
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
GLY67ARG substitution in RSPO4 disrupts the WNT signaling pathway due to an abnormal binding pattern with LGRs leading to anonychia†
Syed Irfan Razaac,
Abdul Khaliq Navide,
Zainab Noorb,
Khadim Shaha,
Nasser Rashid Dard,
Wasim Ahmada and
Sajid Rashid *b
*b
aDepartment of Biochemistry, Faculty of Biological Sciences, Quaid-i-Azam University, Islamabad, Pakistan
bNational Centre for Bioinformatics, Quaid-i-Azam University, Islamabad, 44000, Pakistan. E-mail: sajidrwp@yahoo.co.uk; Tel: +92-51-90644107
cDepartment of Biosciences, University of Wah, Wah, Pakistan
dDepartment of Dermatology, Combined Military Hospital, Rawalpindi, Pakistan
eIslamic Medical College, Riffah University, Islamabad, Pakistan
First published on 20th March 2017
Abstract
R-Spondins regulate the WNT/β-catenin signaling pathway by interacting with leucine rich-repeat containing G-protein coupled receptors (LGR4–6). These receptors share unique sequence and structural similarities with each other. Here, we report comparative binding analysis of R-Spondin-4 (RSPO4) with LGRs through structural characterization of a missense variant (GLY67ARG) identified in two consanguineous families of Pakistani origin. The modeled structure of RSPO4 comprises two contiguous Furin-like cysteine-rich domains that are involved in binding with LGRs. We observed an overall conservation of overlapping interacting residues among LGRs which recognized RSPO4 at two specific parallel positions (sites ‘a’ and ‘b’). The residual contributions of RSPO4 reconciled previously defined interactions of RSPO1 with LGRs. To check the comparative expression pattern of β-catenin, we quantified β-catenin levels in normal and anonychia patients. β-catenin level was significantly reduced in the patients exhibiting mutated RSPO4 as compared to control individuals. These findings confirm that RSPO4 modulates the LGR-dependent WNT/β-catenin signaling pathway and may have therapeutic potential in anonychia patients.
Introduction
R-Spondins are secretary proteins (RSPO1–4) that share approximately 60% of sequence homology and have a close resemblance in domain organization.1–3 Each of the four R-Spondins contains two Furin-like cysteine rich domains at the N-terminal, a trans-membrane domain and a basic C-terminal tail domain.1 Several studies confirmed the involvement of R-Spondins in WNT/β-catenin signaling.2,3 The key outcome of the WNT signaling activation is the stabilization of β-catenin1,4,5 and subsequent formation of nuclear β-catenin/T-cell factor dependent expression of WNT target genes. In the absence of WNT, β-catenin is subjected to multi-protein β-catenin destruction complex that comprises Axin, adenomatous polyposis coli (APC), and glycogen kinase 3 (GSK3). GSK3 constitutively phosphorylates β-catenin which is subsequently degraded through ubiquitin-dependent pathway.6,7WNT signaling pathway is crucial for various biological processes like embryonic development, adult homeostasis and diverse pathogenesis.8–10 R-Spondins (RSPO1–4) regulate WNT/β-catenin signaling by binding to leucine rich-repeat containing G protein-coupled receptor superfamily (LGR4–6).11–14 LGRs are unique in having a large N-terminal extracellular domain (ECD) which contains 17 leucine-rich repeats flanked by cysteine-rich sequences at both N- and C-termini. On the basis of expressions in distinct types of adult tissues including stem cells,15 intestine, colon,16 hair follicles,17 stomach,15 kidney,18 liver19 and mammary glands,20 LGRs are considered as key components of WNT signaling. The junction between ectodomain and first trans-membrane domain is highly conserved between LGR4 and LGR5.21 Several studies reported that WNT/β-catenin signaling is initiated through the phosphorylation of WNT co-receptors LRP5/6 (low-density lipoprotein receptor-related protein) at multiple sites.6,22 It can be speculated that activation of LGR4–6 by R-Spondins may possibly enhance the activity of one or multiple kinases that phosphorylate LRPs through an unknown mechanism.
Defects in WNT/β-catenin signaling have been associated with several disorders such as cancers, bone disorders, diabetes and neurodegenerative diseases.6 Multiple cases have been reported with faulty sex development and abrogated nail formation which are linked with abnormal RSPO1 and RSPO4.23–25 Despite diverse biological functions attributed to R-Spondins, the underlying exact mechanism of WNT activation and subsequent β-catenin stabilization remains elusive.
In the present study, screening of RSPO4 in two consanguineous Pakistani families led to the identification of two previously reported variants including 26 bp deletion (-9- +17del26) and a missense c.199G>C (p.GLY67ARG) mutation. Comparative modeling of GLY67ARG substituted 3D structure of RSPO4 and its interaction with LGR4–6 revealed useful clues to understand regulation of WNT signaling.
Methods
Human subjects
The present research study defines clinical and molecular analyses of two unrelated consanguineous Pakistani families, A and B, manifesting anonychia segregating in autosomal recessive manner. Prior to start of the study, approval was obtained from the Institution Review Board (IRB) of Quaid-i-Azam University Islamabad, Pakistan. In writing, consent was obtained from all the family members who were enrolled in the present study. Five affected (III-6, IV-3, IV-4, IV-5, IV-6) and six unaffected (III-1, III-4, III-5, III-7, IV-1, IV-2) members from family-A and three affected (IV-1, IV-2, IV-3) and four unaffected (III-2, III-3, IV-4, IV-5) members from family B (Fig. 1) participated in the study. Detailed clinical examination was performed at Department of Dermatology Combined Military Hospital (CMH) Rawalpindi, Pakistan. This is to certify that the experiments performed for the subject purpose were performed as per guidelines approved by Institutional Review Board of Quaid-I-Azam University. The study was reviewed and granted approval for implementation by the Institutional Review Board. It was assigned protocol # IRB-QAU-160. Written informed consents were obtained from human subjects participated in the study.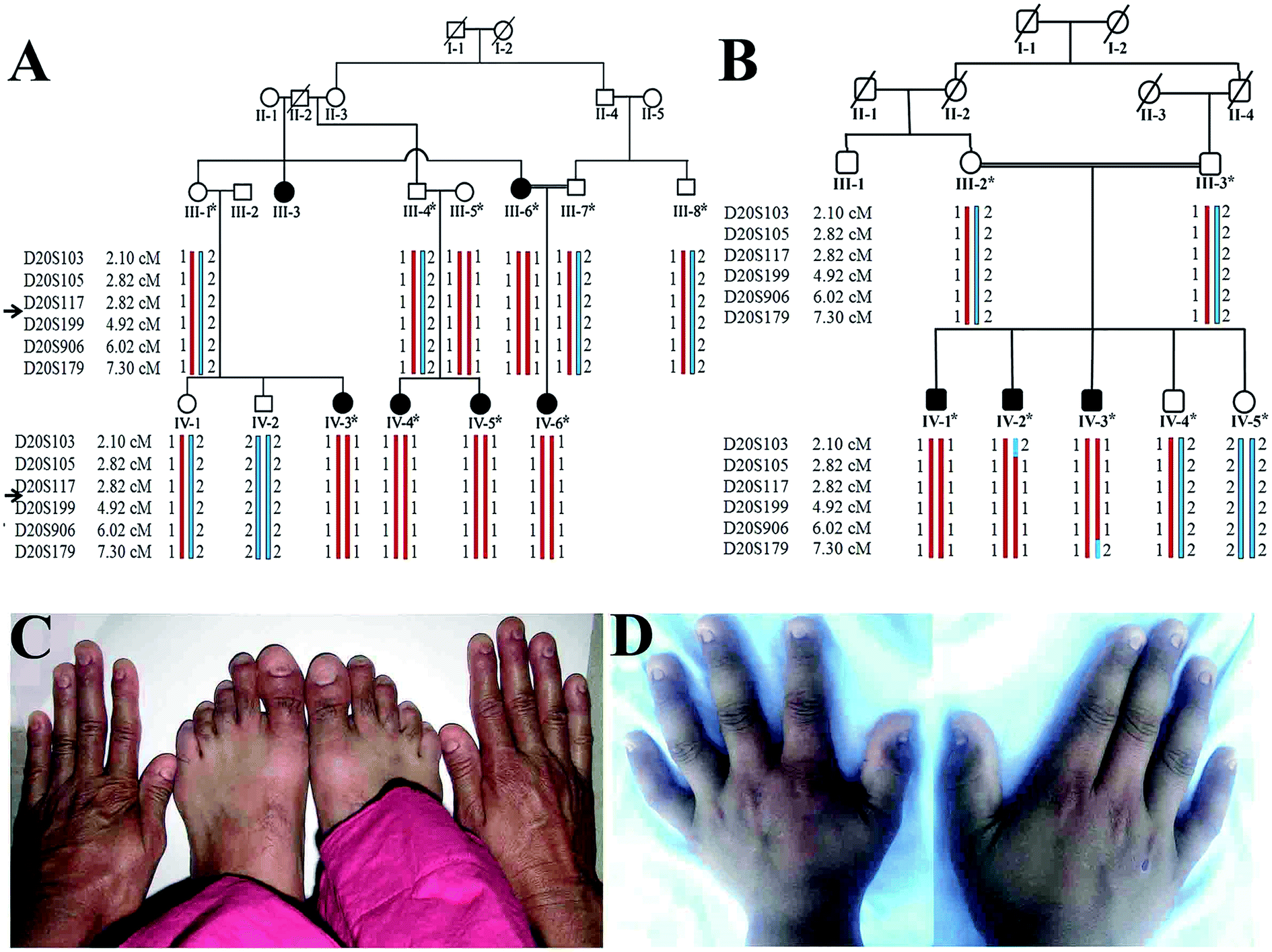 | ||
| Fig. 1 Pedigrees and clinical manifestations of the two families. (A & B) Pedigrees of consanguineous families exhibiting autosomal recessive anonychia. Double lines are indicative of consanguineous union. Clear symbols represent unaffected while filled symbols represent affected individuals. Symbols with bars represent deceased individuals. The individual numbers labeled with asterisks indicate the samples which were available for this study. (C & D) Clinical presentation of autosomal recessive anonychia congenita. Photograph of both hands of affected individuals (III-6 and IV-3) of families A and B, respectively. | ||
Collection of biological samples and genotyping
Peripheral blood samples (3–5 ml) from available affected and unaffected members in each of the two families were collected in EDTA-containing tubes. Genomic DNA was extracted using GenElute Blood Genomic DNA Kit (Sigma-Aldrich, MO, USA). The extracted genomic DNA was quantified by Nano-drop-1000 (Thermo Scientific Wilmington, DE, USA).Nail biopsy
To perform β-catenin expression analysis, biopsy samples of complete proximal nail fold along with eponychium area of toe thumb were collected from a female patient (IV-3, Family A) using localized anesthesia at surgical unit of Department of Dermatology CMH Rawalpindi, Pakistan. The biopsy sample was immediately frozen in liquid nitrogen and stored at −80 °C at the Centre of Excellence Laboratory, Army Medical College Rawalpindi, Pakistan.Genotyping and sequencing of RSPO4
Based on clinical features in affected members in the families (A and B), genotyping was performed using microsatellite markers (D20S103, D20S105, D20S117, D20S199, D20S906, D20S179) mapped in the flanking regions of the gene RSPO4 on chromosome 20p.13 All the selected microsatellite markers were PCR amplified in a total volume of 25 μl in a 0.2 ml PCR tube according to standard procedure.26 The PCR was performed in T3000 thermocycler (Biometra, Goettingen, Germany) and PCR products were resolved on 8% non-denaturing polyacrylamide gel. Gels were stained with ethidium bromide and genotypes were assigned by visual inspection. Haplotype analysis established linkage in both the families to the tested RSPO4 gene.Exons and exon–intron boundaries of the RSPO4 gene were PCR amplified using DNA from available affected and unaffected members of the two families. The amplified PCR products were purified with commercially available kits (Axygen Biosciences Central Avenue, Union City, USA). Bidirectional DNA sequencing was performed using DTCS Quick Start sequencing kit (Beckman Coulter, Fullerton, CA, USA) according to the manufacturer's instructions. Sequence variants were identified via BIOEDIT sequence alignment editor, version 6.0.7.
Western blotting
For analysis of expression of β-catenin, total protein was extracted from nail biopsies of anonychia patients and control individuals. Samples were subjected to SDS-PAGE and transferred to the nitrocellulose membrane, as described elsewhere.27 Briefly, after blocking with nonfat milk, membrane was incubated for 16–20 hours at 4 °C with primary antibody (Rabbit monoclonal anti β-catenin) with a diluting ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1000 (Abcam, Cambridge, MA, USA) and then treated with AP-conjugated goat anti Rabbit IgG (Abcam, Cambridge, MA, USA) for 1 hour at room temperature. The membrane was treated with NBT/BCIP solution (Sigma-Aldrich, MO, USA) for signal development.
:1000 (Abcam, Cambridge, MA, USA) and then treated with AP-conjugated goat anti Rabbit IgG (Abcam, Cambridge, MA, USA) for 1 hour at room temperature. The membrane was treated with NBT/BCIP solution (Sigma-Aldrich, MO, USA) for signal development.
Bioinformatics analysis
Interaction patterns of group B LGRs with wild-type and mutated (GLY67ARG) RSPO4 (mRSPO4) were investigated. High-resolution co-crystallized structures of LGR4 and LGR5 were retrieved through Protein Databank28 with PDB IDs: 4KT1 and 4BSR, respectively. Using sequencing information, tertiary structures of the RSPO4 and LGR6 were generated through comparative modeling approach.29 Primary protein sequences for RSPO4 (ID: ENSP00000217260) and LGR6 (ID: ENSP00000356247) were retrieved through Ensemble genome browser30 and subjected to BLAST31 search against protein data bank28 to find a suitable template. MODELLER 9V8 tool32 was used to predict RSPO4 (normal and mutated) and LGR6 structures through 4KNG-chain A (LGR5) and 4KNG-chain M (RSPO1) as templates, respectively. Templates were chosen on the basis of high sequence identity and query coverage values directed from NCBI31 (Table 1). 3D structures of RSPO4 and LGR6 were energy minimized using VEGA ZZ release 2.0.8 (ref. 33) and refined by adding charges and missing atoms using KoBaMIN server34 and UCSF Chimera35 followed by verification through MolProbity,36 RAMPAGE,37 ERRAT38 and Verify3D39 and ProSA-web40 tools. Root mean square deviation (RMSD) values of predicted proteins were also computed by comparing them with template structures using UCSF Chimera.35 KoBaMIN server34 performs structure refinement through minimization of a knowledge-based potential of mean force, derived from PDB structure. ERRAT evaluates structure by computing non-bonded interactions between carbon, nitrogen and oxygen atoms. The percentage of residues not included within 95 percentile is provided as the overall quality.38 Verify3D computes model quality through 3D–1D profiling of each residue in a 21-residue sliding window. The propensity of each amino acid to exist in each structural environment class (alpha, beta, loop, polar and non-polar) is statistically evaluated through known PDB structures, and the final score given to the protein structure is the sum of propensities of the individual residues.39 The Z-score evaluates model quality by comparing it with the Z-score values of known crystalized protein structures. The Z-score value must be within the range of scores found for protein chains of similar sizes.40| Template | Target-template identity (%) | Query coverage (%) | Template resolution (Å) | Target-template 3D structure RMSD (Å) | |
|---|---|---|---|---|---|
| LGR6 | 4KNG-chain A | 98 | 66 | 2.50 | 0.7 |
| RSPO4 | 4KNG-chain M | 52 | 95 | 2.80 | 0.4 |
Interaction analysis
To monitor the comparative interactions of normal and mutated RSPO4 with LGR4–6, binding site analysis was performed on the basis of known complexes of LGRs and RSPO1.41 Both normal and mutated RSPO4 structures were docked against all three LGR receptors (LGR4–6), respectively. Molecular docking analysis was performed by HADDOCK server with active and passive receptor binding residues as input parameters.42 These parameters indicate the potential binding regions of receptor which were analyzed through literature.41 HADDOCK score is weighted as sum of the following four terms: Electrostatic energy (weight 0.2), Van der Waals energy (weight 1.0), Desolvation energy (weight 1.0) and Restraints violation energies (weight 0.1).42 The more negative HADDOCK score indicates a reliable interaction. HADDDOCK derived Z-score is the quantitative measure of cluster standard from the average score, more negative value of Z-score denotes more significant cluster. HADDOCK RMSD values demonstrate the statistical significance of the top scored clusters, computed on the basis of interface and ligand RMSDs with respect to the best water refined model. RMSD is calculated by fitting the backbone atoms (CA, C, H, O, N) of the receptor protein and reference complexes followed by computing the RMSD of ligand–protein residues.Results
Clinical findings
Available affected members in the two families (A and B) were examined at Government Hospital Punjab Pakistan. At the time of the study in the family A, youngest affected sibling (IV-6) was 3.5 years old while the eldest member (III-3) was 55 years old (Fig. 1). Males and females were equally affected. The detailed clinical report was provided by an expert Dermatologist serving Department of Dermatology, Combined Military Hospital (CMH) Rawalpindi Pakistan. Anonychia representing complete absence of finger- and toe-nails was observed in all affected members of both the families. Nail bed was normal in appearance and structure; however, nail matrix was swollen (Fig. 1). Teeth and sweat glands were normal in the affected members of both the families. Skin disorders including ichthyosis, palmar keratosis, hyperhidrosis and any type of pigmentation were not observed in the family members. Results of routine laboratory tests including white blood cell count and granulocyte function were normal. Limb and girdle analyses failed to reveal any abnormality. The heterozygous carriers showed normal nail phenotype indistinguishable from healthy individuals.Genotyping and sequencing analysis
Based on disease phenotype observed in the affected members in two families, linkage was tested by genotyping microsatellite markers (D20S117, D20S199, D20S906) mapped in the vicinity of RSPO4 located on chromosome 20p13. Exons and exon–intron boundaries of the RSPO4 were PCR amplified and sequenced in all available affected and unaffected members of families. DNA sequence analysis of RSPO4 was performed using a control reference sequence obtained through the Ensemble database30 (Ensemble accession ID: ENSG00000139679).Sequence analysis revealed a previously reported 26 bp deletion (-9- +17del26) including start codon (ATG) in exon 1 in the family A, and a known missense variant (c.199G>C, p.GLY67ARG) in family B. Both the variants were present in the heterozygous state in obligate carriers of the family. Polymorphic nature of both deletion and missense variants was excluded by screening 100 ethnically matched control individuals.
β-catenin expression studies
Western blot analysis revealed significantly reduced levels of β-catenin in anonychia patient samples compared to control individuals (Fig. 2). Monoclonal anti β-tubulin was used to monitor loading samples. The data suggested that p.GLY67ARG substitution in RSPO4 resulted in abrogated WNT signaling thereby affecting β-catenin expression.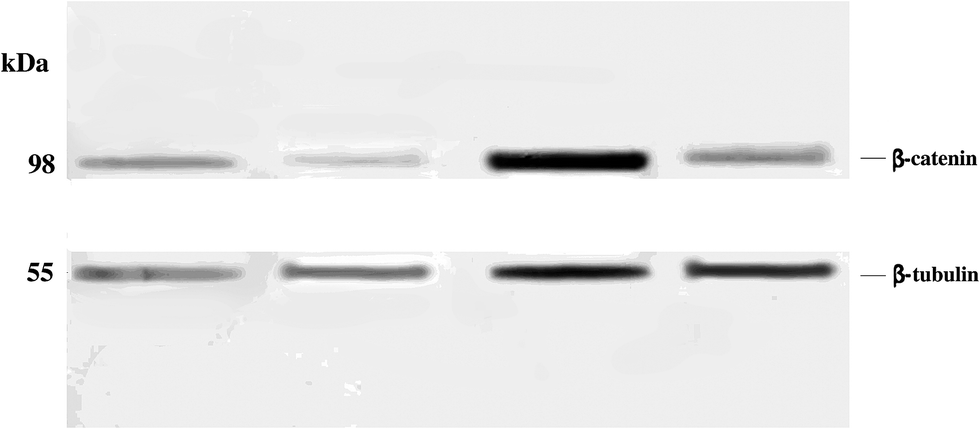 | ||
| Fig. 2 Western blot to detect β-catenin expression. Protein samples isolated from the nail biopsies of normal individuals (lanes 1 and 3) and anonychia patients (lanes 2 and 4) were resolved by 10% SDS PAGE. 5 μl protein samples were loaded in the first two lanes, while in the 3rd and 4th lanes, 12 μl protein samples was loaded in each lane. The filter was probed with anti-β-catenin and anti-β-tubulin antibodies. | ||
Structural characterization
LGR6 shares high sequence and structural similarity with other group B coupled receptors (LGR4, LGR5) by comprising an extracellular domain (ECD). The 456 amino acid long (Pro28–GLY483) solenoid-like structure encompasses 18 Leucine-Rich Repeats (LRRs) accompanying cysteine-rich N-terminal (LRRNT) and C-terminal (LRRCT) flanking regions (Fig. 3a). LRRs together with N–C termini generate 20 β-strands and 4 α-helices. R-Spondin comprises of two Furin-like Cysteine-Rich Domains (Fu-CRDs), which are involved in binding to LGRs (Fig. 3b). Moreover, 100 amino acid (GLY32–ASN131) long domain of RSPO4 with six β-strands (β1–β6) adopted an elongated architecture and formed adjacent β-hairpins (β1–β2, β4–β3, β5–β6) (Fig. 3b). Such structural composition has been shown to be conserved among R-Spondin family (RSPO1–4) members due to their high sequence similarity. | ||
| Fig. 3 Structural analysis of LGR6. (a) 3D structure of LGR6. LRRs are shown in green and N–C termini are shown in brown. (b) Structure of RSPO4. Furin-like domains 1 and 2 are represented in purple and blue colors, respectively. | ||
In Ramachandran plots, 93%, 97% and 95% non-glycine residues of normal RSPO4, mutated RSPO4 and LGR6 structures were lying in the most favored regions (core areas), respectively (Table 2). The average fraction of outliers and residual distribution in the generously allowed region, for all structures, are indicated in Table 2. The Verify3D analysis suggested that 92.76%, 81% and 97% residues had an average 3D–1D score higher than 0.2 for normal RSPO4, mutated RSPO4 and LGR6 structures, respectively. For predicted models used in this study, Z-scores were also within the range of scores calculated from experimentally known structures, which is 0 to −5 for all three structures (Fig. S1†). Overall, the evaluation data indicated that predicted models with ERRAT values of 73%, 88% and 76% are of good quality for further analysis (Table 2; Fig. S2†).
| Core (%) | Allowed (%) | Disallowed (%) | Bad backbone bonds (%) | Bad backbone angles (%) | Z-Score | Poor rotamers (%) | ERRAT Score | Verify3D% | |
|---|---|---|---|---|---|---|---|---|---|
| LGR6 | 95 | 4 | 1 | <1 | <2 | −5 | <1 | 73 | 92.76 |
| RSPO4 (normal) | 93 | 6 | 1 | <0.5 | <0.5 | −4.21 | 0 | 88 | 81 |
| RSPO4 (mutated) | 97 | 2 | 1 | <1 | <1 | −4.53 | 0 | 76 | 97 |
| R-Spondin-4 | Site | Binding residues | |||||
|---|---|---|---|---|---|---|---|
| LGR4 | RSPO4 | LGR5 | RSPO4 | LGR6 | RSPO4 | ||
| Normal R-Spondin | A | Asn114 | Arg81 | Asn123 | Arg81 | Asn123 | Arg81 |
| Asp161 | Ser100 | Gln125 | Gln83 | Gln125 | Gln83 | ||
| Asp162 | Lys89 | Asp170 | Ser100 | Arg144 | Cys104 | ||
| Asn210 | Arg60 | Asp171 | Lys89 | Asp170 | Ser100 | ||
| Tyr234 | Lys90 | Tyr243 | Arg60 | Asp171 | Arg87 | ||
| Tyr243 | Gln101 | ||||||
| B | Thr229 | Lys115 | Gln141 | Tyr112 | Gln189 | Tyr114 | |
| Glu252 | Lys117 | Glu237 | Lys115 | Glu237 | Lys115 | ||
| Thr238 | Lys117 | Glu261 | Lys117 | ||||
| Glu261 | Lys117 | ||||||
| Mutated R-Spondin | A | Asp162 | Arg87 | Asn123 | Arg87 | Asn123 | Glu84 |
| Asn210 | Lys89 | Asp171 | Gln83 | Gln125 | Glu84 | ||
| Asp281 | Ile63 | Arg144 | Ser100 | ||||
| Asp281 | Gln65 | Asp170 | Lys89 | ||||
| Asp171 | Arg87 | ||||||
| Tyr243 | Arg60 | ||||||
| B | Thr229 | Lys115 | Gln141 | Tyr112 | Gln189 | Tyr114 | |
| Glu252 | Lys117 | Glu237 | Lys115 | Glu237 | Lys115 | ||
| Thr238 | Lys117 | Glu261 | Lys117 | ||||
| Glu261 | Lys117 | ||||||
Molecular docking studies
The details of docking scores, RMSD, the number of clusters and Z-scores for LGR4–6 and RSPO4 complexes are given in Table S1.†RSPO4–LGR4/5/6
LGRs exhibited a similar binding pattern for RSPO4.41 A detailed comparative binding overview of RSPO4 with LGR4–6 depicted multiple overlapping residues at the interaction sites of LGRs (Fig. S3†). These residues were found to be conserved among all group B LGRs and associated with RSPO4 at two parallel positions (site ‘a’ and ‘b’) (Fig. S3†). Site ‘a’ specific residues were more frequently involved in the formation of complexes as compared to site ‘b’. Docking studies demonstrated LGR binding at Furin-like domains (Fu-CRD) of RSPO4, revealing the importance of these domains in interactions. At site ‘a’, LGR specific N-terminal loop residues exhibited binding to the RSPO4 specific N-terminal β-hairpin. At site ‘b’, LGR specific N-terminal β-sheets were binding with C-terminal β-hairpin of RSPO4 (Fig. S3†).In LGR4–RSPO4 complex, at site ‘a’, ASN114, ASP161, ASP162, ASN210 and TYR234 of LGR4 formed hydrogen bonds with ARG81, SER100, LYS89, ARG60 and LYS90 of RSPO4, respectively (Fig. S3a†). Site ‘b’ exhibited binding of THR229 and GLU252 of LGR4 with LYS115 and LYS117 of RSPO4, respectively (Fig. S3b†).
Hydrogen bonds involved in the formation of site ‘a’ of LGR5–RSPO4 complex included ASN123, GLN125, ASP170, ASP171 and TYR243 of LGR5 with ARG81, GLN83, SER100, LYS89 and ARG60 residues of RSPO4 (Fig. S3a†). At site ‘b’, GLN141, GLU237, THR238 and GLU261 residues of LGR5 were involved in hydrogen bonding with TYR112, LYS115 and LYS117 of RSPO4, respectively (Fig. S3b†).
Subsequently, in LGR6–RSPO4 complex, LGR6 specific residues of site ‘a’ involved in bonding were ASN123, GLN125, ARG144, ASP170, ASP171 and TYR243 with ARG81, GLN83, CYS104, SER100, ARG87 and GLN101 of RSPO4, respectively (Fig. S3a†). Site ‘b’ was established through bonds between GLN189, GLU237, GLU261 of LGR6 and TYR114, LYS115, LYS117 of RSPO4, respectively (Fig. S3b†). The detailed residual contribution for RSPO4 and LGRs is described in Table 3.
mRSPO4–LGR4/5/6
Docking analysis of mRSPO4 and LGRs showed multiple structural transitions in the vicinity of ARG67 at site ‘a’ as it is located in β1-sheet of β1–β2 hairpin (Fig. S4†), while site ‘b’ remained unaffected and showed similar results as observed in normal RSPO4. Minor binding pose transformations were detected in mutated RSPO4 complexes with LGRs. Particularly, at site ‘a’ of LGR, binding alterations were more prominent.In LGR4–mRSPO4 (Fig. S4,† top), three hydrogen bonds were detected between ASP162, ASN210 and ASP281 of LGR4 and ARG87, LYS89 and ILE63/GLN65 of mRSPO4, respectively (Table 3). In LGR5–mRSPO4 complex, ASN123 and ASP171 of LGR5 showed binding to the GLN83 and ARG87 of mRSPO4, respectively (Fig. S4,† middle). Similarly, in LGR6–mRSPO4 complex, ASN123/GLN125, ARG144, ASP170, ASP171 and TYR243 of LGR6 formed hydrogen bonds with GLU84, SER100, LYS89, ARG87 and ARG60 of mRSPO4, respectively (Fig. S4,† bottom). Thus, GLY67ARG substitution in Fu-CRD domain 1 of RSPO4 resulted in significant binding alterations with LGRs (Fig. S4†).
Discussion
Isolated congenital anonychia with total absence of finger and toe-nails segregates in both autosomal recessive and dominant modes. Since, Blaydon et al.25 mapped first case of anonychia on human chromosome 20p13, several disease-causing variants have been reported in the RSPO4 gene.25,43–46 The mammalian family of R-Spondins includes four independent gene products (RSPO1–4) that share 40–60% sequence similarity.1 R-Spondins potentiate WNT/β-catenin pathway through orphan receptors LGR4, LGR5 and LGR6.11–14 Clinical features exhibited by the patients in the present study are coherent to those reported previously. Mutation analysis in the present study reported a 26 bp deletion (-9-+17del26) and a missense variant (c.199G>C, p.GLY67ARG) in two separate families. In order to evaluate the effect of mutated RSPO4 on WNT/β-catenin pathway, in silico analysis was performed to delineate a comparative binding pattern of RSPO4 with leucine-rich orphan receptors (LGR4–6).In this study, we have modeled 456 amino acid (PRO28–GLY483) long solenoid-like structure harboring 18 Leucine-Rich Repeats (LRRs) with flanking four cysteine-rich (CYS29, CYS33, CYS35, CYS43) N-terminal (LRRNT) domain and C-terminal (LRRCT) region. These cysteine residues are highly conserved in LGR4–6 and play crucial roles in maintaining LRRNT structure. Clinical studies have shown that amino acid variations in these cysteine residues completely disrupt RSPO4 function and result in anonychia in human.22,23 These findings substantiate the recently modeled LGR4–6 structures.41,47–50 LRRs together with N- and C-termini generate 20 β-strands and 4 α-helices which lie in the linking regions of β-sheets. In the modeled structure of RSPO4, two contiguous Furin-like Cysteine-Rich Domains (Fu-CRDs) are evident which are responsible for binding to LGRs. These Furin-like domains are highly conserved in the members of R-Spondin family.2,47,51
LGRs play important role in ZNRF3 (Zinc and Ring Finger 3) clearance by cross-bridge formation through R-Spondin. These observations are well-supported by the crystal structure of LGR5–RSPO1–RNF43, where LGR5 contact with RNF43 is mediated by RSPO1.48 These findings are in good agreement with our observations of RSPO4 and LGR4–6. The linking loops of R-Spondin β-hairpins (β1–β2, β4–β3, β5–β6) participate in interaction with LGRs. The present study reveals the binding of RSPO4 to the concaved N-terminal LRRs of LGRs at two adjacent positions, as evident in the crystallized LGR4–RSPO1 complex.41,47 At one site, residues of LGR N-terminal loop exhibited binding to N-terminal β-hairpin linking loops and β4-strand, while the other site of LGR showed binding to the C-terminal β-hairpin loop of RSPO4.
In LGR4 and mutated RSPO4 complex, a slight alteration in the binding pose was evident due to which ARG60, ARG81, LYS90 and SER100 residues of mutated RSPO4 (Fig. 4a) were not detected in binding with LGR4. Instead, three residues ASP162, ASN210, ASP281 of LGR4 were involved in binding ARG87, LYS89, GLN65/ILE63 of mutated RSPO4, respectively (Fig. 4b). Similarly, in LGR5–mutated RSPO4 complex, interactions were detected between ASN123 and ASP171 of LGR5, and GLN83 and ARG87 of mutated RSPO4, respectively (Fig. 5). Another complex (LGR6–mutated RSPO4) encountered loss of previous interactions (Fig. 6a). Interactions of ARG81, GLN83, GLN101 and CYS104 residues were disrupted in the mutated state. In addition, ASN123/GLN125, ARG144, ASP170, ASP171 and TYR243 residues of LGR6 showed interactions with GLU84, SER100, LYS89, ARG87 and ARG60 of mutated RSPO4, respectively (Fig. 6b). These outcomes validated the earlier experimental findings of Xu et al.47 where almost similar effects were observed upon modeling of p.Arg87Ala RSPO1 with LGR4 which resulted into sufficiently abrogated WNT signaling.
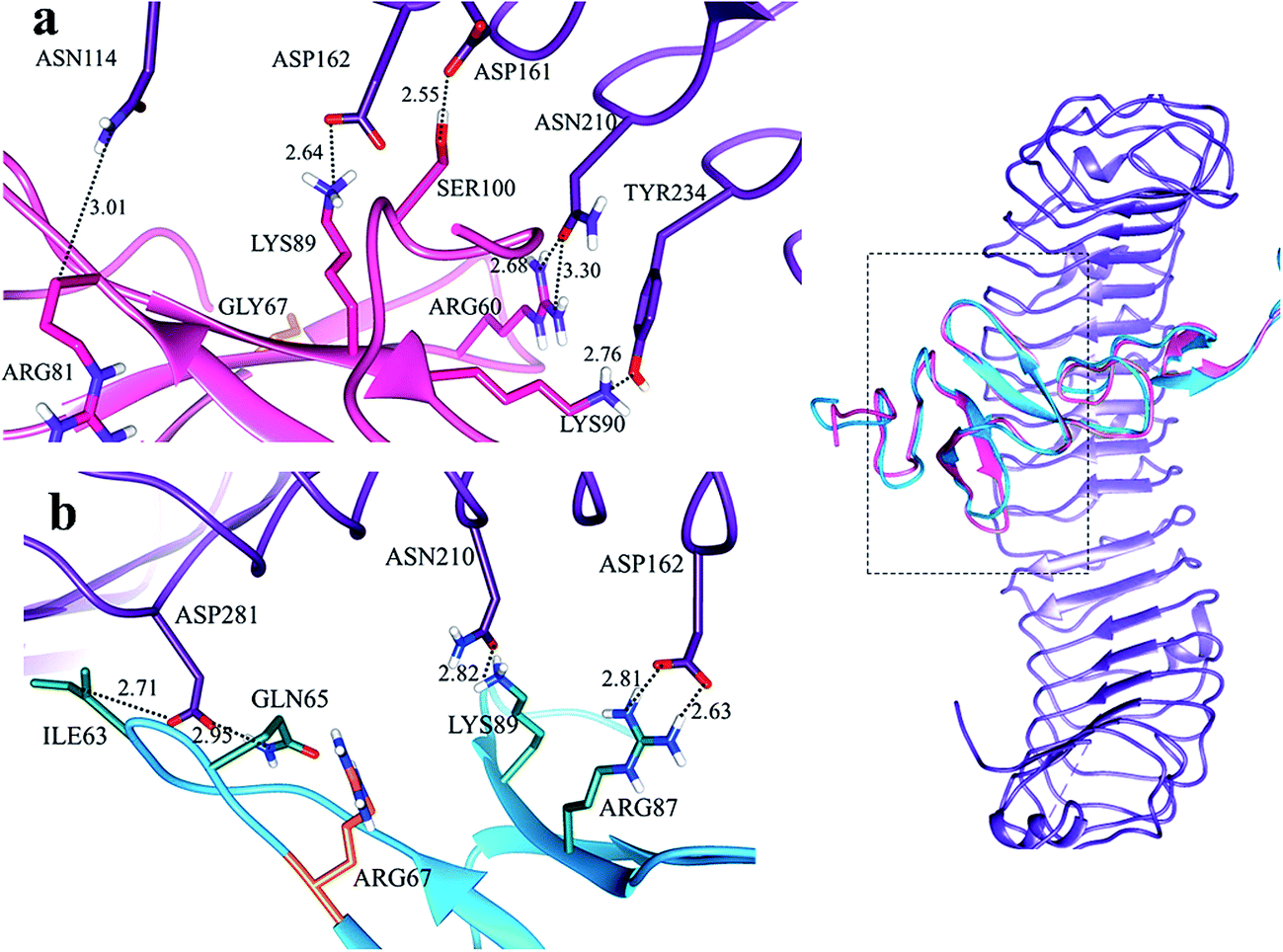 | ||
| Fig. 4 Comparative binding view of normal (pink) and mutated (blue) RSPO4 with LGR4 (purple). | ||
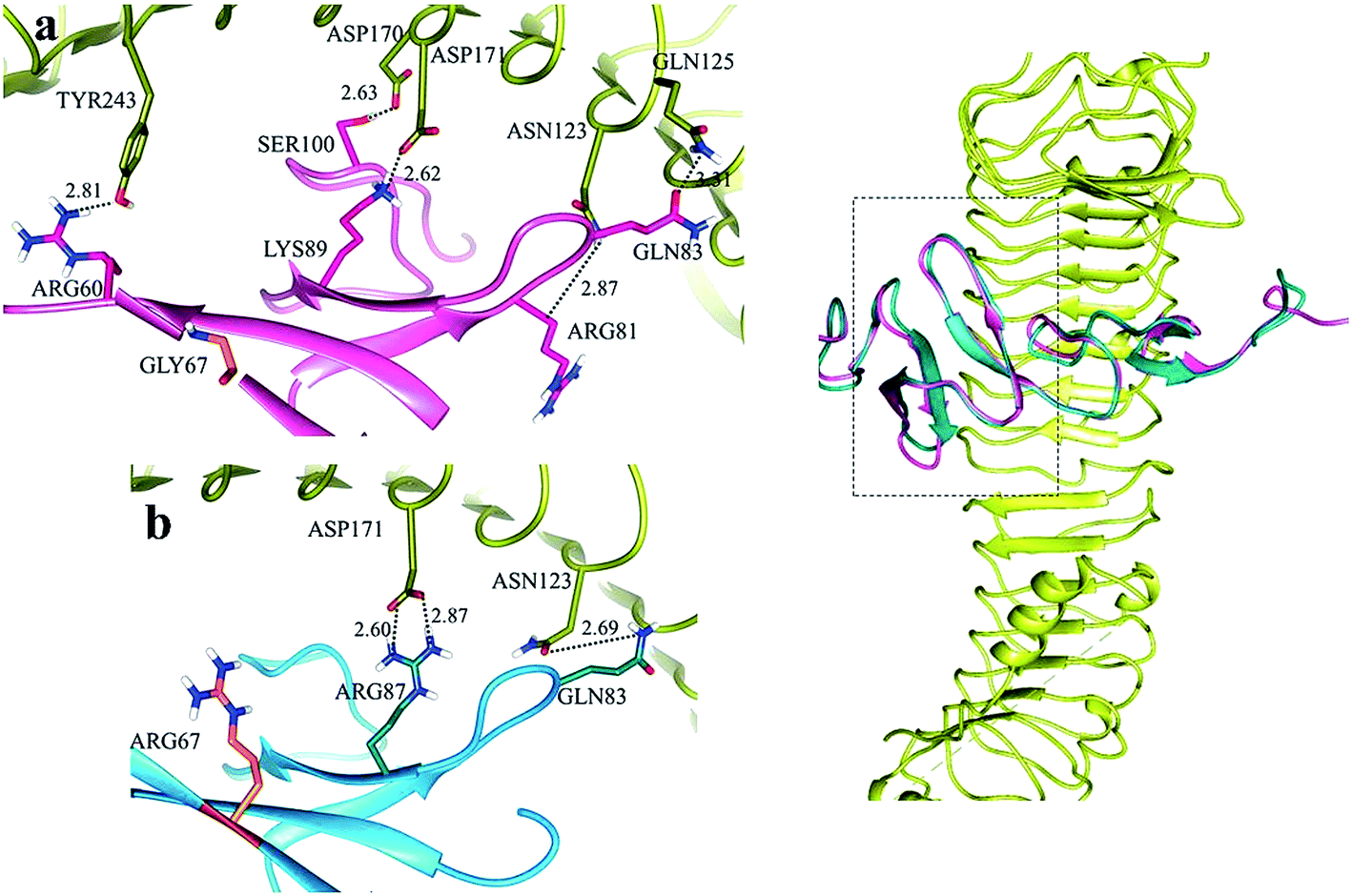 | ||
| Fig. 5 Comparative binding view of normal (pink) and mutated (blue) RSPO4 with LGR5 (yellow). | ||
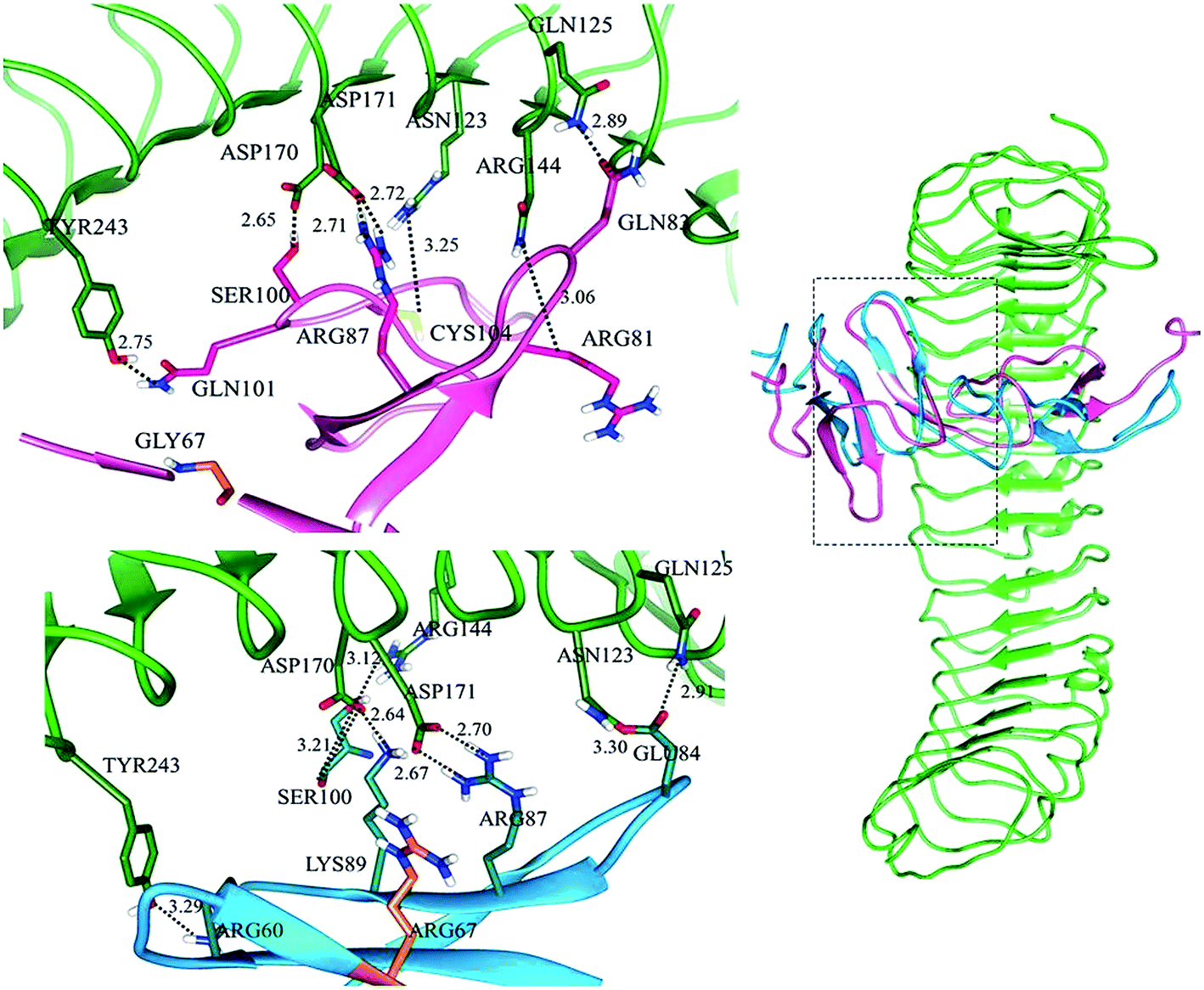 | ||
| Fig. 6 Comparative binding view of normal (pink) and mutated (blue) RSPO4 with LGR6 (green). | ||
WNT LGRs efficiently recruit R-Spondins and adjust their positions to interact with transmembrane E3 ubiquitin ligase ZNRF3 and related RNF43 (Ring Finger Protein 43).41,52 The Furin1 domain of R-Spondin binds with PA (protease-associated) domain of ZNRF3, while its Furin2 domain binds to LGR4–6.48 As a result of this attachment, membrane clearance of ZNRF3 occurs which leads to enhanced WNT signaling. In the absence of R-Spondin, PA-domain of ZNRF3 recognizes Frizzled, leading to poly-ubiquitination of its 7TM domain via intracellular E3 ligase RING domain. As a result, Frizzled is degraded via endocytosis52 leading to reduced WNT signaling (Fig. 7). Possibly, p.GLY67ARG mutation in the Furin1 domain of RSPO4 may hinder its binding with ZNRF3. The conserved Furin repeat residues (SER48, ASN51, ARG66 and GLN71) of RSPO1 involved in ZNRF3 interaction lie at a similar region corresponding to mutated residue of RSPO4.41 Another report confirms that substitution of ARG66 and GLN71 residues into ALA in the Furin1 domain of RSPO1 completely abolishes its binding with ZNRF3.53 Intriguingly, known mutations in RSPO4 of anonychia patients reside in the dimerization interface of ZNRF3.41,48,52,53 The ZNRF3 interaction site on R-Spondin overlaps with the dimerization interface, including residues related to anonychia mutations. This overlap implies that dimerization would compete with ZNRF3 binding. Thus GLY67ARG substitution in RSPO4 may disturb the balance of cross-bridge formation among ZNRF3, RSPO4 and LGRs resulting in the subsequent membrane clearance of ZNRF3.
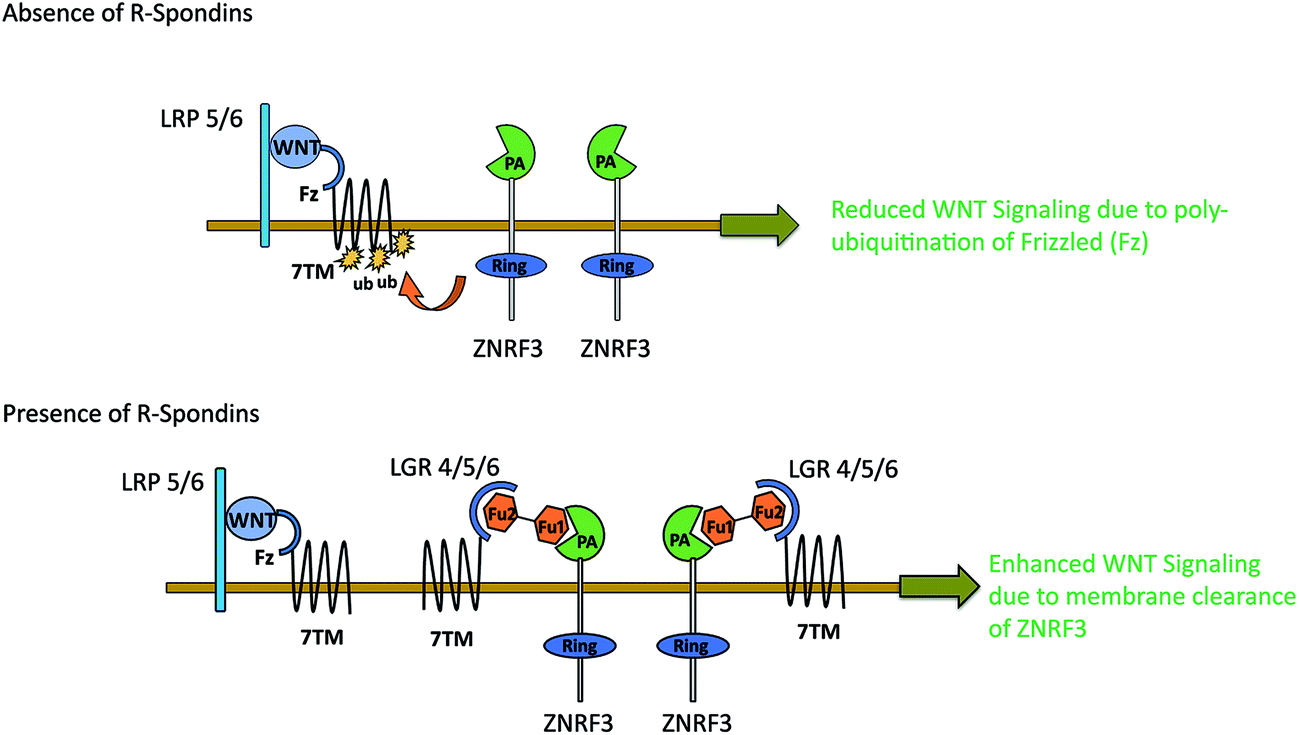 | ||
| Fig. 7 Action mechanism of R-Spondin–LGR–ZNRF3 complex on WNT signaling. | ||
Conclusion
Since R-Spondin family of proteins is well documented to enhance β-catenin signaling in WNT-dependent manner,1,3,51,54,55 we further hypothesized that abrogated RSPO4–LGRs mediated singling may result in insufficiently stabilized cytosolic β-catenin to regulate the underlying LEF1/TCF-dependent gene expression. To check this hypothesis, we quantified β-catenin level in patients suffering from anonychia through western blotting. β-catenin was significantly reduced in patients with mRSPO4 as compared to healthy control individuals. Thus, our findings in conjunction with earlier reports confirm that R-Spondins modulate LGR-dependent WNT β-catenin signaling and that GLY67ARG substituted RSPO4 causes a significant reduction in cytosolic β-catenin. As documented for bone osteolytic lesions,56–59 RSPO4 may prove to be a valuable therapeutic agent in anonychia patients.Competing interests
The authors declare that they have no competing interests.Author contributions
WA conceived and designed the wet lab experimentation, contributed reagents and proofread the manuscript. SR designed Bioinformatics analysis and drafted Bioinformatics section of the manuscript. SIR performed sequencing and analyzed genetic data and helped in manuscript drafting. AKN assisted in the analysis of genetic data. NRD performed nail biopsy. ZN assisted in computational analysis and figure preparation. KS helped in sample collection and genotyping. All authors read and approved the final manuscript.Abbreviations
| RSPO4 | R-Spondin-4 |
| LGR | Leucine-rich-repeat containing G-protein coupled receptors |
| LRRNT | Leucine-rich repeats N-terminal |
| LRRCT | Leucine-rich repeats C-terminal |
| LEF1 | Lymphoid enhancing factor |
| TCF | T-cell factor |
| LRR | Leucine-Rich Repeats |
| CRD | Cysteine-rich domain |
| Fu-CRDs | Furin-like Cysteine-Rich Domains |
Acknowledgements
We are obliged to all members of the two families for participating in the study.References
- O. Kazanskaya, A. Glinka, I. del Barco Barrantes, P. Stannek, C. Niehrs and W. Wu, Dev. Cell, 2004, 7, 525–534 CrossRef CAS PubMed.
- K. A. Kim, J. Zhao, S. Andarmani, M. Kakitani, T. Oshima, M. E. Binnerts, A. Abo, K. Tomizuka and W. D. Funk, Cell Cycle, 2006, 5, 23–26 CrossRef CAS PubMed.
- J. S. Nam, T. J. Turcotte, P. F. Smith, S. Choi and J. K. Yoon, J. Biol. Chem., 2006, 281, 13247–13257 CrossRef CAS PubMed.
- M. E. Binnerts, K. A. Kim, J. M. Bright, S. M. Patel, K. Tran, M. Zhou and A. Abo, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 14700–14705 CrossRef CAS PubMed.
- Q. Wei, C. Yokota, M. V. Semenov, B. Doble, J. Woodgett and X. He, J. Biol. Chem., 2007, 282, 15903–15911 CrossRef CAS PubMed.
- B. T. MacDonald, K. Tamai and X. He, Dev. Cell, 2009, 17, 9–26 CrossRef CAS PubMed.
- H. Clevers, Cell, 2006, 127, 469–480 CrossRef CAS PubMed.
- C. Y. Logan and R. Nusse, Annu. Rev. Cell Dev. Biol., 2004, 20, 781–810 CrossRef CAS PubMed.
- R. T. Moon, A. D. Kohn, G. V. De Ferrari and A. Kaykas, Nat. Rev. Genet., 2004, 5, 691–701 CrossRef CAS PubMed.
- R. Nusse, Cell Res., 2005, 15, 28–32 CrossRef CAS PubMed.
- W. De Lau, N. Barker, T. Y. Low, B. K. Koo, V. S. Li, H. Teunissen, P. Kujala, A. Haegebarth, P. J. Peters, M. van de Wetering, D. E. Stange, J. E. van Es, D. Guardavaccaro, R. B. Schasfoort, Y. Mohri, K. Nishimori, S. Mohammed, A. J. Heck and H. Clevers, Nature, 2011, 476, 293–297 CrossRef CAS PubMed.
- K. S. Carmon, X. Gong, Q. Lin, A. Thomas and Q. Liu, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 11452–11457 CrossRef CAS PubMed.
- A. Glinka, C. Dolde, N. Kirsch, Y. L. Huang, O. Kazanskaya, D. Ingelfinger, M. Boutros, C. M. Cruciat and C. Niehrs, EMBO Rep., 2011, 12, 1055–1061 CrossRef CAS PubMed.
- H. Ruffner, J. Sprunger, O. Charlat, J. Leighton-Davies, B. Grosshans, A. Salathe, S. Zietzling, V. Beck, M. Therier, A. Isken, Y. Xie, Y. Zhang, H. Hao, X. Shi, D. Liu, Q. Song, I. Clay, G. Hintzen, J. Tchorz, L. C. Bouchez, G. Michaud, P. Finan, V. E. Myer, T. Bouwmeester, J. Porter, M. Hild, F. Bassilana, C. N. Parker and F. Cong, PLoS One, 2012, 7, e0976, DOI:10.1371/journal.pone.0040976.
- N. Barker and H. Clevers, Gastroenterology, 2010, 138, 1681–1696 CrossRef CAS PubMed.
- N. Barker, J. H. van Es, J. Kuipers, P. Kujala, M. van den Born, M. Cozijnsen, A. aegebarth, J. Korving, H. Begthel, P. J. Peters and H. Clevers, Nature, 2007, 449, 1003–1007 CrossRef CAS PubMed.
- V. Jaks, N. Barker, M. Kasper, J. H. van Es, H. J. Snippert, H. Clevers and R. Toftgård, Nat. Genet., 2008, 40, 1291–1299 CrossRef CAS PubMed.
- N. Barker, M. B. Rookmaaker, P. Kujala, A. Ng, M. Leushacke, H. Snippert, M. van de Wetering, S. Tan, J. H. Van Es, M. Huch, R. Poulsom, M. C. Verhaar, P. J. Peters and H. Clevers, Cell Rep., 2012, 2, 540–552 CrossRef CAS PubMed.
- M. Huch, C. Dorrell, S. F. Boj, J. H. van Es, V. S. Li, M. van de Wetering, T. Sato, K. Hamer, N. Sasaki, M. J. Finegold, A. Haft, R. G. Vries, M. Grompe and H. Clevers, Nature, 2013, 494, 247–250 CrossRef CAS PubMed.
- V. Plaks, A. Brenot, D. A. Lawson, J. R. Linnemann and E. C. Van Kappel, Cell Rep., 2013, 3, 70–78 CrossRef CAS PubMed.
- J. Schuijers and H. Clevers, EMBO J., 2012, 31, 2685–2696 CrossRef CAS PubMed.
- C. Niehrs and J. Shen, Cell. Mol. Life Sci., 2010, 67, 2551–2562 CrossRef CAS PubMed.
- P. Parma, O. Radi, V. Vidal, M. C. Chaboissier, E. Dellambra and S. Valentini, Nat. Genet., 2006, 38, 1233–1244 CrossRef PubMed.
- C. Bergmann, J. Senderek, D. Anhuf, C. T. Thiel, A. B. Ekici, P. Poblete-Gutierrez, M. van Steensel, D. Seelow, G. Nürnberg, H. H. Schild, P. Nürnberg, A. Reis, J. Frank and K. Zerres, Am. J. Hum. Genet., 2006, 79, 1105–1109 CrossRef CAS PubMed.
- D. C. Blaydon, Y. Ishii, E. A. O'Toole, H. C. Unsworth, M. T. Teh, F. Ruschendorf, C. Sinclair, V. K. Hopsu-Havu, N. Tidman, C. Moss, R. Watson, D. de Berker, M. Wajid, A. M. Christiano and D. P. Kelsell, Nat. Genet., 2006, 38, 1245–1247 CrossRef CAS PubMed.
- S. I. Raza, R. Nasser Dar, A. A. Shah and W. Ahmad, Ann. Hum. Genet., 2015, 79, 92–98 CrossRef CAS PubMed.
- S. Rashid, I. Pilecka, A. Torun, M. Olchowik, B. Bielinska and M. Miaczynska, J. Biol. Chem., 2009, 284, 18115–18128 CrossRef CAS PubMed.
- H. M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat, H. Weissig, I. N. Shindyalov and P. E. Bourne, Nucleic Acids Res., 2000, 28, 235–242 CrossRef CAS PubMed.
- C. N. Cavasotto and S. S. Phatak, Drug Discovery Today, 2009, 14, 676–683 CrossRef CAS PubMed.
- T. J. Hubbard, B. L. Aken, S. Ayling, B. Ballester, K. Beal, E. Bragin and V. Curwen, Nucleic Acids Res., 2009, 37, D690–D697 CrossRef CAS PubMed.
- M. Johnson, I. Zaretskaya, Y. Raytselis, Y. Merezhuk, S. Mcginnis and T. L. Madden, Nucleic Acids Res., 2008, 36(2), W5–W9 CrossRef CAS PubMed.
- N. Eswar, B. Webb, M. A. Marti-Renom, M. S. Madhusudhan, D. Eramian, M. Y. Shen and A. Sali, Current Protocols in Bioinformatics, 2006, ch. 5, Unit 5.6 Search PubMed.
- A. Pedretti, L. Villa and G. Vistoli, J. Comput.-Aided Mol. Des., 2004, 18, 167–173 CrossRef CAS PubMed.
- J. P. Rodrigues, M. Levitt and G. Chopra, Nucleic Acids Res., 2012, W323–W328, DOI:10.1093/nar/gks376.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS PubMed.
- V. B. Chen, W. B. Arendall, J. J. Headd, D. A. Keedy, R. M. Immormino, G. J. Kapral, L. W. Murray, J. S. Richardson and D. C. Richardson, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 66, 12–21 CrossRef PubMed.
- S. C. Lovell, I. W. Davis, W. B. Arendall, P. I. W. de Bakker, J. M. Word, M. G. Prisant, J. S. Richardson and D. C. Richardson, Proteins: Struct., Funct., Bioinf., 2003, 50, 437–450 CrossRef CAS PubMed.
- C. Colovos and T. O. Yeates, Protein Sci., 1993, 2, 1511–1519 CrossRef CAS PubMed.
- D. Eisenberg, L. Roland and U. B. James, Methods Enzymol., 1997, 277, 396–404 CAS.
- M. Wiederstein and J. S. Manfred, Nucleic Acids Res., 2007, 2, 407–410 CrossRef PubMed.
- P. Chicago, W. de-Lau, F. Forneris, J. C. Granneman, M. Huch, H. Clevers and P. Gros, Cell Rep., 2013, 3, 1885–1892 CrossRef PubMed.
- S. J. De Vries, M. van Dijk and A. M. Bonvin, Nat. Protoc., 2010, 5, 883–897 CrossRef CAS PubMed.
- Y. Ishii, M. Wajid, H. Bazzi, K. A. Fantauzzo, A. G. Barber, D. C. Blaydon, J. S. Nam, J. K. Yoon, D. P. Kelsell and A. M. Christiano, J. Invest. Dermatol., 2008, 128, 867–870 CrossRef CAS PubMed.
- M. S. Chishti, N. Kausar, M. A. Rafiq, M. Amin and W. Ahmad, Br. J. Dermatol., 2008, 158, 621–623 CrossRef CAS PubMed.
- N. Wasif and W. Ahmad, Pediatric Dermatology, 2013, 30, 139–141 CrossRef PubMed.
- T. N. Khan, J. Klar, S. Nawaz, M. Jameel, M. Tariq, N. A. Malik, S. M. Baig and N. Dahl, BMC Med. Genet., 2012, 13, 120, DOI:10.1186/1471-2350-13-120.
- J. G. Xu, C. Huang, Z. Yang, M. Jin, P. Fu, N. Zhang and Y. Zhu, J. Biol. Chem., 2015, 290, 2455–2465 CrossRef CAS PubMed.
- P. H. Chen, X. Chen, Z. Lin, D. Fang and X. He, Genes Dev., 2013, 27, 1345–1350 CrossRef CAS PubMed.
- D. Wang, B. Huang, S. Zhang, X. Yu, W. Wu and X. Wang, Genes Dev., 2013, 27, 1339–1344 CrossRef CAS PubMed.
- K. Xu, Y. Xu, K. R. Rajashankar, D. Robev and D. B. Nikolov, 2013, 21, 1683–1689.
- K. A. Kim, M. Wagle, K. Tran, X. Zhan, M. A. Dixon, S. Liu, D. Gros, W. Korver, S. Yonkovich, N. Tomasevic, M. Binnerts and A. Abo, Mol. Biol. Cell, 2008, 19, 2588–2596 CrossRef CAS PubMed.
- M. Zebisch, Y. Xu, C. Krastev, B. T. MacDonald, M. Chen, R. J. Gilbert, X. He and E. Y. Jones, Nat. Commun., 2013, 4, 2787 Search PubMed.
- Y. Xie, R. Zamponi, O. Charlat, M. Ramones, S. Swalley, X. Jiang, D. Rivera, W. Tschantz, B. Lu, L. Quinn, C. Dimitri, J. Parker, D. Jeffery, S. K. Wilcox, M. Watrobka, P. LeMotte, B. Granda, J. A. Porter, V. E. Myer, A. Loew and F. Cong, EMBO Rep., 2013, 14, 1120–1126 CrossRef CAS PubMed.
- K. A. Kim, M. Kakitani, J. Zhao, T. Oshima, T. Tang, M. Binnerts, Y. Liu, B. Boyle, E. Park, P. Emtage, W. D. Funk and K. Tomizuka, Science, 2005, 309, 1256–1259 CrossRef CAS PubMed.
- M. T. Veeman, J. D. Axelrod and R. T. Moon, Dev. Cell, 2003, 5, 367–377 CrossRef CAS PubMed.
- E. Tian, F. Zhan, R. Walker, E. Rasmussen, Y. Ma and B. Barlogie, N. Engl. J. Med., 2003, 349, 2483–2494 CrossRef CAS PubMed.
- S. Colla, F. Zhan, W. Xiong, X. Wu, H. Xu, O. Stephens, S. Yaccoby, J. Epstein, B. Barlogie and J. D. Shaughnessy Jr, Blood, 2007, 109, 4470–4477 CrossRef CAS PubMed.
- D. Diarra, M. Stolina, K. Polzer, J. Zwerina, M. S. Ominsky, D. Dwyer, A. Korb, J. Smolen, M. Hoffmann, C. Scheinecker, D. van der Heide, R. Landewe, D. Lacey, W. G. Richards and G. Schett, Nat. Med., 2007, 13, 156–163 CrossRef CAS PubMed.
- S. Yaccoby, W. Ling, F. Zhan, R. Walker, B. Barlogie and J. D. Shaughnessy, Blood, 2007, 109, 2106–2111 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra00762k |
| This journal is © The Royal Society of Chemistry 2017 |