcox and N. E. Grossoehme, Dalton Trans., 2015, 44, 16494–16505 RSC.
- B. H. Geierstanger, T. F. Kagawa, S. L. Chen, G. J. Quigley and P. S. Ho, J. Biol. Chem., 1991, 266, 20185–20191 CAS.
- S. Oikawa and S. Kawanishi, Biochemistry, 1996, 35, 4584–4590 CrossRef CAS PubMed.
- V. Hong, S. I. Presolski, C. Ma and M. G. Finn, Angew. Chem., Int. Ed., 2009, 48, 9879–9883 CrossRef CAS PubMed.
- V. Hong, N. F. Steinmetz, M. Manchester and M. G. Finn, Bioconjugate Chem., 2010, 21, 1912–1916 CrossRef CAS PubMed.
- S. I. Presolski, V. Hong, S.-H. Cho and M. G. Finn, J. Am. Chem. Soc., 2010, 132, 14570–14576 CrossRef CAS PubMed.
- A. E. Pegg, Mutat. Res., Rev. Mutat. Res., 2000, 462, 83–100 CrossRef CAS.
- K. Bender, M. Federwisch, U. Loggen, P. Nehls and M. F. Rajewsky, Nucleic Acids Res., 1996, 24, 2087–2094 CrossRef CAS PubMed.
- L.-K. Liem, C.-W. Wong, A. Lim and B. F. L. Li, J. Mol. Biol., 1993, 231, 950–959 CrossRef CAS PubMed.
- J. J. Rasimas, S. R. Kar, A. E. Pegg and M. G. Fried, J. Biol. Chem., 2007, 282, 3357–3366 CrossRef CAS PubMed.
- E. M. Duguid, P. A. Rice and C. He, J. Mol. Biol., 2005, 350, 657–666 CrossRef CAS PubMed.
- Y. Mishina, E. Duguid and C. He, Chem. Rev., 2006, 106, 215–232 CrossRef CAS PubMed.
- D. E. G. Shuker and G. P. Margison, Cancer Res., 1997, 57, 366–369 CAS.
- P. Senthong, C. L. Millington, O. J. Wilkinson, A. S. Marriott, A. J. Watson, O. Reamtong, C. E. Eyers, D. M. Williams, G. P. Margison and A. C. Povey, Nucleic Acids Res., 2013, 41, 3047–3055 CrossRef CAS PubMed.
- C. C. Nawrat and C. J. Moody, Nat. Prod. Rep., 2011, 28, 1426–1444 RSC.
- Y. Katoh, M. Maekawa and Y. Sano, Mutat. Res., Rev. Genet. Toxicol., 1995, 342, 37–41 CrossRef CAS.
- D. Bhattacharyya, R. S. Foote, A. M. Boulden and S. Mitra, Eur. J. Biochem., 1990, 193, 337–343 CrossRef CAS PubMed.
- S. A. Moore and D. E. G. Shuker, J. Labelled Compd. Radiopharm., 2011, 54, 855–858 CrossRef CAS.
- K. Abdu, M. K. Aiertza, O. J. Wilkinson, J. A. Grasby, P. Senthong, A. C. Povey, G. P. Margison and D. M. Williams, Chem. Commun., 2012, 48, 11214–11216 RSC.
- C. L. Millington, A. J. Watson, A. S. Marriott, G. P. Margison, A. C. Povey and D. M. Williams, Nucleosides, Nucleotides Nucleic Acids, 2012, 31, 328–338 CAS.
- T. Shibata, N. Glynn, T. B. H. McMurry, R. S. McElhinney, G. P. Margison and D. M. Williams, Nucleic Acids Res., 2006, 34, 1884–1891 CrossRef CAS PubMed.
- M. Hollenstein, Molecules, 2012, 17, 13569–13591 CrossRef CAS PubMed.
- C. Gloeckner, K. B. M. Sauter and A. Marx, Angew. Chem., Int. Ed., 2007, 46, 3115–3117 CrossRef CAS PubMed.
- S. Obeid, A. Schnur, C. Gloeckner, N. Blatter, W. Welte, K. Diederichs and A. Marx, ChemBioChem, 2011, 12, 1574–1580 CrossRef CAS PubMed.
- L. A. Wyss, A. Nilforoushan, F. Eichenseher, U. Suter, N. Blatter, A. Marx and S. J. Sturla, J. Am. Chem. Soc., 2015, 137, 30–33 CrossRef CAS PubMed.
- E. T. Kool, Acc. Chem. Res., 2002, 35, 936–943 CrossRef CAS PubMed.
- H. O. Sintim and E. T. Kool, Angew. Chem., Int. Ed., 2006, 45, 1974–1979 CrossRef CAS PubMed.
- S. Ulrich and E. T. Kool, Biochemistry, 2011, 50, 10343–10349 CrossRef CAS PubMed.
- I. Hirao, M. Kimoto and R. Yamashige, Acc. Chem. Res., 2012, 45, 2055–2065 CrossRef CAS PubMed.
- A. Pegg, Chem. Res. Toxicol., 2011, 24, 618–639 CrossRef CAS PubMed.
- A. Gautier, A. Juillerat, C. Heinis, I. R. Corrêa Jr, M. Kindermann, F. Beaufils and K. Johnsson, Chem. Biol., 2008, 15, 128–136 CrossRef CAS PubMed.
- P. M. Takahara, A. C. Rosenzweig, C. A. Frederick and S. J. Lippard, Nature, 1995, 377, 649–652 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: For complete experimental details and characterization data. See DOI: 10.1039/c6sc03502g |
| This journal is © The Royal Society of Chemistry 2017 |