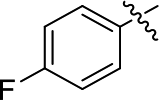
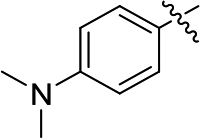
DOI:
10.1039/C7RA01795B
(Paper)
RSC Adv., 2017,
7, 18120-18131
Regioselective synthesis of dipyrrolopyrazine (DPP) derivatives via metal free and metal catalyzed amination and investigation of their optical and thermal properties†
Received
13th February 2017
, Accepted 6th March 2017
First published on 24th March 2017
Abstract
Pyrazine is an important molecular scaffold employed in organic optoelectronic materials. Here we report efficient methods for the synthesis of dipyrrolopyrazine, and pyrrolothieno-pyrazine derivatives that involve regio-selective amination reactions of dihalo-pyrrolopyrazines. The developed protocol readily affords either 2-amino- or 3-amino-pyrrolopyrazines from the corresponding 2-bromo-3-chloro-5H-pyrrolo[2,3-b]pyrazines. When the amination reactions are carried under metal free under microwave irradiation, 3-amino-pyrrolopyrazines are obtained exclusively. In contrast, Buchwald cross coupling of the 2-bromo-3-chloro-5H-pyrrolo[2,3-b]pyrazines affords only 2-amino-pyrrolopyrazine. The pyrrolopyrazine scaffolds were converted to the respective 1,7- and 1,5-dihydrodipyrrolo[2,3-b]pyrazines derivatives using Sonogashira reactions. A comprehensive study of the optical properties, thermal properties, and molecular packing of the synthesized compounds was carried out. The results indicate that the 1,7-derivatives may be promising organic materials for optoelectronic applications.
1. Introduction
Fused heterocyclic compounds are important and valuable substances, especially in the field of optoelectronics.1 In particular, multicyclic pyrazine fused aromatic compounds are attracting great interest as promising electronic materials that are used to construct field effect transistors,2,3 light-emitting diodes,4,5 and photovoltaic cells.6,7 During the past decade, substances containing the interesting pyrrolo[2,3-b]pyrazine molecular scaffold have been prepared, albeit in poor yields. The first efficient synthesis of pyrrolo[2,3-b]pyrazine starting from a halo-pyrazine derivative was described by Corey and co-workers. The key step route involved palladium catalysed Sonogashira cross-coupling followed by base induce C–N cyclization to produce pyrrolo[2,3-b]pyrazine.8,9 Interestingly, Simpson et al. described a one-pot protocol for the preparation of pyrrolo[2,3-b]pyrazine starting with a halo-pyrazine derivative.10,11 More recently, a strategy relying on a crucial C–N nucleophilic aromatic substitution reaction under microwave (MW) irradiation conditions was developed to prepare this substance.12–15 It is well known that (metal catalysed) reactions, like the one employed in this sequence, carried out under MW conditions proceed with enhanced regio- and stereo-selectivities.16
Benzodithiophene (BDT) is another intriguing heterocyclic scaffold that has been used as the foundation for highly efficient optoelectronic devices.17–22 Although it has been shown that materials containing 2,6-diphenyl BDT analogues are good organic field effect transistors, these substances in polymers undergo rapid degradation caused by to photo-oxidation in air.12 Substitution of the benzene ring in BDT by a pyrazine moiety suppresses the photo-oxidation process. As a result, pyrazine fused dithiophenes serve as the basis for the development of new n-type transistors. Like BDT, dithienopyrazine (DTP) is planar and it undergoes molecular π–π stacking in the solid state.23,24 In an effort to prepare new pyrazine fused polycyclic aromatic compounds and explore their potential use as optoelectronics materials, we have conducted an investigation exploring the synthesis of substituted dipyrrolopyrazine derivatives (DPP). At the outset, we hypothesized that the DPP derivatives would be less prone to photo-oxidation than their BDT analogues. Furthermore, it was expected that DPPs would be planar molecule in which the pyrrole ring participates in electron delocalization leading to potentially beneficial electronic properties.
In the studies described below, we developed efficient routes for the synthesis of new 2- and 3-amine functionalised pyrrolopyrazine which serve as advanced precursors to variety of new DPP derivatives. The routes for this purpose utilize key, regioselective 2- and 3-C–N functionalization reactions of 2-bromo-3-chloro-5H-pyrrolo[2,3-b]pyrazines 2a,b using MW irradiation (metal free conditions) and metal catalysed conditions, respectively. The products of these processes were employed to prepare 1,7- and 1,5-dihydrodipyrrolo[3,2-b:3′,2′-e]pyrazine derivatives (DPP), whose optical, thermal and morphological properties were evaluated. Finally, to assess the applicability of the DPP derivatives to optoelectronics, a systematic investigation of the photo physical and thermal properties of these substances were performed.
2. Results and discussion
2.1 Synthesis and characterization
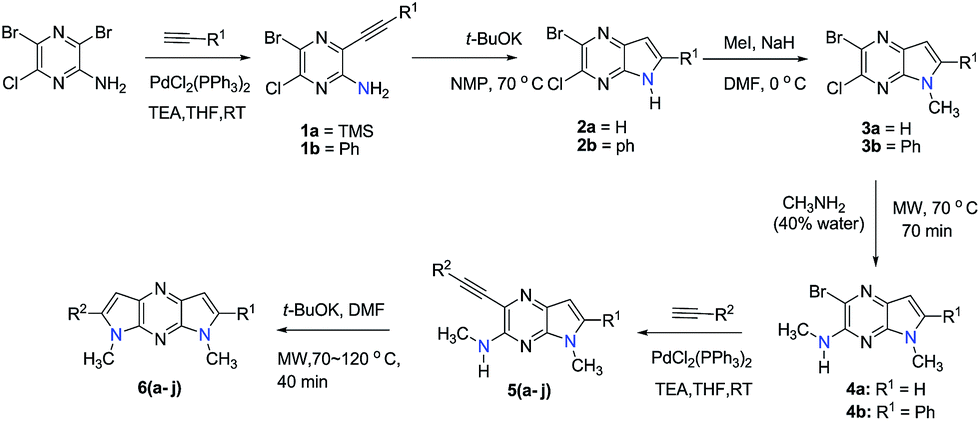
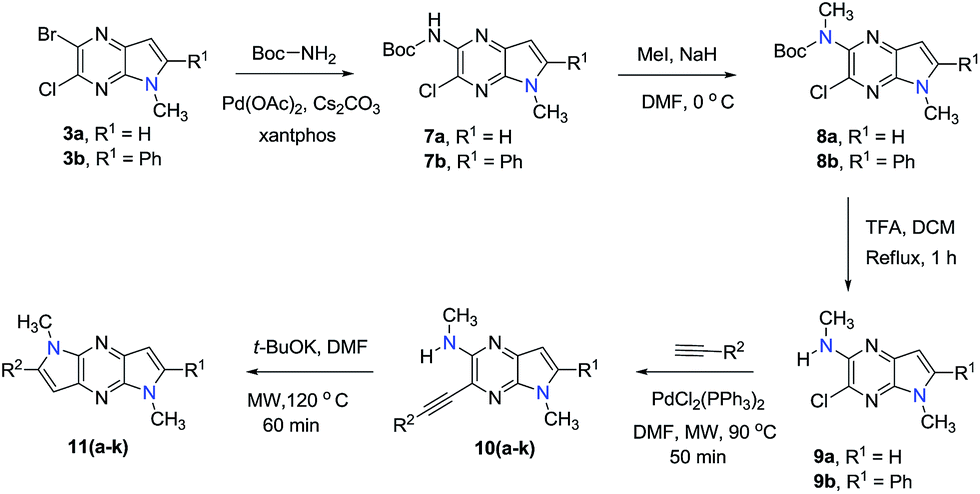
The 2-bromo-3-chloro-5H-pyrrolo[2,3-b]pyrazines 2a,b, key intermediates in the routes for synthesis of the 1,7- and 1,5-dihydrodipyrrolo[3,2-b:3′,2′-e]pyrazines, were prepared in two steps starting with commercially available 3,5-dibromo-6-chloropyrazin-2-amine. The sequences commenced with Sonogashira cross-coupling of a dibromo-chloropyrazine using appropriate alkynes (Scheme 1). t-BuOK promoted intramolecular cyclization then produces 2a,b in good yields. In addition to the 5-NH substrates 3c,d, pyrrolopyrazines containing the electron donating 5-NMe (3a,b) and electron withdrawing 5-NTs (3e) substituents were prepared from 2a,b to explore the challenging C–N coupling process.
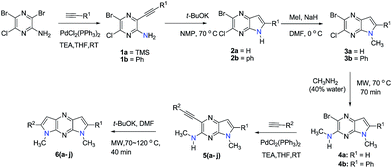 |
| | Scheme 1 Synthesis of 1,7-dihydrodipyrrolo[2,3;3′2′-e]pyrazine derivatives. | |
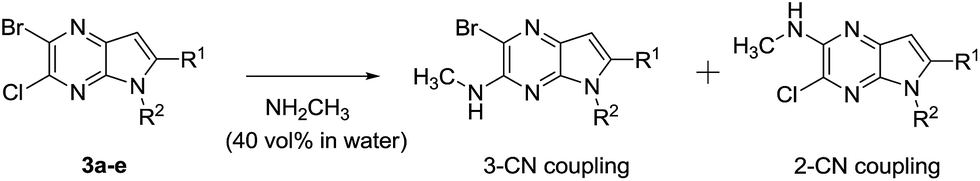
Under normal conditions (40 vol% in water, 60 °C), the pyrrolopyrazine derivatives 3a–e did not react with methylamine to produce the corresponding amination products. Exploratory studies showed that the use of MW conditions with THF as the solvent facilitate the C–N bond forming substitution reactions of these pyrrolopyrazine derivatives and methylamine, although little selectivity was observed to occur between C–N bond formation at the 2-bromo and 3-chlorosubstituted centers. Specifically, reactions of the free amines 3c,d and N-tosyl derivative 3e react under these conditions form mixtures of 2- and 3-N-methylamine coupling products (Table 1). Interestingly, under solvent free conditions, the N-Me derivatives 3a,b react to produce exclusively the 3-N-methylamine products 4a,b in 69% and 39% yield, respectively. The 5-N-methylamine derivatives 4a,b, formed in the manner described above were then used to prepare the 1,7-dihydrodipyrrolo[2,3-b;3′2′-e]pyrazines 6a–j. To this end, Pd(PPh3)2Cl2 catalysed Sonogashira reaction of 4a,b with a series of terminal alkynes were carried out to generate the respective 2-ethynyl-N,5-dimethyl-5H-pyrrolo[2,3-b]pyrazin-3-amines 5a–j (59–74%, Table 2). Finally, t-BuOK mediated intramolecular cyclization of 5a–j form the corresponding target 1,7-dihydrodipyrrolo[2,3-b:3′,2′-e]pyrazines 6a–j in good to excellent yields (63–96%).
Table 1 The influence of 5-N-substituents on C–N coupling reactions of pyrrolopyrazinesa
Table 2 Substrate scope for preparation 1,7-dihydrodipyrrolo[3,2-b:3′,2′-e]pyrazine derivatives
| No |
R1 |
R2 |
Yield (%) |
No |
R1 |
R2 |
Yield (%) |
| Trace amounts of cyclization product was observed. The TMS group was displaced during the reaction due to high basicity of the t-BuOK. |
| 5a |
 |
 |
66 |
6a |
 |
 |
78b |
| 5b |
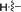 |
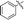 |
68 |
6b |
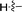 |
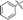 |
96 |
| 5c |
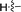 |
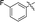 |
65 |
6c |
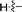 |
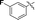 |
90 |
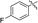
| 5d |
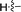 |
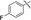 |
69 |
6d |
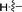 |
 |
88 |
| 5e |
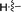 |
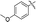 |
70 |
6e |
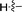 |
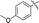 |
92 |
| 5f |
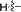 |
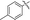 |
71 |
6f |
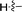 |
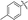 |
89 |
| 5g |
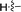 |
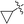 |
74 |
6g |
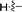 |
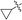 |
91 |
| 5h |
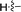 |
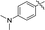 |
59a |
6h |
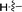 |
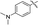 |
76 |
| 5i |
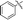 |
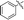 |
67 |
6i |
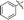 |
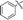 |
63 |
| 5j |
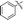 |
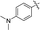 |
64a |
6j |
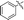 |
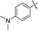 |
74 |
Our attention was next focussed at the synthesis of the 1,5-dihydrodipyrrolo[3,2-b:3′,2′-e]pyrazine derivatives. For this purpose, 3a,b were subjected to reactions using the standard Buchwald coupling conditions. Pd(OAc)2 with xantphos as the ligand was selected as the catalyst together with t-butyl carbamate as the coupling partner in the process.25 As expected, these reactions takes place exclusively at the 2-bromo positions in 3a,b to give the respective 2-N-Boc protected derivatives 7a,b in good yields. Subsequent N-methylation of 7a,b followed by Boc removal using TFA produces the corresponding 2-N-methylamine derivatives 9a,b. Sonogashira cross coupling reactions of 9a,b with a variety of different terminal alkynes were investigated. In accordance with the earlier observations, under non-MW conditions the starting materials remained unreactive. Fortunately, under MW (Scheme 2), the desired products 10a–k were detected and could be isolated in average to good yields were detected and could be isolated in average to good yields (41–82%) from 9a,b. Attempts at preparation of the target 1,5-dihydrodipyrrolo[3,2-b:3′,2′-e]pyrazine derivatives from 10a–k by using intramolecular cyclization under non-MW conditions proved to be futile. However, under optimized condition developed above (MW irradiation for 50 min at 120 °C), the corresponding 1,5-DPP 11a–k are obtained in excellent yields (Table 3).
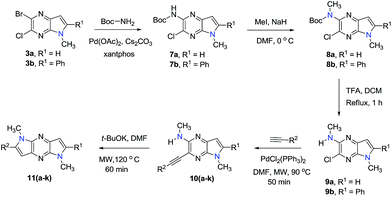 |
| | Scheme 2 Synthesis of 1,5-dihydrodipyrrolo[2,3-b;3′2′-e]pyrazine derivative. | |
Table 3 Substrate scope for preparation of 1,5-dihydrodipyrrolo[3,2-b:3′,2′-e]pyrazine derivatives
| No |
R1 |
R2 |
Yield (%) |
No |
R1 |
R2 |
Yield (%) |
| Trace amount of cyclization product was also observed. TMS group was displaced during reaction due to high basicity of the reagent. |
| 10a |
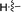 |
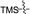 |
53a |
11a |
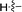 |
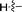 |
87b |
| 10b |
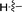 |
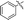 |
62a |
11b |
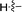 |
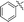 |
91 |
| 10c |
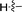 |
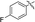 |
41 |
11c |
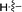 |
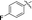 |
84 |
| 10d |
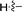 |
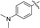 |
52a |
11d |
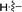 |
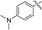 |
78 |
| 10e |
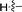 |
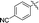 |
61a |
11e |
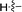 |
 |
82 |
| 10f |
 |
 |
68 |
11f |
 |
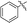 |
92 |
| 10g |
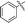 |
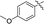 |
74 |
11g |
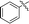 |
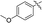 |
90 |
| 10h |
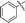 |
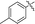 |
78 |
11h |
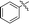 |
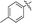 |
90 |
| 10i |
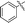 |
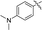 |
66 |
11i |
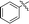 |
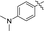 |
83 |
| 10j |
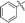 |
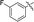 |
69 |
11j |
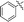 |
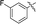 |
85 |
| 10k |
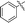 |
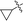 |
82 |
11k |
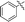 |
 |
91 |
The structures of the 1,7- and 1,5-dihydrodipyrrolo[3,2-b:3′,2′-e]pyrazines, synthesized using the sequences described above, were confirmed by using NMR spectroscopy, LCMS and HRMS analysis (see ESI†). It is noteworthy that the NMR spectra of 6a–j and 11a–f exhibit similar features.
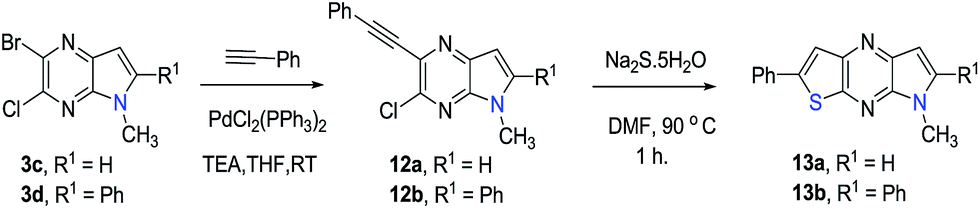
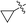
Finally, the pyrrolo[3,2-e]thieno[2,3-b]pyrazines 13a,b were prepared by using the methodology developed in the effort described above. Accordingly, palladium catalysed reactions of 3a,b and phenyl acetylene followed by intramolecular cyclization26 gives 13a,b in moderate yields (Scheme 3).
 |
| | Scheme 3 Annulation sequence for synthesis of pyrrolo[3,2-e]thieno[2,3-b]pyrazine. | |
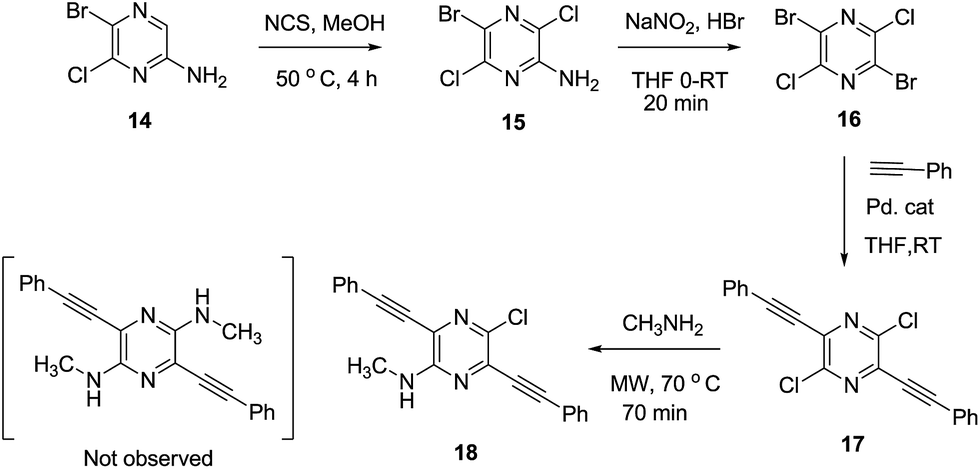
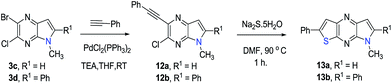
In an attempt to develop a more efficient routes for synthesis of the highly interesting symmetric 1,5-dipyrrolopyrazines, the double cyclization strategy was depicted in Scheme 4 was explored. For this purpose, symmetric 2,5-dibromo-3,6-dichloro pyrazine (16) was prepared using literature procedures and subjected to a double Sonogashira coupling process. The bis-acetylene derivative 17 was produced in only low yield along with unreacted starting material in this reaction. Next, displacement of both chlorides in 17 with methyl amine groups was examined. However, only the mono-aminated product 18 is produced. Therefore this route was abandoned in favour of the aforementioned protocol.
 |
| | Scheme 4 Double cyclization approach to prepare 1,5-dihydrodipyrrolo[3,2-b:3′,2′-e]pyrazine. | |
The solubility and stabilities of the dipyrrolopyrazine derivatives in diverse organic solvents is an important criteria for their applications in optoelectronic devices. Therefore, the solubilities of these crystalline substances were assessed. All of the tested dipyrrolopyrazine proved to be highly soluble in CH2Cl2, MeOH, THF and CHCl3. Additionally, all of the crystalline in nature. Pyrrolopyrazines have long shelf lives, ranging up to one year at room temperature. Next, a comparative study of the optical and thermal properties of the three different pyrazine cores was undertaken.
2.2 Crystallographic analysis
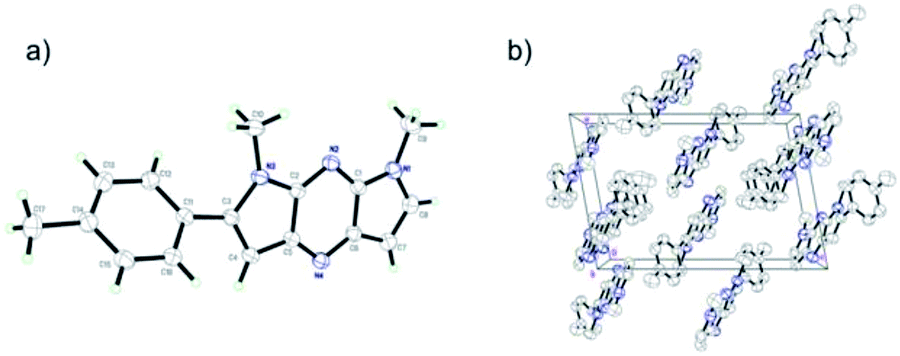
A single crystal of pyrrolopyrazine 6f was grown by slow evaporation of a mixture of ether and dichloromethane. X-ray crystallographic analysis revealed that 6f forms monoclinic crystal system with space group P2(1)/n and is planar with the exception of slight twist of the phenyl substituent. Furthermore, possibly as a consequence of its planar backbone, pyrrolopyrazine derivative 6f adopt a near perfect orthogonal orientation in the crystalline state27 (Fig. 1).
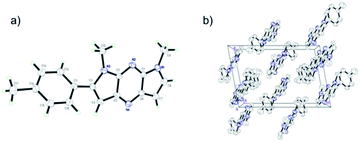 |
| | Fig. 1 (a) Crystal structure of 6f. (b) Packing diagram of 6f viewed along b axis in a bulk single crystal. | |
3. Photophysical properties
3.1 Thermal properties
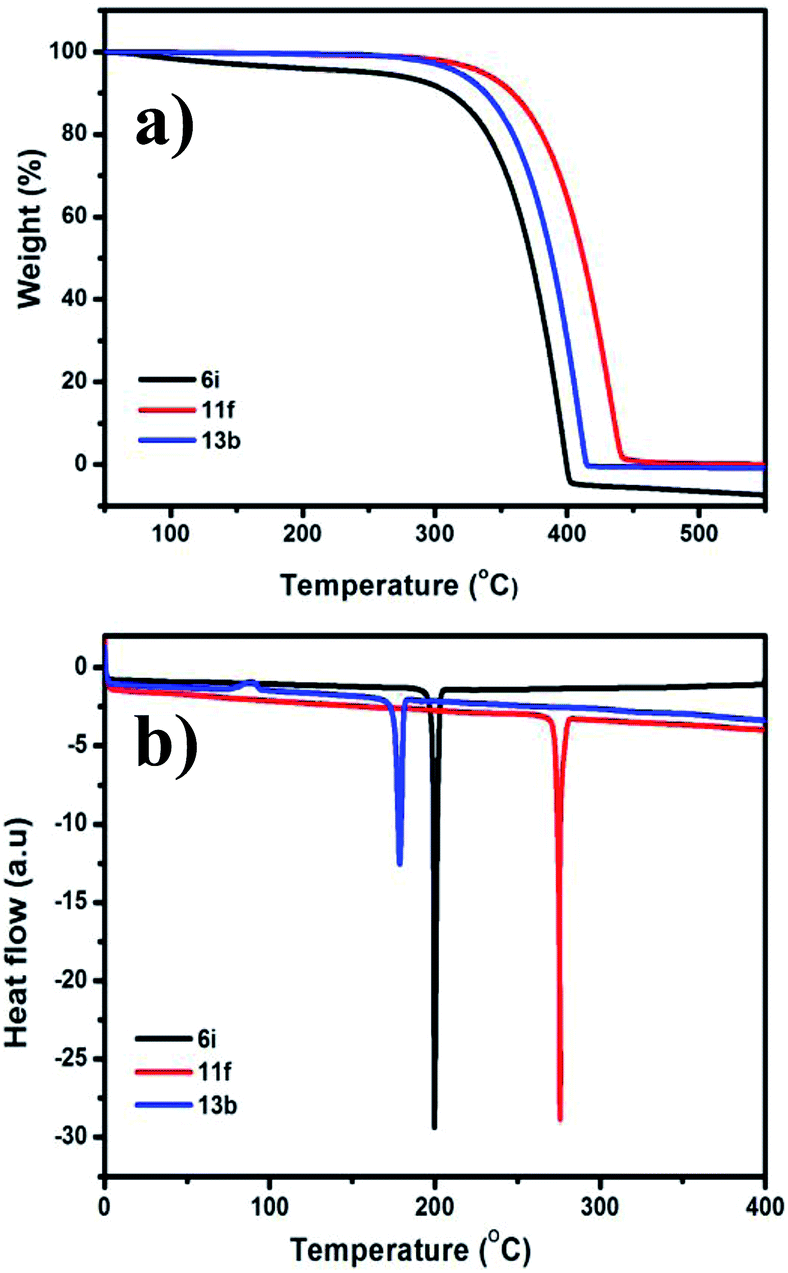
The thermal properties of one member of each of the three types of pyrazine derivative prepared above (6i, 11f, and 13b) were subjected to thermogravimetric analysis and differential scanning calorimetry (Fig. 2). The phase transitions of the selected compounds were analysed using DSC under a nitrogen atmosphere with a heating rate of 10 °C min−1. The results of both the TGA and DSC analyses reveal that all of these substances possess high thermal stabilities. A sharp endothermic melting peak was observed in each case, which confirms the highly crystalline nature of the compounds (Table 5). Furthermore, the high Td and Tm of 6i, 11f, and 13b indicate that they have remarkable thermal stabilities.
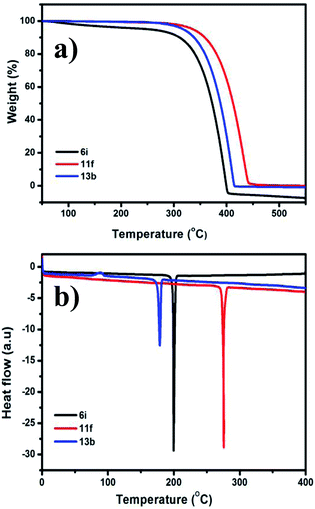 |
| | Fig. 2 (a) TGA and (b) DSC thermograms of the 6i, 11f, and 13b recorded with a heating rate of 10 °C min−1 under nitrogen atmosphere. | |
3.2 Optical properties
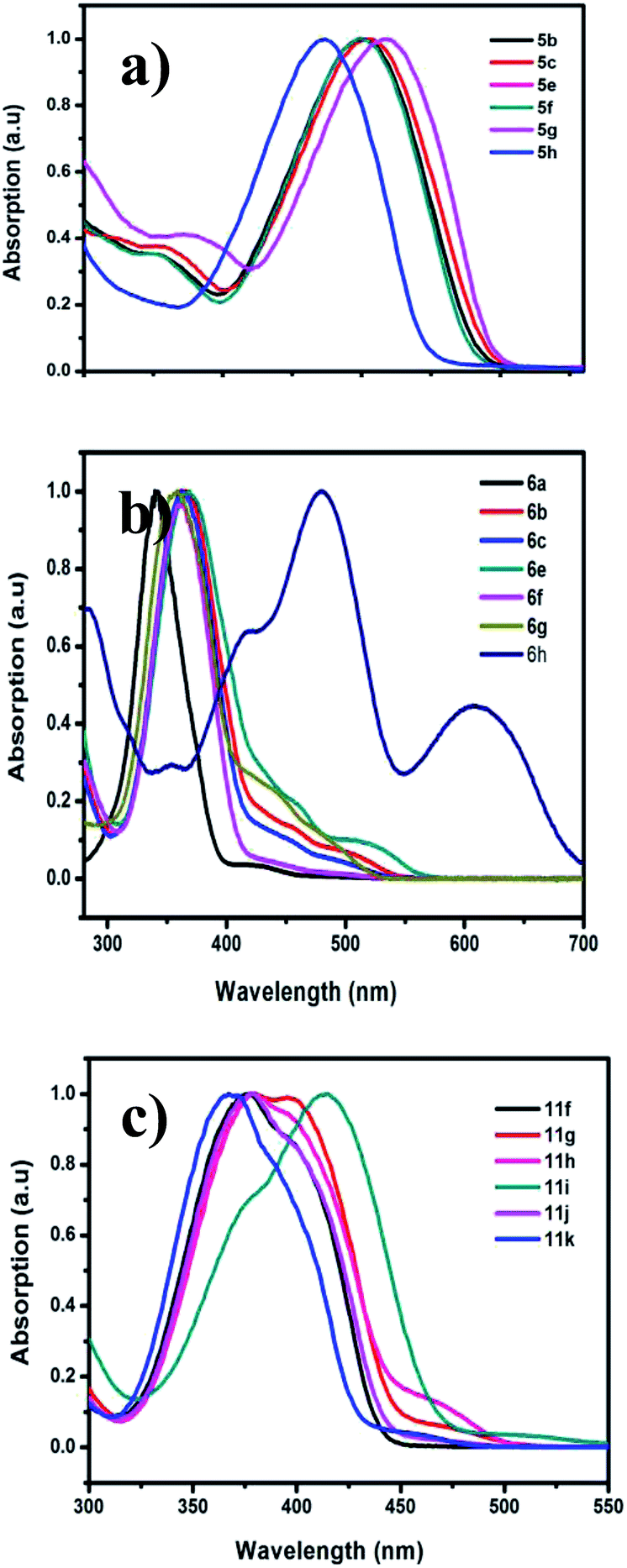
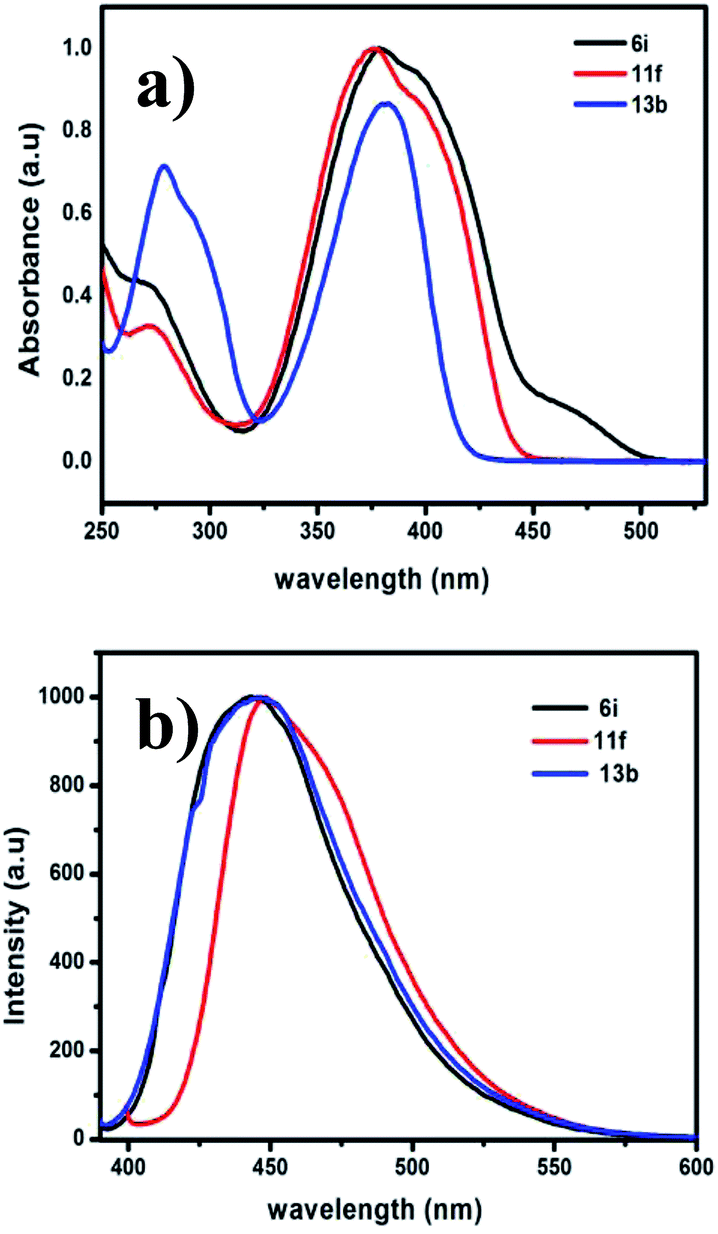
The optical properties of the prepared pyrazine derivatives in dichloromethane were evaluated by using absorption and emission spectroscopy (Fig. 3). The results show that the presence of the phenyl substituent (i.e., in 6i,j) and acetylene (5a–j) groups on the pyrrolopyrazine core result in a significant bathochromic shift with respect to those of unsubstituted pyrrolopyrazine derivatives. The extended conjugation in the substituted compounds is the likely reasons for the observed bathochromic shift. The absorption maxima (λmax) occur between 386–410 nm for the acyclic compounds 5a–j and at 358–380 nm for the cyclic analogues 6a–j, respectively. A similar trend is observed for the 1,5-dihydrodipyrrolo[3,2-b:3′,2′-e]pyrazine derivatives 11a–k (Table 4). Substitution at positions 2 and 6 of the DPP core (i.e., 11i,g) leads to a longer conjugation length, thereby resulting in red shift of the absorption maximum. The absorption spectrum of 6h is especially remarkable in that it displays an unusually large bathochromic shift, presumably as a result of intramolecular charge transfer excitation (ICT), in addition to a localised π–π* transition as observed in Fig. 3(b). Comparisons of the results of the spectroscopy studies reveal that 6i and 13b have similar absorption and emission properties (Table 5). Interestingly, 11f shows a significant red shift in its absorption spectrum (Fig. 4). The optical band gap energies (Eg) for 6i, 11f, and 13b approximated using the onset of their absorption bands, are 2.7, 2.78, and 2.94 eV, respectively.
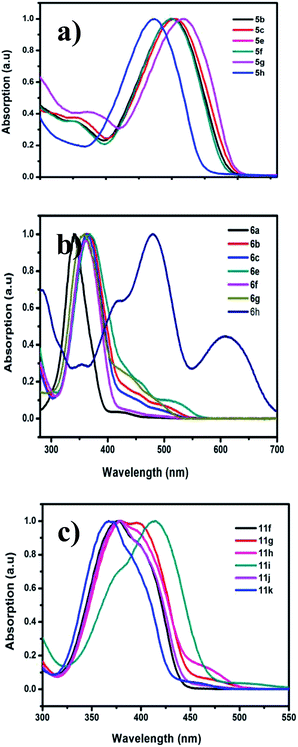 |
| | Fig. 3 UV-vis absorption spectra of DPP derivatives in DCM. (a) Acyclic compounds 5, (b) cyclic compounds 6, (c) cyclic 2,6-substituted DPP series 11. | |
Table 4 UV-visible spectroscopic data of DPP derivatives
| Molecules |
λmaxa (nm) |
Eoptgb (eV) |
Molecules |
λmaxa (nm) |
Eoptgb (eV) |
| Absorption spectra measured in DCM solvent. Optical band gap calculated from the UV-vis absorption onset in solution. |
| 5b |
400 |
2.76 |
6f |
366 |
2.81 |
| 5c |
402 |
2.73 |
6g |
479, 613 |
1.75 |
| 5e |
400 |
2.76 |
6h |
358 |
2.65 |
| 5f |
400 |
2.76 |
11f |
378 |
2.76 |
| 5g |
410 |
2.72 |
11g |
385 |
2.66 |
| 5h |
386 |
2.88 |
11h |
381 |
2.61 |
| 6b |
360 |
2.81 |
11i |
413 |
2.66 |
| 6c |
360 |
2.96 |
11j |
380 |
2.78 |
| 6e |
367 |
2.66 |
11k |
367 |
2.81 |
Table 5 Optical and thermal properties of selected scaffolds
| Molecules |
λmaxa (nm) |
λmaxb (nm) |
Eoptgc (eV) |
Tdd (°C) |
Tme (°C) |
| Absorption spectra. Emission spectra both measured in DCM solvent. Optical band gap calculated from the UV-vis absorption onset in solution. Degradation temperature observed from TGA corresponding to 5% weight loss at 10 °C min−1 under a nitrogen atmosphere. Melting temperature observed from DSC at 10 °C min−1 under nitrogen atmosphere. |
| 6i |
378 |
443 |
2.70 |
298 |
200 |
| 11f |
377 |
450 |
2.78 |
342 |
280 |
| 13b |
384 |
446 |
2.94 |
340 |
184 |
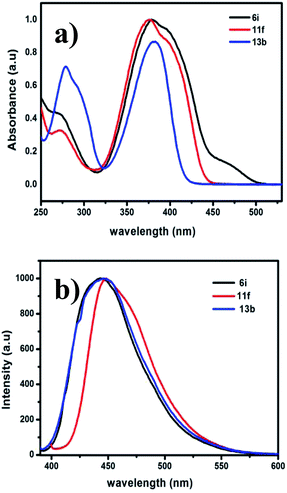 |
| | Fig. 4 (a) UV-vis absorption spectra of 6i, 11f, and 13b in DCM. (b) Emission spectra of 6i, 11f, and 13b in DCM when excited at 375 nm. | |
3.3 Structural and morphological characteristics
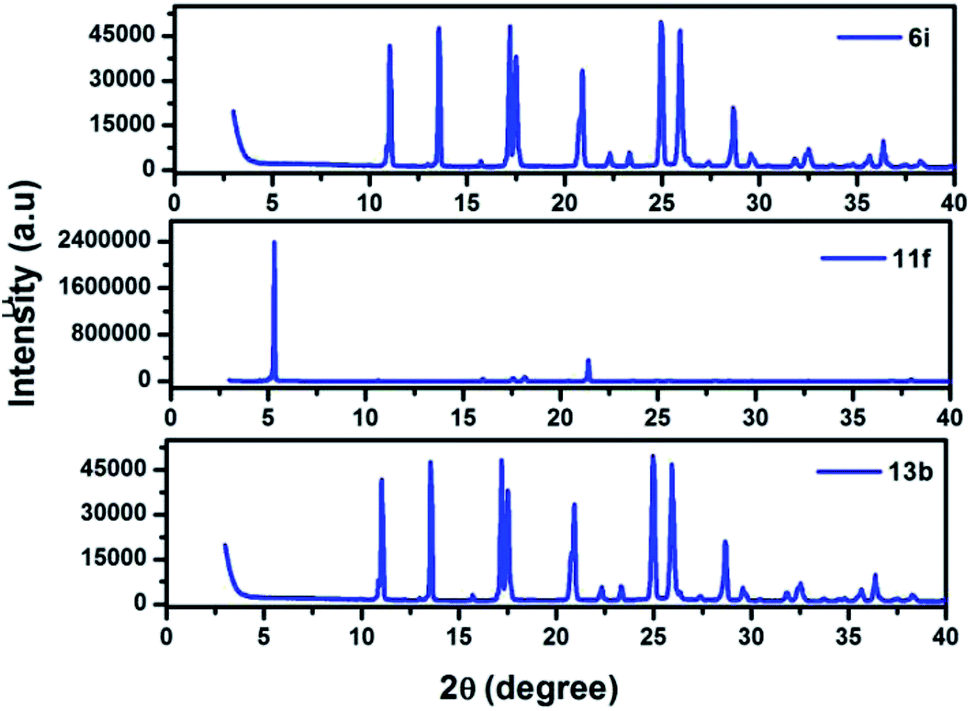
The structural and morphological properties of 6i, 11f, and 13b were evaluated by using X-ray diffraction analysis. Each molecule displays a series of sharply resolved diffraction peaks, indicating that all the compounds possess a high degree of ordering in the solid state (Fig. 5).
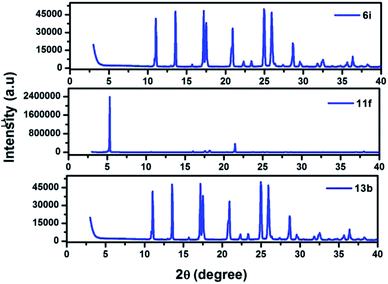 |
| | Fig. 5 Powder X-ray diffraction patterns of 6i, 11f, and 13b at room temperature. | |
4. Experimental section
4.1 General method and materials
All chemicals and solvents are of reagent grade unless otherwise indicated. Reactions with air sensitive materials were carried out under a nitrogen atmosphere. Melting points are uncorrected. 1H NMR and 13C NMR spectra were recorded using a Bruker 500 MHz NMR spectrometer and are given in ppm (δ) units relative to protons and carbons in the deuterated solvent. Mass spectra were recorded using 6400 Triple Quadrupole LC/MS (Agilent). High-resolution mass spectrometry was performed using a 6550 iFunnel Q-TOF LC/MS system (Agilent). UV-vis spectra were recorded using Varian cary-50 spectrophotometer. TGA and DSC were performed with TGA 3 plus and DSC 2 STAR system, respectively. X-ray diffraction and single crystal XRD analysis was performed using a Smartlab and Bruker D8 Discover X-ray Diffractometer with GADDS, respectively.
5-Bromo-6-chloro-3-((trimethylsilyl)ethynyl)pyrazin-2-amine (1a). To a stirred solution of 3,5-dibromo-6-chloro-pyrazin-2-ylamine (2.0 g, 7.04 mmol) in anhydrous THF (20 mL) at 0 °C, TEA (2.1 g, 21 mmol) and CuI (0.13 g, 0.7 mmol) were added subsequently. The mixture was purged thoroughly with nitrogen for 20 min followed by the addition of Pd(PPh3)2Cl2 (0.49 g, 0.7 mmol). Next, TMS–acetylene (1.0 mL, 7.04 mmol) was added drop wise and the mixture was allowed to slowly warm to 15 °C over a period of 3 h. Then the mixture was diluted with water and extracted with EtOAc (3×). The combined organic extracts were concentrated in vacuum and the residue was subjected to silica gel column chromatography to give title compound as a yellow solid. Yield: 81%. The NMR data was found to be identical to that reported in literature. Yield: 81%, LC-MS (ESI): m/z = 306.10 [M + 2]+.
5-Bromo-6-chloro-3-(phenylethynyl)pyrazin-2-amine (1b). The above procedure was followed and the reaction mixture is stirred at room temperature for 2 h. Yield: 83%, 1H NMR (500 MHz, CDCl3) δ 7.63–7.55 (m, 2H), 7.50–7.38 (m, 3H), 5.27 (s, 2H). 13C NMR (126 MHz, CDCl3) δ 153.3, 145.9, 132.0, 129.9, 128.6, 123.9, 122.4, 120.9, 98.0, 82.4. LC-MS (ESI): m/z = 310.1 [M + 2]+. HRMS (ESI) m/z: [M + H]+ calculated for C12H7BrClN3, 307.9585; found 307.9606.
2-Bromo-3-chloro-5H-pyrrolo[2,3-b]pyrazine (2a). To a solution of 5-bromo-6-chloro-3-((trimethylsilanyl)ethynyl)pyrazin-2-amine (1.0 g, 3.2 mmol) in anhydrous NMP (20 mL) a solution of t-BuOK in NMP (0.7 g, 6.5 mmol) was added slowly under a nitrogen atmosphere. The mixture was refluxed at 80 °C for 1 h, cooled to ambient temperature, and diluted with EtOAc and water. The organic layer was separated, dried over MgSO4, concentrated to give a light brown solid. This material was used directly without further purification. Yield: 72%, 1H NMR (500 MHz, DMSO) δ 12.52 (s, 1H), 8.1 (d, J = 1.8 Hz, 1H), 6.70 (d, J = 1.8 Hz, 1H). 13C NMR (126 MHz, DMSO) δ 139.1, 139.1, 138.5, 134.4, 129.9, 100.7. LC-MS (ESI): m/z = 234.0 [M + 2]+.
2-Bromo-3-chloro-6-phenyl-5H-pyrrolo[2,3-b]pyrazine (2b). Yield: 78%, 1H NMR (500 MHz, DMSO) δ 12.97 (s, 1H), 8.02 (d, J = 7.7 Hz, 2H), 7.55 (t, J = 7.5 Hz, 2H), 7.48 (t, J = 7.2 Hz, 1H), 7.24 (s, 1H). 13C NMR (126 MHz, DMSO) δ 145.8, 140.8, 139.6, 139.0, 130.5, 130.3, 130.3, 129.6, 126.5, 97.6. LC-MS (ESI): m/z = 310.0 [M + 2]+. HRMS (ESI) m/z: [M + H]+ calculated for C12H7BrClN3, 307.9585; found 307.9604.
2-Bromo-3-chloro-5-methyl-5H-pyrrolo[2,3-b]pyrazine (3a). To a solution of 2-bromo-3-chloro-5H-pyrrolo[2,3-b]pyrazine (3.0 g, 12.9 mmol) in DMF (40 mL) at 0 °C, NaH (60% dispersion in mineral oil, 0.36 g, 12.9 mmol) was added carefully under nitrogen atmosphere. After stirring for 30 min, MeI (0.8 mL, 12.9 mmol) was added and the mixture was warmed slowly to room temperature. After 3 h, the mixture is poured into ice-coldwater and the precipitate was collected by vacuum filtration. The crude solid is dissolved in EtOAc and subjected to silica gel chromatography to give title compound. (20% EA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :hexane). Yield: 90%, 1H NMR (500 MHz, CDCl3) δ 7.46 (d, J = 3.6 Hz, 1H), 6.65 (d, J = 3.5 Hz, 1H), 3.88 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 140.6, 138.6, 138.0, 134.7, 131.0, 100.6, 31.8. LC-MS (ESI): m/z = 248.0 [M + 2]+. HRMS (ESI) m/z: [M + H]+ calcd for C7H5BrClN3, 245.9428; found 245.9436.
:hexane). Yield: 90%, 1H NMR (500 MHz, CDCl3) δ 7.46 (d, J = 3.6 Hz, 1H), 6.65 (d, J = 3.5 Hz, 1H), 3.88 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 140.6, 138.6, 138.0, 134.7, 131.0, 100.6, 31.8. LC-MS (ESI): m/z = 248.0 [M + 2]+. HRMS (ESI) m/z: [M + H]+ calcd for C7H5BrClN3, 245.9428; found 245.9436.
2-Bromo-3-chloro-5-methyl-6-phenyl-5H-pyrrolo[2,3-b]pyrazine (3b). Yield: 88%, 1H NMR (500 MHz, CDCl3) δ 7.55 (dd, J = 8.3, 4.4 Hz, 5H), 6.70 (s, 1H), 3.86 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 147.9, 140.2, 140.0, 138.2, 131.2, 130.6, 129.6, 129.1, 128.9, 100.1, 30.2. LC-MS (ESI): m/z = 324.0 [M + 2]+. HRMS (ESI) m/z: [M + H]+ calculated for C13H9BrClN3, 321.9741; found 321.9762.
2-Bromo-N,5-dimethyl-5H-pyrrolo[2,3-b]pyrazin-3-amine (4a). To 2-bromo-3-chloro-5-methyl-5H-pyrrolo[2,3-b]pyrazine (0.1 g, 0.4 mmol) is added methylamine (2 mL, 40% solution in water). The mixture was stirred at reflux for 40 min in a microwave vial. After cooling, the mixture was extracted with EtOAc. The organic layer was washed with water and dried over MgSO4, and concentrated in vacuum giving a residue that was subjected to silica gel chromatography to give desired product. Yield: 69%, 1H NMR (500 MHz, CDCl3) δ 6.99 (d, J = 3.6 Hz, 1H), 6.43 (d, J = 3.6 Hz, 1H), 5.13 (s, 1H), 3.75 (s, 3H), 3.07 (d, J = 5.0 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 147.6, 139.3, 129.5, 127.0, 121.4, 100.1, 31.1, 28.7. LC-MS (ESI): m/z = 241.1 [M + H]+. HRMS (ESI) m/z: [M + H]+ calculated for C8H9BrN4, 241.0083; found 241.0090.
2-Bromo-N,5-dimethyl-6-phenyl-5H-pyrrolo[2,3-b]pyrazin-3-amine (4b). Yield: 41%, 1H NMR (500 MHz, CDCl3) δ 7.54 (d, J = 7.6 Hz, 2H), 7.49 (t, J = 7.3 Hz, 2H), 7.42 (t, J = 7.1 Hz, 1H), 6.54 (s, 1H), 5.19 (s, 1H), 3.79 (s, 3H), 3.13 (d, J = 4.4 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 147.6, 141.1, 140.2, 132.2, 129.8, 128.8, 128.6, 128.1, 121.6, 100.0, 29.7, 28.7. LC-MS (ESI): m/z = 317.0 [M + H]+. HRMS (ESI) m/z: [M + H]+ calculated for C14H13BrN4, 317.0396; found 317.0406.
4.2 General procedure for Sonogashira reactions
To a stirred solution of 2-bromo-N,5-dimethyl-5H-pyrrolo[2,3-b]pyrazin-3-amine (0.2 g, 0.8 mmol) in THF (15 mL) was added TEA (0.25 mL, 24 mmol), Pd(PPh3)2Cl2 (0.058 g, 0.08 mmol), CuI (0.015 g, 0.08 mmol) under a nitrogen atmosphere. After 10 min, TMS–acetylene (0.11 g, 0.8 mmol) was added dropwise. The mixture was stirred at room temperature for 1.5 h and concentrated to give a residue that was diluted with water and extracted with EtOAc. The organic layer was washed with water and dried over MgSO4, and concentrated in vacuum to give a residue that was subjected to silica gel chromatography to afford the desired compound.
N,5-Dimethyl-2-((trimethylsilyl)ethynyl)-5H-pyrrolo[2,3-b]pyrazin-3-amine (5a). Mp: 126–128 °C. 1H NMR (500 MHz, CDCl3) δ 7.03 (s, 1H), 6.45 (s, 1H), 5.30 (s, 1H), 3.76 (d, J = 8.2 Hz, 3H), 3.11 (d, J = 4.7 Hz, 3H), 0.32 (s, 9H). 13C NMR (126 MHz, CDCl3) δ 152.6, 139.6, 130.1, 128.0, 119.4, 101.1, 101.0, 100.1, 30.8, 28.2, 0.3. LC-MS (ESI): m/z = 259.2 [M + H]+. HRMS (ESI) m/z: [M + H]+ calcd for C13H18N4Si, 259.1373; found 259.1378.
N,5-Dimethyl-2-(phenylethynyl)-5H-pyrrolo[2,3-b]pyrazin-3-amine (5b). Mp: 136–138 °C. 1H NMR (500 MHz, CDCl3) δ 7.60 (dd, J = 6.5, 2.9 Hz, 2H), 7.43–7.29 (m, 3H), 7.03 (d, J = 3.6 Hz, 1H), 6.47 (d, J = 3.6 Hz, 1H), 5.35 (d, J = 3.8 Hz, 1H), 3.76 (s, 3H), 3.12 (d, J = 5.0 Hz, 3H). 13C NMR (126 CDCl3) δ 152.4, 139.6, 131.7, 130.3, 128.7, 128.4, 127.9, 122.5, 119.6, 100.9, 94.3, 85.7, 30.9, 28.2. LC-MS (ESI): m/z = 263.1 [M + H]+. HRMS (ESI) m/z: [M + H]+ calcd for C16H14N4, 263.1291; found 263.124.
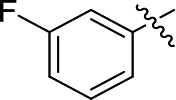
2-((3-Fluorophenyl)ethynyl)-N,5-dimethyl-5H-pyrrolo[2,3-b]pyrazin-3-amine (5c). Mp: 110–112 °C. 1H NMR (500 MHz, CDCl3) δ 7.33 (ddd, J = 21.2, 15.5, 7.8 Hz, 3H), 7.09 (d, J = 8.3 Hz, 1H), 7.05 (d, J = 3.5 Hz, 1H), 6.48 (d, J = 3.5 Hz, 1H), 5.33 (d, J = 3.3 Hz, 1H), 3.76 (s, 3H), 3.13 (d, J = 4.9 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 162.3 (d, J = 246.8 Hz), 161.3, 152.5, 139.7, 130.2, 130.0 (d, J = 8.7 Hz), 128.3, 127.6 (d, J = 3.0 Hz), 124.3 (d, J = 9.5 Hz), 119.0, 118.4, 118.3, 116.1, 115.9, 100.9, 92.9 (d, J = 3.4 Hz), 86.6, 30.9, 28.2. LC-MS (ESI): m/z = 281.2 [M + H]+. HRMS (ESI) m/z: [M + H]+ calculated for C16H13FN4, 281.1197; found 281.1202.
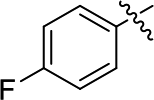
2-((4-Fluorophenyl)ethynyl)-N,5-dimethyl-5H-pyrrolo[2,3-b]pyrazin-3-amine (5d). 1H NMR (500 MHz, CDCl3) δ 7.60 (dd, J = 7.6, 5.9 Hz, 2H), 7.32–7.27 (m, 1H), 7.08 (t, J = 8.4 Hz, 2H), 6.42 (d, J = 3.0 Hz, 1H), 5.21 (s, 1H), 3.82 (s, 3H), 3.12 (d, J = 4.9 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 162.9 (d, J = 251.0 Hz), 154.1, 136.6, 135.3, 133.8 (d, J = 8.5 Hz), 132.9, 118.4, 118.4, 115.8 (d, J = 22.2 Hz), 98.9, 94.4, 85.5, 31.6, 28.7. LC-MS (ESI): m/z = 281.2 [M + H]+. HRMS (ESI) m/z: [M + H]+ calculated for C16H13FN4, 281.1197; found 281.1201.
2-((4-Methoxyphenyl)ethynyl)-N,5-dimethyl-5H-pyrrolo[2,3-b]pyrazin-3-amine (5e). Mp: 148–150 °C. 1H NMR (500 MHz, CDCl3) δ 7.55 (d, J = 8.3 Hz, 2H), 7.03 (d, J = 3.0 Hz, 1H), 6.90 (d, J = 8.3 Hz, 2H), 6.47 (d, J = 2.9 Hz, 1H), 5.37 (d, J = 4.0 Hz, 1H), 3.84 (s, 3H), 3.76 (s, 3H), 3.13 (d, J = 4.7 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 160.0, 152.2, 139.4, 133.2, 130.1, 127.7, 120.1, 114.5, 114.1, 100.8, 94.4, 84.4, 55.3, 30.9, 28.2. LC-MS (ESI): m/z = 293.2 [M + H]+. HRMS (ESI) m/z: [M + H]+ calculated for C17H16N4O, 293.1397; found 293.1403.
N,5-Dimethyl-2-(p-tolylethynyl)-5H-pyrrolo[2,3-b]pyrazin-3-amine (5f). Mp: 132–134 °C. 1H NMR (500 MHz, CDCl3) δ 7.51 (d, J = 6.8 Hz, 2H), 7.19 (d, J = 7.0 Hz, 2H), 7.04 (s, 1H), 6.48 (s, 1H), 5.38 (s, 1H), 3.77 (s, 3H), 3.14 (s, 3H), 2.40 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 152.3, 139.5, 139.0, 131.6, 130.2, 129.2, 127.8, 119.9, 119.4, 100.9, 94.6, 85.1, 30.9, 28.2, 21.5. LC-MS (ESI): m/z = 277.2 [M + H]+. HRMS (ESI) m/z: [M + H]+ calculated for C17H16N4, 277.1448; found 277.1452.
2-(Cyclopropylethynyl)-N,5-dimethyl-5H-pyrrolo[2,3-b]pyrazin-3-amine (5g). Mp: 122–124 °C. 1H NMR (500 MHz, CDCl3) δ 6.99 (d, J = 3.1 Hz, 1H), 6.42 (d, J = 3.1 Hz, 1H), 5.30 (s, 1H), 3.74 (s, 3H), 3.09 (d, J = 4.9 Hz, 3H), 1.62–1.43 (m, 1H), 0.94 (d, J = 8.4 Hz, 4H). 13C NMR (126 MHz, CDCl3) δ 152.4, 139.1, 129.6, 127.3, 120.5, 100.7, 98.9, 72.2, 30.8, 28.2, 9.0, 0.3. LC-MS (ESI): m/z = 227.2 [M + H]+. HRMS (ESI) m/z: [M + H]+ calculated for C13H14N4, 227.1291; found 227.126.
2-((4-(Dimethylamino)phenyl)ethynyl)-N,5-dimethyl-5H-pyrrolo[2,3-b]pyrazin-3-amine (5h). Mp: 180–182 °C. LC-MS (ESI): m/z = 306.3 [M + H]+. HRMS (ESI) m/z: [M + H]+ calculated for C18H19N5, 306.1713; found 306.1717.
N,5-Dimethyl-6-phenyl-2-(phenylethynyl)-5H-pyrrolo[2,3-b]pyrazin-3-amine (5i). Mp: 199–200 °C. 1H NMR (500 MHz, CDCl3) δ 7.64 (d, J = 3.4 Hz, 2H), 7.57 (d, J = 6.9 Hz, 2H), 7.50 (t, J = 6.9 Hz, 2H), 7.45–7.38 (m, 4H), 6.58 (s, 1H), 5.41 (s, 1H), 3.81 (s, 3H), 3.18 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 152.4, 141.4, 141.1, 132.2, 131.7, 130.6, 128.8, 128.7, 128.6, 128.4, 128.1, 122.5, 119.8, 100.7, 94.5, 85.8, 29.5, 28.3. LC-MS (ESI): m/z = 339.2 [M + H]+. HRMS (ESI) m/z: [M + H]+ calculated for C22H18N4, 339.1604; found 339.1607.
2-((4-(Dimethylamino)phenyl)ethynyl)-N,5-dimethyl-6-phenyl-5H-pyrrolo[2,3-b]pyrazin-3-amine (5j). Mp: 129–131 °C. LC-MS (ESI): m/z = 382.3 [M + H]+. HRMS (ESI) m/z: [M + H]+ calculated for C24H23N5, 382.2026; found 382.2026.
4.3 General procedure for cyclization of DPP derivatives
A solution of N,5-dimethyl-2-((trimethylsilyl)ethynyl)-5H-pyrrolo[2,3-b]pyrazin-3-amine (0.09 g, 0.34 mmol) in NMP (5 mL) containing t-BuOK (0.078 g, 0.7 mmol) was stirred at reflux for 2 h. The mixture was quenched with water and extracted with EtOAc. The combined organic layers were concentrated in vacuum to give a residue that was subjected to silica gel chromatography eluting with 20% EtOAc to afford the titled compound as a pale yellow solid.
1,7-Dimethyl-1,7-dihydrodipyrrolo[2,3-b:3′,2′-e]pyrazine (6a). Mp: 168–170 °C. 1H NMR (500 MHz, CDCl3) δ 7.35 (d, J = 3.5 Hz, 1H), 6.69 (d, J = 3.6 Hz, 1H), 3.90 (s, 3H). 13CNMR (126 MHz, CDCl3) δ 138.4, 136.2, 130.8, 99.7, 31.3. LC-MS (ESI): m/z = 187.1 [M + H]+. HRMS (ESI) m/z: [M + H]+ calculated for C10H10N4, 187.0978; found 187.0988.
1,7-Dimethyl-2-phenyl-1,7-dihydrodipyrrolo[2,3-b:3′,2′-e]pyrazine (6b). Mp: 165–166 °C. 1H NMR (500 MHz, CDCl3) δ 7.61 (d, J = 7.3 Hz, 2H), 7.51 (t, J = 7.5 Hz, 2H), 7.44 (d, J = 7.4 Hz, 1H), 7.35 (d, J = 3.6 Hz, 1H), 6.77 (s, 1H), 6.69 (d, J = 3.6 Hz, 1H), 3.92 (s, 3H), 3.91 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 143.8, 140.3, 138.3, 136.6, 136.4, 132.3, 130.6, 129.1, 128.6, 128.4, 99.9, 99.8, 31.4, 29.8. LC-MS (ESI): m/z = 263.2 [M + H]+. HRMS (ESI) m/z: [M + H]+ calculated for C16H14N4, 263.1291; found 263.1303.
2-(3-Fluorophenyl)-1,7-dimethyl-1,7-dihydrodipyrrolo[2,3-b:3′,2′-e]pyrazine (6c). Mp: 187–188 °C. 1H NMR (500 MHz, CDCl3) δ 7.47 (dd, J = 14.2, 7.7 Hz, 1H), 7.43–7.36 (m, 2H), 7.32 (d, J = 8.7 Hz, 1H), 7.14 (t, J = 7.2 Hz, 1H), 6.81 (d, J = 4.6 Hz, 1H), 6.71 (t, J = 5.0 Hz, 1H), 3.93 (dd, J = 9.7, 4.5 Hz, 6H). 13C NMR (126 MHz, CDCl3) δ 162.7 (d, J = 246.8 Hz), 142.2, 140.3, 138.5, 136.9, 136.0, 134.3 (d, J = 8.1 Hz), 131.0, 130.2 (d, J = 8.5 Hz), 124.7, 115.9 (d, J = 22.4 Hz), 115.2 (d, J = 21.0 Hz), 100.5, 99.8, 31.3, 29.9. LC-MS (ESI): m/z = 281.1 [M + H]+. HRMS (ESI) m/z: [M + H]+ calculated for C16H13FN4, 281.1197; found 281.1206.
2-(4-Fluorophenyl)-1,7-dimethyl-1,7-dihydrodipyrrolo[2,3-b:3′,2′-e]pyrazine (6d). Mp: 158–159 °C. 1H NMR (500 MHz, CDCl3) δ 7.60–7.51 (m, 2H), 7.35 (d, J = 3.6 Hz, 1H), 7.23–7.12 (m, 2H), 6.73 (s, 1H), 6.69 (d, J = 3.7 Hz, 1H), 3.92 (s, 3H), 3.87 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 163.8, 161.8, 142.6, 140.1, 138.3, 136.7, 136.3, 130.8 (d, J = 8.2 Hz), 130.7, 128.4 (d, J = 3.3 Hz), 115.7 (d, J = 21.7 Hz), 99.8 (d, J = 15.8 Hz), 31.3, 29.7. LC-MS (ESI): m/z = 281.1 [M + H]+. HRMS (ESI) m/z: [M + H]+ calculated for C16H13FN4, 281.1197; found 281.1209.
2-(4-Methoxyphenyl)-1,7-dimethyl-1,7-dihydrodipyrrolo[2,3-b:3′, 2′-e]pyrazine (6e). Mp: 155–156 °C. 1H NMR (500 MHz, CDCl3) δ 7.50 (d, J = 8.7 Hz, 2H), 7.29 (d, J = 3.6 Hz, 1H), 7.00 (d, J = 8.7 Hz, 2H), 6.70 (s, 1H), 6.67 (d, J = 3.6 Hz, 1H), 3.87 (s, 3H), 3.85 (s, 3H), 3.84 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 159.8, 143.8, 140.1, 138.1, 136.5, 136.4, 130.4, 130.3, 124.6, 114.1, 99.6, 99.0, 55.3, 31.3, 29.7. LC-MS (ESI): m/z = 293.2 [M + H]+. HRMS (ESI) m/z: [M + H]+ calculated for C17H16N4O, 293.1397; found 293.1406.
1,7-Dimethyl-2-p-tolyl-1,7-dihydrodipyrrolo[2,3-b:3′,2′-e]pyrazine (6f). Mp: 174–176 °C. 1H NMR (500 MHz, CDCl3) δ 7.52 (d, J = 8.0 Hz, 2H), 7.36 (d, J = 3.6 Hz, 1H), 7.33 (d, J = 7.9 Hz, 2H), 6.76 (s, 1H), 6.71 (d, J = 3.6 Hz, 1H), 3.94 (s, 3H), 3.92 (s, 3H), 2.46 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 144.0, 140.2, 138.4, 138.2, 136.6, 136.5, 130.51, 129.4, 129.4, 129.0, 99.7, 99.5, 31.3, 29.8, 21.3. LC-MS (ESI): m/z = 277.3 [M + H]+. HRMS (ESI) m/z: [M + H]+ calculated for C17H16N4, 277.1448; found 277.1455.
2-Cyclopropyl-1,7-dimethyl-1,7-dihydrodipyrrolo[2,3-b:3′,2′-e]pyrazine (6g). Mp: 163–165 °C. 1H NMR (500 MHz, CDCl3) δ 6.65 (d, J = 3.1 Hz, 1H), 6.26 (s, 1H), 3.93 (d, J = 4.5 Hz, 3H), 3.91 (d, J = 4.5 Hz, 3H), 1.99 (d, J = 5.1 Hz, 1H), 1.07 (d, J = 6.5 Hz, 2H), 0.85 (d, J = 3.2 Hz, 2H). 13C NMR (126 MHz, CDCl3) δ 146.5, 139.3, 137.8, 136.5, 135.7, 129.6, 99.6, 95.2, 31.3, 28.0, 8.1, 6.9. LC-MS (ESI): m/z = 227.2 [M + H]+. HRMS (ESI) m/z: [M + H]+ calculated for C13H14N4, 227.1291; found 227.1304.
4-(1,7-Dimethyl-1,7-dihydrodipyrrolo[2,3-b:3′,2′-e]pyrazin-2-yl)-N,N-dimethylaniline (6h). Mp: 188–190 °C. 1H NMR (500 MHz, CDCl3) δ 7.50 (d, J = 8.8 Hz, 2H), 7.36 (d, J = 3.6 Hz, 1H), 6.83 (d, J = 8.8 Hz, 2H), 6.66 (d, J = 3.6 Hz, 1H), 6.65 (s, 1H), 3.94 (d, J = 3.7 Hz, 6H), 3.04 (s, 6H). 13C NMR (126 MHz, CDCl3) δ 150.4, 146.1, 141.5, 139.8, 135.7, 133.8, 130.82, 130.0, 119.7, 112.0, 98.9, 97.5, 40.3, 31.7, 30.0. LC-MS (ESI): m/z = 306.2 [M + H]+. HRMS (ESI) m/z: [M + H]+ calculated for C18H19N5, 306.1713; found 306.1717.
1,7-Dimethyl-2,6-diphenyl-1,7-dihydrodipyrrolo[2,3-b:3′,2′-e]pyrazine (6i). Mp: 197–199 °C. 1H NMR (500 MHz, CDCl3) δ 7.66 (d, J = 7.2 Hz, 2H), 7.55 (t, J = 7.5 Hz, 2H), 7.48 (d, J = 7.4 Hz, 1H), 6.81 (s, 1H), 3.97 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 144.7, 141.8, 134.9, 132.2, 129.1, 128.7, 128.5, 99.3, 30.1. LC-MS (ESI): m/z = 339.2 [M + H]+. HRMS (ESI) m/z: [M + H]+ calculated for C22H18N4, 339.1604; found 339.1607.
4-(1,7-Dimethyl-6-phenyl-1,7-dihydrodipyrrolo[2,3-b:3′,2′-e]pyrazin-2-yl)-N,N-dimethylaniline (6j). Mp: 231–233 °C. 1H NMR (500 MHz, CDCl3) δ 7.62 (s, 2H), 7.51 (s, 4H), 7.44 (d, J = 6.2 Hz, 1H), 6.84 (d, J = 6.9 Hz, 2H), 6.77 (s, 1H), 6.67 (s, 1H), 3.93 (s, 6H), 3.05 (s, 6H). 13C NMR (126 MHz, CDCl3) δ 150.3, 144.8, 142.9, 140.2, 139.8, 137.4, 136.3, 132.5, 129.9, 129.0, 128.6, 128.2, 119.9, 112.1, 99.9, 98.2, 40.3, 29.9. LC-MS (ESI): m/z = 382.2 [M + H]+. HRMS (ESI) m/z: [M + H]+ calculated for C24H23N5, 382.2026; found 382.2037.
4.4 General method for Buchwald reaction
tert-Butyl 3-chloro-5-methyl-5H-pyrrolo[2,3-b]pyrazin-2-ylcarbamate (7a). An air-dried RB flask was charged sequentially with 1,4-dioxane (50 mL), t-amyl alcohol (10 mL), 2-bromo-3-chloro-5-methyl-5H-pyrrolo[2,3-b]pyrazine (2.2 g, 8.9 mmol), Pd(OAc)2 (0.2 g, 0.89 mmol), xantphos (0.51 g, 0.89 mmol), Cs2CO3 (5.82 g, 17 mmol) and t-butyl carbamate (1.04 g, 8.9 mmol). The resultant suspension was stirred at reflux at 90 °C for 3 h. Once the reaction was determined to be complete by using TLC, the mixture was cooled to room temperature, diluted with EtOAc and filtered through a pad of Celite. The filtrate was concentrated in vacuum. The residue was subjected to silica gel column chromatography to afford desired product as a crystalline solid. Yield: 78%, 1H NMR (500 MHz, CDCl3) δ 7.41 (s, 1H), 7.13 (s, 1H), 6.64 (s, 1H), 3.87 (s, 3H), 1.50 (s, 9H). 13C NMR (126 MHz, CDCl3) δ 151.4, 138.7, 136.9, 136.0, 133.6, 132.2, 100.8, 81.5, 31.7, 28.2. LC-MS (ESI): m/z = 305.2 [M + Na]+. HRMS (ESI) m/z: [M + H]+ calculated for C12H15ClN4O2, 283.0956; found 283.0961.
tert-Butyl3-chloro-5-methyl-6-phenyl-5H-pyrrolo[2,3-b]pyrazin-2-ylcarbamate (7b). Yield: 76%, 1H NMR (500 MHz, CDCl3) δ 7.53 (m, J = 15.4, 7.1 Hz, 5H), 7.22 (s, 1H), 6.74 (s, 1H), 3.83 (s, 3H), 1.57 (s, 9H). 13C NMR (126 MHz, CDCl3) δ 151.5, 146.6, 139.0, 138.5, 136.2, 131.6, 131.1, 129.1, 129.1, 128.8, 100.6, 81.4, 30.1, 28.3. LC-MS (ESI): m/z = 381.2 [M + Na]+. HRMS (ESI) m/z: [M + H]+ calculated for C18H19ClN4O2, 359.1269; found 359.1290.
tert-Butyl3-chloro-5-methyl-5H-pyrrolo[2,3-b]pyrazin-2-yl(methyl) carbamate (8a). To a solution of 2-bromo-3-chloro-5H-pyrrolo[2,3-b]pyrazine (2.76 g, 9.7 mmol) in DMF (35 mL) under nitrogen atmosphere at 0 °C was added NaH (60% dispersion in mineral oil, 0.44 g, 10 mmol) carefully. After 30 min, MeI (0.6 mL, 9.7 mmol) was added and the mixture is warmed slowly to ambient temperature. After 3 h, the mixture was poured into ice-cold water and the precipitate is collected by vacuum filtration. The crude solid was subjected to silica gel chromatography. Yield: 92%, 1H NMR (500 MHz, CDCl3) δ 7.49 (s, 1H), 6.65 (s, 1H), 3.90 (s, 3H), 3.29 (d, J = 0.8 Hz, 3H), 1.37 (s, 9H). 13C NMR (126 MHz, CDCl3) δ 154.1, 143.1, 139.6, 138.5, 136.5, 134.2, 100.8, 80.9, 35.1, 31.8, 28.1. HRMS (ESI) m/z: [M + H]+ calculated for C13H17ClN4O2, 297.1113; found 297.1118.
tert-Butyl 3-chloro-5-methyl-6-phenyl-5H-pyrrolo[2,3-b]pyrazin-2-yl(methyl)carbamate (8b). Yield: 95%, 1H NMR (500 MHz, CDCl3) δ 7.56 (dt, J = 10.7, 7.0 Hz, 5H), 6.73 (s, 1H), 3.89 (s, 3H), 3.32 (s, 3H), 1.41 (s, 9H). 13C NMR (126 MHz, CDCl3) δ 154.1, 147.3, 143.5, 140.1, 139.0, 136.7, 130.9, 129.4, 129.1, 128.9, 100.4, 80.9, 30.9, 30.2, 28.2. LC-MS (ESI): m/z = 317.1 [M − butyl]+. HRMS (ESI) m/z: [M + H]+ calculated for C19H21ClN4O2, 373.1426; found 373.1445.
3-Chloro-N,5-dimethyl-5H-pyrrolo[2,3-b]pyrazin-2-amine (9a). A solution of tert-butyl 3-chloro-5-methyl-5H-pyrrolo[2,3-b]pyrazin-2-yl(methyl)carbamate (2.6 g, 8.7 mmol) in DCM (50 mL) containing TFA (5.0 mL, 43 mmol) was stirred at room temperature for 10 min then refluxed for 50 min. The mixture was made basic by addition of 1 M NaOH solution and extracted with DCM. The organic layer was dried over MgSO4, and concentrated in vacuum to get the desired product. Yield: 96%, 1H NMR (500 MHz, CDCl3) δ 7.20 (d, J = 3.3 Hz, 1H), 6.44 (d, J = 5.0 Hz, 1H), 4.98 (s, 1H), 3.80 (d, J = 1.8 Hz, 3H), 3.11 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 147.9, 135.2, 133.5, 130.7, 128.0, 98.7, 31.6, 29.0. LC-MS (ESI): m/z = 197.0 [M + H]+. HRMS (ESI) m/z: [M + H]+ calculated for C8H9ClN4, 197.0589; found 197.0595.
3-Chloro-N,5-dimethyl-5H-pyrrolo[2,3-b]pyrazin-2-amine (9b). Yield: 98%, 1H NMR (500 MHz, CDCl3) δ 7.55 (d, J = 7.2 Hz, 2H), 7.51 (t, J = 7.4 Hz, 2H), 7.45 (t, J = 7.2 Hz, 1H), 6.55 (s, 1H), 5.00 (s, 1H), 3.81 (s, 3H), 3.13 (d, J = 4.7 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 148.0, 143.6, 135.5, 135.2, 131.9, 129, 128.7, 128.5, 128.0, 99.0, 30.0, 28.9; LC-MS (ESI): m/z = 273.2 [M + H]+. HRMS (ESI) m/z: [M + H]+ calculated for C14H13ClN4, 273.0902; found 273.0919.
4.5 General procedure for Sonogashira reaction
To a stirred solution of 3-chloro-N,5-dimethyl-5H-pyrrolo[2,3-b]pyrazin-2-amine (0.2 g, 1.0 mmol) in DMF (2 mL) was added TEA (0.3 mL, 3.0 mmol), CuI (0.019 g, 0.1 mmol), Pd(PPh3)2Cl2 (0.07 g, 0.1 mmol) and TMS–acetylene (0.1 mL, 1.0 mmol). The mixture subjected to microwave irradiation for 50 min at 90 °C. Then the reaction mixture was cooled to ambient temperature and diluted with water and extracted with EtOAc twice. The combined organic layer was concentrated in vacuum to give a residue that was subjected to silica column chromatography to give desired compound as a brown solid.
3-((4-Fluorophenyl)ethynyl)-N,5-dimethyl-5H-pyrrolo[2,3-b]pyrazin-2-amine (10c). 1H NMR (500 MHz, CDCl3) δ 7.47 (dd, J = 14.2, 7.7 Hz, 1H), 7.43–7.36 (m, 2H), 7.32 (d, J = 8.7 Hz, 1H), 7.14 (t, J = 7.2 Hz, 1H), 6.81 (d, J = 4.6 Hz, 1H), 6.71 (t, J = 5.0 Hz, 1H), 3.93 (dd, J = 9.7, 4.5 Hz, 6H). 13C NMR (126 MHz, CDCl3) δ 162.9 (d, J = 251.0 Hz), 152.8, 136.6, 135.6, 133.8 (d, J = 8.5 Hz), 132.9, 119.0, 118.1, 115.8 (d, J = 22.2 Hz), 98.9, 95.3, 86.3, 33.1, 29.88. LC-MS (ESI): m/z = 281.1 [M + H]+. HRMS (ESI) m/z: [M + H]+ calculated for C16H13FN4, 281.1197; found 281.1201.
N,5-Dimethyl-6-phenyl-3-(phenylethynyl)-5H-pyrrolo[2,3-b]pyrazin-2-amine (10f). Mp: 171–173 °C, 1H NMR (500 MHz, CDCl3) δ 7.66–7.63 (m, 2H), 7.59–7.56 (m, 2H), 7.52 (d, J = 7.2 Hz, 2H), 7.47–7.44 (m, 1H), 7.40 (dd, J = 4.2, 2.3 Hz, 3H), 6.56 (s, 1H), 5.31 (d, J = 5.5 Hz, 1H), 3.85 (s, 3H), 3.17 (d, J = 5.0 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 153.2, 145.7, 137.1, 136.8, 131.9, 131.8, 129.0, 128.9, 128.7, 128.6, 128.4, 122.3, 117.8, 99.4, 95.7, 86.1, 30.0, 29.7, 28.8; LC-MS (ESI): m/z = 339.3 [M + H]+. HRMS (ESI) m/z: [M + H]+ calculated for C22H18N4, 339.1604; found 339.1624.
3-((4-Methoxyphenyl)ethynyl)-N,5-dimethyl-6-phenyl-5H-pyrrolo[2,3-b]pyrazin-2-amine (10g). LC-MS (ESI): m/z = 369.2 [M + H]+. HRMS (ESI) m/z: [M + H]+ calculated for C23H20N4O, 369.1710; found 369.1715.
N,5-Dimethyl-3-(p-tolylethynyl)-5H-pyrrolo[2,3-b]pyrazin-2-amine (10h). Mp: 144–146 °C, 1H NMR (500 MHz, CDCl3) δ 7.58 (d, J = 7.2 Hz, 2H), 7.56–7.50 (m, 4H), 7.46 (t, J = 7.3 Hz, 1H), 7.22 (d, J = 7.9 Hz, 2H), 6.56 (s, 1H), 5.31 (d, J = 4.8 Hz, 1H), 3.85 (s, 3H), 3.17 (d, J = 5.0 Hz, 3H), 2.41 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 153.2, 145.5, 139.2, 137.0, 136.6, 131.9, 131.7, 129.2, 129.0, 128.7, 128.6, 119.2, 118.0, 99.4, 96.1, 85.4, 30.0, 28.8, 21.6. LC-MS (ESI): m/z = 353.2 [M + H]+. HRMS (ESI) m/z: [M + H]+ calculated for C23H20N4, 353.1761; found 353.1766.
3-((4-(Dimethylamino)phenyl)ethynyl)-N,5-dimethyl-6-phenyl-5H-pyrrolo[2,3-b]pyrazin-2-amine (10i). LC-MS (ESI): m/z = 382.3 [M + H]+.
3-((3-Fluorophenyl)ethynyl)-N,5-dimethyl-6-phenyl-5H-pyrrolo[2,3-b]pyrazin-2-amine (10j). Mp: 158–160 °C, 1H NMR (500 MHz, CDCl3) δ 7.58 (d, J = 7.2 Hz, 2H), 7.53 (t, J = 7.5 Hz, 2H), 7.47 (t, J = 7.3 Hz, 1H), 7.43 (d, J = 7.7 Hz, 1H), 7.40–7.33 (m, 2H), 7.14–7.08 (m, 1H), 6.56 (s, 1H), 5.25 (d, J = 4.7 Hz, 1H), 3.85 (s, 3H), 3.17 (d, J = 5.0 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 162.3 (d, J = 247.0 Hz), 153.3, 146.1, 137.1 (d, J = 4.0 Hz), 131.8, 130.0 (d, J = 8.6 Hz), 129.2, 129.0, 128.7, 127.7 (d, J = 3.1 Hz), 124.2, 124.1, 118.54 (d, J = 23.0 Hz), 117.1, 116.26 (d, J = 21.2 Hz), 99.4, 94.3 (d, J = 3.4 Hz), 86.9, 30.0, 28.8. LC-MS (ESI): m/z = 357.2 [M + H]+. HRMS (ESI) m/z: [M + H]+ calculated for C22H17FN4, 357.1510; found 357.1515.
3-(Cyclopropylethynyl)-N,5-dimethyl-6-phenyl-5H-pyrrolo[2,3-b]pyrazin-2-amine (10k). LC-MS (ESI): m/z = 303.2 [M + H]+. HRMS (ESI) m/z: [M + H]+ calculated for C19H18N4, 303.1604; found 303.1610.
4.6 General procedure for cyclization
A suspension containing N,5-dimethyl-3-((trimethylsilyl)ethynyl)-5H-pyrrolo[2,3-b]pyrazin-2-amine (0.125 g, 0.48 mmol) in DMF (2 mL) and t-BuOK (0.1 g, 9.6 mmol) was stirred at 120 °C for 1 h under microwave. The reaction mixture was cooled to room temperature, diluted with EtOAc and water. The organic layer was separated, washed with water and dried over MgSO4, and concentrated in vacuum to give a pale yellow crystal which was subjected to silica gel column chromatography to give desired compound.
1,5-Dimethyl-1,5-dihyrodipyrrolo[3,2-b:3′,2′-e]pyrazine (11a). Mp: 192–193 °C, 1H NMR (500 MHz, CDCl3) δ 7.42 (d, J = 3.6 Hz, 2H), 6.66 (d, J = 3.6 Hz, 2H), 3.94 (s, 6H). 13C NMR (126 MHz, CDCl3) δ 139.6, 134.9, 132.0, 98.7, 31.6. LC-MS (ESI): m/z = 187.1 [M + H]+. HRMS (ESI) m/z: [M + H]+ calculated for C10H10N4, 187.0978; found 187.0988.
1,5-Dimethyl-2-phenyl-1,5-dihyrodipyrrolo[3,2-b:3′,2′-e]pyrazine (11b). Mp: 156–157 °C, 1H NMR (500 MHz, CDCl3) δ 7.61 (d, J = 7.4 Hz, 2H), 7.51 (t, J = 7.0 Hz, 2H), 7.47–7.43 (m, 1H), 7.41 (s, 1H), 6.75 (s, 1H), 6.68 (s, 1H), 3.95 (s, 3H), 3.94 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 144.9, 141.4, 140.0, 135.1, 134.6, 132.2, 131.7, 129.1, 128.6, 128.5, 99.1, 98.9, 31.7, 30.0. LC-MS (ESI): m/z = 263.1 [M + H]+. HRMS (ESI) m/z: [M + H]+ calculated for C16H14N4, 263.1291; found 263.1309.
2-(4-Fluorophenyl)-1,5-dimethyl-1,5-dihydrodipyrrolo[3,2-b:3′,2′-e]pyrazine (11c). 1H NMR (500 MHz, CDCl3) δ 7.60–7.51 (m, 2H), 7.35 (d, J = 3.6 Hz, 1H), 7.23–7.12 (m, 2H), 6.73 (s, 1H), 6.69 (d, J = 3.7 Hz, 1H), 3.92 (s, 3H), 3.87 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 162.8 (d, J = 248.8 Hz), 142.6, 140.1, 138.3, 136.7, 136.3, 130.9, 130.8, 130.7, 128.4 (d, J = 3.3 Hz), 115.7 (d, J = 21.7 Hz), 99.8 (d, J = 15.8 Hz), 31.3, 29.7. HRMS (ESI) m/z: [M + H]+ calculated for C16H13FN4, 281.1197; found 284.1212.
4-(1,5-Dimethyl-1,5-dihydrodipyrrolo[3,2-b:3′,2′-e]pyrazin-2-yl)-N,N-dimethylaniline (11d). Mp: 154–155 °C, 1H NMR (500 MHz, CDCl3) δ 7.50 (d, J = 8.8 Hz, 2H), 7.36 (d, J = 3.6 Hz, 1H), 6.83 (d, J = 8.8 Hz, 2H), 6.66 (d, J = 3.6 Hz, 1H), 6.65 (s, 1H), 3.94 (d, J = 3.7 Hz, 6H), 3.04 (s, 6H). 13C NMR (126 MHz, CDCl3) δ 150.4, 146.1, 141.5, 139.8, 135.7, 133.8, 130.8, 130.0, 119.7, 112.0, 98.9, 97.5, 40.3, 31.7, 30.0. LC-MS (ESI): m/z = 306.2 [M + H]+. HRMS (ESI) m/z: [M + H]+ calculated for C18H19N5, 306.1713; found 306.1722.
4-(1,5-Dimethyl-1,5-dihydrodipyrrolo[3,2-b:3′,2′-e]pyrazin-2-yl)benzonitrile (11e). Mp: 268–269 °C, 1H NMR (500 MHz, CDCl3) δ 7.80 (d, J = 7.9 Hz, 2H), 7.73 (d, J = 8.0 Hz, 2H), 7.48 (d, J = 3.2 Hz, 1H), 6.84 (s, 1H), 6.69 (d, J = 3.3 Hz, 1H), 3.97 (s, 6H). 13C NMR (126 MHz, CDCl3) δ 142.1, 141.8, 140.3, 136.6, 135.6, 134.4, 132.9, 132.4, 129.4, 118.5, 111.9, 100.9, 99.0, 31.7, 30.3. LC-MS (ESI): m/z = 288 [M + H]+. HRMS (ESI) m/z: [M + H]+ calculated for C17H13N5, 288.1244; found 288.1257.
1,5-Dimethyl-2,6-diphenyl-1,5-dihyrodipyrrolo[3,2-b:3′,2′-e]pyrazine (11f). Mp: 258–260 °C, 1H NMR (500 MHz, CDCl3) δ 7.67–7.63 (m, 2H), 7.55 (t, J = 7.5 Hz, 2H), 7.48 (t, J = 7.4 Hz, 1H), 6.80 (s, 1H), 3.99 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 144.7, 141.8, 134.9, 132.2, 129.1, 128.7, 128.5, 99.3, 30.1; LC-MS (ESI): m/z = 339.2 [M + H]+. HRMS (ESI) m/z: [M + H]+ calculated for C22H18N4, 339.1604; found 339.1610.
2-(4-Methoxyphenyl)-1,5-dimethyl-6-phenyl-1,5-dihydrodipyrrolo[3,2-b:3′,2′-e]pyrazine (11g). Mp: 228–230 °C. 1H NMR (500 MHz, CDCl3) δ 7.65 (d, J = 7.2 Hz, 2H), 7.60–7.52 (m, 4H), 7.47 (t, J = 7.4 Hz, 1H), 7.07 (d, J = 8.6 Hz, 2H), 6.79 (s, 1H), 6.73 (s, 1H), 3.98 (s, 3H), 3.96 (s, 3H), 3.91 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 159.9, 144.7, 144.3, 141.7, 135.1, 134.5, 132.3, 130.4, 129.1, 128.7, 128.4, 124.6, 114.7, 114.2, 99.3, 98.6, 55.4, 30.1, 30.0. LC-MS (ESI): m/z = 369.2 [M + H]+. HRMS (ESI) m/z: [M + H]+ calculated for C23H20N5O, 369.1710; found 369.1717.
1,5-Dimethyl-2-phenyl-6-p-tolyl-1,5-dihydrodipyrrolo[3,2-b:3′,2′-e]pyrazine (11h). Mp: 204–206 °C. 1H NMR (500 MHz, CDCl3) δ 7.67–7.64 (m, 2H), 7.56–7.53 (m, 4H), 7.48 (d, J = 7.4 Hz, 1H), 7.36 (d, J = 7.9 Hz, 2H), 6.79 (s, 1H), 6.76 (s, 1H), 3.99 (s, 3H), 3.98 (s, 3H), 2.48 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 144.9, 144.4, 141.8, 141.8, 138.6, 135.0, 134.7, 132.3, 129.4, 129.3, 129.1, 129.0, 128.7, 128.5, 99.3, 98.9, 30.15, 30.1, 21.3. LC-MS (ESI): m/z = 353.3 [M + H]+. HRMS (ESI) m/z: [M + H]+ calculated for C23H20N4, 353.1761; found 353.1774.
4-(1,5-Dimethyl-6-phenyl-1,5-dihydrodipyrrolo[3,2-b:3′,2′-e]pyrazin-2-yl)-N,N-dimethylaniline (11i). Mp: 219–220 °C. 1H NMR (500 MHz, CDCl3) δ 7.65 (d, J = 7.2 Hz, 2H), 7.54 (dd, J = 8.2, 4.9 Hz, 4H), 7.47 (d, J = 7.4 Hz, 1H), 6.87 (d, J = 8.7 Hz, 2H), 6.79 (s, 1H), 6.70 (s, 1H), 3.98 (s, 6H), 3.08 (s, 6H). 13C NMR (126 MHz, CDCl3) δ 150.4, 145.9, 143.7, 141.9, 141.7, 135.5, 134.1, 132.4, 130.0, 129.1, 128.7, 128.3, 119.7, 112.1, 99.3, 97.7, 40.3, 30.1, 30.1. LC-MS (ESI): m/z = 382.3 [M + H]+. HRMS (ESI) m/z: [M + H]+ calculated for C24H23N5, 382.2026; found 382.2035.
2-(3-Fluorophenyl)-1,5-dimethyl-6-phenyl-1,5-dihydrodipyrrolo[3,2-b:3′,2′-e]pyrazine (11j). Mp: 234–236 °C. 1H NMR (500 MHz, CDCl3) δ 7.67–7.64 (m, 2H), 7.55 (t, J = 7.5 Hz, 2H), 7.53–7.47 (m, 2H), 7.44 (d, J = 7.7 Hz, 1H), 7.36 (dd, J = 9.4, 1.9 Hz, 1H), 7.18 (d, J = 1.7 Hz, 1H), 6.81 (s, 1H), 6.80 (s, 1H), 4.00 (s, 3H), 3.99 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 162.7 (d, J = 246.8 Hz), 145.1, 143.0 (d, J = 2.4 Hz), 141.9 (d, J = 10.2 Hz), 135.3, 134.5, 134.3 (d, J = 8.2 Hz), 132.1, 130.3, 130.3, 129.1, 128.7, 128.6, 124.8 (d, J = 2.9 Hz), 115.9 (d, J = 22.4 Hz), 115.4 (d, J = 21.1 Hz), 99.9, 99.3, 30.2, 30.1. LC-MS (ESI): m/z = 357.3 [M + H]+. HRMS (ESI) m/z: [M + H]+ calculated for C22H17FN4, 357.1510; found 357.1527.
2-Cyclopropyl-1,5-dimethyl-6-phenyl-1,5-dihydrodipyrrolo[3,2-b:3′,2′-e]pyrazine (11k). Mp: 120–121 °C. 1H NMR (500 MHz, CDCl3) δ 7.62–7.58 (m, 2H), 7.49 (dd, J = 10.7, 4.2 Hz, 2H), 7.42 (td, J = 7.1, 1.1 Hz, 1H), 6.74 (s, 1H), 6.26 (s, 1H), 3.97 (d, J = 1.3 Hz, 3H), 3.93 (d, J = 0.8 Hz, 3H), 2.03–1.96 (m, 1H), 1.09 (d, J = 8.2 Hz, 2H), 0.86 (d, J = 5.1 Hz, 2H). 13C NMR (126 MHz, CDCl3) δ 147.6, 143.5, 141.4, 140.9, 135.0, 133.8, 132.4, 129.1, 128.6, 128.2, 99.2, 94.6, 30.0, 28.2, 8.2, 7.1. LC-MS (ESI): m/z = 303.3 [M + H]+. HRMS (ESI) m/z: [M + H]+ calculated for C19H18N4, 303.1604; found 303.1620.
3-Chloro-5-methyl-2-(phenylethynyl)-5H-pyrrolo[2,3-b]pyrazine (12b). The procedure described above for 5a was used. The titled compound was obtained as pale yellow solid with 72% yield. LC-MS (ESI): m/z = 344.1 [M + H]+. HRMS (ESI) m/z: [M + H]+ calculated for C21H14ClN4, 344.0949; found 344.0954.
4.7 General procedure for preparing pyrrolothienopyrazine
To a solution of 3-chloro-5-methyl-2-(phenylethynyl)-5H-pyrrolo[2,3-b]pyrazine (0.12 g 0.4 mmol) in DMF (4 mL) was added Na2S·5H2O (0.14 g, 1.2 mmol). The mixture was refluxed for 1 h, cooled to ambient temperature then diluted with EtOAc and water. The organic layer was separated, dried over MgSO4, and concentrated in vacuum to give a residue that was subjected to silica gel column chromatography to give a desired compound.
7-Methyl-2-phenyl-7H-pyrrolo[3,2-e]thieno[2,3-b]pyrazine (13a). Yield: 74%, light brown solid. Mp: 196–197 °C, 1H NMR (500 MHz, CDCl3) δ 7.82–7.78 (m, 2H), 7.75 (s, 1H), 7.52 (d, J = 3.6 Hz, 1H), 7.49 (t, J = 7.6 Hz, 2H), 7.42 (d, J = 7.3 Hz, 1H), 6.74 (d, J = 3.6 Hz, 1H), 3.97 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 149.9, 146.3, 144.1, 139.4, 138.9, 134.1, 133.9, 129.1, 128.9, 126.3, 117.8, 100.3, 31.6. LC-MS (ESI): m/z = 266.1 [M + H]+. HRMS (ESI) m/z: [M + H]+ calculated for C15H11N4S, 266.0746; found 266.0751.
7-Methyl-2,6-phenyl-7H-pyrrolo[3,2-e]thieno[2,3-b]pyrazine (13b). Yield: 69%, light brown solid. Mp: 194–196 °C, 1H NMR (500 MHz, CDCl3) δ 7.78 (d, J = 7.5 Hz, 2H), 7.74 (s, 1H), 7.63–7.60 (m, 2H), 7.54 (t, J = 7.3 Hz, 2H), 7.50–7.45 (m, 3H), 7.39 (t, J = 7.4 Hz, 1H), 6.78 (s, 1H), 3.93 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 149.5, 146.9, 146.7, 143.9, 141.2, 139.1, 134.1, 131.4, 129.1, 129.1, 129.0, 128.8, 128.8, 126.2, 117.8, 100.1, 30.0. LC-MS (ESI): m/z = 342.2 [M + H]+. HRMS (ESI) m/z: [M + H]+ calculated for C21H16N4S, 342.1059; found 342.1068.
5-Bromo-3,6-dichloropyrazine-2-amine (15). Yield: 90%, pale yellow solid. Mp: 129–130 °C, 1H NMR (500 MHz, CDCl3) δ 5.24 (s, 2H). 13C NMR (126 MHz, CDCl3) δ 149.7, 145.5, 129.4, 121.5. LC-MS (ESI): m/z = 339.9 [M − 1]−.
2,5-Dibromo-3,6-dichloropyrazine (16). To a solution of 5-bromo-3,6-dichloropyrazin-2-amine (0.1 g, 0.4 mmol) in THF (1 mL), HBr (2 mL) at 0 °C was added NaNO2 (0.07 g, 1.0 mmol) slowly. The mixture was stirred at 0 °C for 20 min and then made basic by using 1 M NaOH and extracted with EtOAc (3×). The combined organic layers were dried over MgSO4, and concentrated to give a residue that was subjected to silica gel column chromatography to give desired compound as pale a yellow solid. Yield: 77%, mp: 93–94 °C, 13C NMR (126 MHz, CDCl3) δ 146.6, 136.3.
2,5-Dichloro-3,6-bis(phenylethynyl)pyrazine (17). To a stirred solution of 2,5-dibromo-3,6-dichloropyrazine (0.6 g, 1.97 mmol) in THF (10 mL), then TEA (1.3 mL, 13 mmol), Pd(PPh3)2Cl2 (0.27 g, 0.34 mmol) and CuI (75 mg, 0.34 mmol) were added subsequently under nitrogen atmosphere. After 10 min, phenyl acetylene (0.42 g, 4 mmol) was added drop wise and the mixture was stirred at room temperature for 1.5 h. The mixture was concentrated in a vacuum to give a residue that was diluted with water and extracted with EtOAc (2×). The combined organic layer was washed with water and dried over MgSO4, and concentrated in vacuum to give a residue that was subjected to silica gel column chromatography to yield the desired compound as pale yellow solid. Yield 38%, 1H NMR (500 MHz, CDCl3) δ 7.66 (d, J = 7.0 Hz, 4H), 7.43 (dt, J = 14.5, 7.1 Hz, 6H). 13C NMR (126 MHz, CDCl3) δ 147.5, 135.8, 132.4, 130.4, 128.6, 120.9, 100.3, 84.3. LC-MS (ESI): m/z = 349.0 [M + H]+.
5. Conclusions
In the study described above, we developed a convenient method for the synthesis of 1,5- and 1,7-dihydrodipyrrolo[2,3-b;3′2′-e]pyrazine derivatives. Protocols were devised to generate either 2-amino- or 3-amino-pyrrolopyrazines from the corresponding 2-bromo-3-chloro-5H-pyrrolo[2,3-b]pyrazines. Specifically, amination reactions of the dihalo substrates carried under metal free conditions in the presence of methylamine under MW irradiation produce 3-amino-pyrrolopyrazine exclusively. In contrast, Buchwald cross coupling affords 2-amino-pyrrolopyrazine. The pyrrolo pyrazine scaffolds were converted to the respective 1,7- and 1,5-DPP derivatives using the Sonogashira coupling reactions. All the synthesized compounds were found to be highly crystalline and readily soluble in organic solvents. Furthermore, the prepared pyrazine derivatives have high thermal stabilities, suggesting that they have high potential for use in organic electronic devices. Ongoing research in our laboratory is aimed at exploring interesting features of the 1,5-dihydrodipyrrolo[3,2-b:3′,2′-e]pyrazine molecular scaffold.
Acknowledgements
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (No. 2016R1D1A1B04932654), and Sabbatical Research Program of the Dongguk University in Seoul, Korea.
References
- J. Zhang, J. Wang, X. Xu, S. Chen, Q. Zhang, C. Yao, X. Zhuang, A. Pan and L. Li, J. Mater. Chem. C, 2015, 3, 5933–5939 RSC.
- Z. Liang, Q. Tang, R. Mao, D. Liu, J. Xu and Q. Miao, Adv. Mater., 2011, 23, 5514–5518 CrossRef CAS PubMed.
- K. Liu, C.-L. Song, Y.-C. Zhou, X.-Y. Zhou, X.-J. Pan, L.-Y. Cao, C. Zhang, Y. Liu, X. Gong and H.-L. Zhang, J. Mater. Chem. C, 2015, 3, 4188–4196 RSC.
- Y.-I. Park, J.-H. Son, J.-S. Kang, S.-K. Kim, J.-H. Lee and J.-W. Park, Chem. Commun., 2008, 2143–2145, 10.1039/b718873k.
- L. Yan, D. Zhao, J. Lan, Y. Cheng, Q. Guo, X. Li, N. Wu and J. You, Org. Biomol. Chem., 2013, 11, 7966–7977 CAS.
- P. Henriksson, C. Lindqvist, B. Abdisa, E. Wang, Z. George, R. Kroon, C. Müller, T. Yohannes, O. Inganäs and M. R. Andersson, Sol. Energy Mater. Sol. Cells, 2014, 130, 138–143 CrossRef CAS.
- X. Lu, G. Zhou, H. Wang, Q. Feng and Z.-S. Wang, Phys. Chem. Chem. Phys., 2012, 14, 4802–4809 RSC.
- C. R. Hopkins and N. Collar, Tetrahedron Lett., 2004, 45, 8087–8090 CrossRef CAS.
- C. R. Hopkins and N. Collar, Tetrahedron Lett., 2004, 45, 8631–8633 CrossRef CAS.
- I. Simpson, S. A. St-Gallay, S. Stokes, D. T. E. Whittaker and R. Wiewiora, Tetrahedron Lett., 2015, 56, 1492–1495 CrossRef CAS.
- A. Keivanloo, M. Bakherad, H. Nasr-Isfahani and S. Esmaily, Tetrahedron Lett., 2012, 53, 3126–3130 CrossRef CAS.
- G. Bashiardes, I. Safir, A. S. Mohamed, F. Barbot and J. Laduranty, Org. Lett., 2003, 5, 4915–4918 CrossRef CAS PubMed.
- G. Xu and Y.-G. Wang, Org. Lett., 2004, 6, 985–987 CrossRef CAS PubMed.
- D. R. Sauer, D. Kalvin and K. M. Phelan, Org. Lett., 2003, 5, 4721–4724 CrossRef CAS PubMed.
- L. Shi, M. Wang, C.-A. Fan, F.-M. Zhang and Y.-Q. Tu, Org. Lett., 2003, 5, 3515–3517 CrossRef CAS PubMed.
- F. Y. Kwong and S. L. Buchwald, Org. Lett., 2003, 5, 793–796 CrossRef CAS PubMed.
- C. Bathula, C. E. Song, S. Badgujar, S.-J. Hong, I.-N. Kang, S.-J. Moon, J. Lee, S. Cho, H.-K. Shim and S. K. Lee, J. Mater. Chem., 2012, 22, 22224–22232 RSC.
- E. Zhu, G. Ge, J. Shu, M. Yi, L. Bian, J. Hai, J. Yu, Y. Liu, J. Zhou and W. Tang, J. Mater. Chem. A, 2014, 2, 13580–13586 CAS.
- Q. Meng, L. Jiang, Z. Wei, C. Wang, H. Zhao, H. Li, W. Xu and W. Hu, J. Mater. Chem., 2010, 20, 10931–10935 RSC.
- H. Wang, Y. Xu, X. Yu, R. Xing, J. Liu and Y. Han, Polymers, 2013, 5, 1272 CrossRef.
- H. Dong, C. Wang and W. Hu, Chem. Commun., 2010, 46, 5211–5222 RSC.
- C. Bathula, M. Kim, C. E. Song, W. S. Shin, D.-H. Hwang, J.-C. Lee, I.-N. Kang, S. K. Lee and T. Park, Macromolecules, 2015, 48, 3509–3515 CrossRef CAS.
- Y. Geng, J. Wang, S. Wu, H. Li, F. Yu, G. Yang, H. Gao and Z. Su, J. Mater. Chem., 2011, 21, 134–143 RSC.
- Z. Liang, Q. Tang, J. Liu, J. Li, F. Yan and Q. Miao, Chem. Mater., 2010, 22, 6438–6443 CrossRef CAS.
- S. Van Epps, B. Fiamengo, J. Edmunds, A. Ericsson, K. Frank, M. Friedman, D. George, J. George, E. Goedken, B. Kotecki, G. Martinez, P. Merta, M. Morytko, S. Shekhar, B. Skinner, K. Stewart, J. Voss, G. Wallace, L. Wang, L. Wang and N. Wishart, Bioorg. Med. Chem. Lett., 2013, 23, 693–698 CrossRef CAS PubMed.
- S. K. Kolli, A. Nakhi, R. Medishetti, S. Yellanki, P. Kulkarni, R. Ramesh Raju and M. Pal, Bioorg. Med. Chem. Lett., 2014, 24, 4460–4465 CrossRef CAS PubMed.
- CCDC deposit number 1536706.
Footnote |
| † Electronic supplementary information (ESI) available: Single XRD data of 6f is available in CIF formate. CCDC 1536706. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra01795b |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *
*