
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA bioinspired heterogeneous catalyst based on the model of the manganese-dependent dioxygenase for selective oxidation using dioxygen†
Jérémy Chaignonab,
Marie Gourguesac,
Lhoussain Khrouza,
Nicolás Molinerb,
Laurent Bonneviota,
Fabienne Fachec,
Isabel Castro*b and
Belén Albela *a
*a
aLaboratoire de Chimie UMR CNRS 5182, Ecole Normale Supérieure de Lyon, University of Lyon, 46, Allée d'Italie, 69364 Lyon, Cedex-07, France. E-mail: belen.albela@ens-lyon.fr
bInstituto de Ciencia Molecular, Universitat de València, C/Catedrático José Beltrán 2, 46980 Paterna, València, Spain. E-mail: isabel.castro@uv.es
cInstitut de Chimie et Biochimie Moléculaires et Supramoléculaires (ICBMS) UMR CNRS 5246, University of Lyon, Bd. du 11 Novembre 1918, 69622 Villeurbanne, Cedex, France
First published on 20th March 2017
Abstract
A hybrid bioinspired material with manganese(II) complexes grafted on the surface of a mesostructured porous silica is investigated. The Mn sites mimic the manganese-dependent dioxygenase (MndD), which is an enzyme that catalyses the oxidation of catechol derivatives. The metal complexes were introduced in the silica using a dinuclear complex [Mn2L2(Cl)2(μ-Cl)2] as a precursor with a clickable ligand N,N′-bis[(pyridin-2-yl)methyl]prop-2-yn-1-amine (L). Azide moieties covalently grafted on MCM-41 type mesoporous silica were utilised to anchor the manganese complex through Huisgen cycloaddition using CuBr(PPh3)3 as a catalyst. A second functional group – trimethylsilyl or pyridine—was grafted on the silica to bring, together with nanopore size confinement, a similar metal environment as in MndD. The mesostructure of the materials was maintained after incorporation of the Mn complex. Catalytic oxidation of 3,5-di-t-butyl-catechol (3,5-DTBC) into quinone occurred without the need of an additional base when the metal complex was confined in the porous solid. In comparision, the oxidation of 4-t-butyl-catechol (4-TBC) that always required a basic media led to a total oxidation into the ortho-quinone, contrary to the molecular analogue.
Introduction
Many industrial processes involve catalytic reactions, since they allow working under milder conditions with high selectivity and short reaction time, decreasing, therefore, the cost of the final product.1 However, these enhanced processes are not a prerogative for industry, for they existed in nature long before chemists could understand them. A class of natural metalloproteins called metalloenzymes is able to carry out extremely specific reactions under physiological conditions (between 10 and 40 °C for most of them).2 In particular, several metalloenzymes called oxygenases perform the oxidation of organic compounds by activating molecular dioxygen. Various metal ions can be found in these oxygenases, mainly copper, iron and manganese.3–9 Although the reaction with dioxygen is thermodynamically favourable, the electronic configuration of the O2 molecule creates a strong kinetic barrier that requires activation, which is useful to prevent any spontaneous reaction of O2 with organic molecules.Structural and mechanistic research on metalloenzymes in the past decades has allowed the development of numerous bioinspired systems that can be used for catalysis. Homogeneous biomimetic complexes are often not optimal for reactions involving dioxygen activation. Indeed, the activation of oxygen can partially oxidise the catalyst. Grafting on a solid support can stabilise the catalyst and confer confinement as in the enzymatic pocket.
Amongst oxygenases, a recurrent metal coordination triad is observed, composed by two histidines (His) and one carboxylic acid, coming either from glutamate (Glu) or aspartate (Asp) moieties. This widespread structural motif is called the 2-His-1-carboxylate facial triad, and is found in many metalloproteins.6 This is the case of catechol dioxygenase, which catalyses the cleavage of catechol derivatives. Two kinds of catechol dioxygenases can be found, which differ on the nature of the metal ion involved at the active site: FeIII allows an intradiol cleavage while FeII or MnII performs the extradiol one.6–9 In particular, the manganese-dependent dioxygenase (MndD) is a system that has attracted much attention since its catalytic mechanism is still under discussion.7,10–12 Therefore, the study of bioinspired systems that mimic the structural and/or catalytic function of MndD could help to understand its reaction mechanism.
Molecular models that contain a manganese centre are however much less frequent than those containing iron. Only few examples in the literature report manganese(II) complexes as mimics of the catechol dioxygenase. The first reported model to our knowledge was synthesised by Sakurai et al. two years after the discovery of MndD.13 Their report describes the use of penicillamine with manganese(II) in the oxidation of catechol to the corresponding muconic acid derivatives resulting from a true extradiol dioxygenase activity. However, very low conversion rates were achieved (<2%). Ramadan and El-Mehasseb subsequently investigated the oxidative properties of manganese(II) with tridentate pyridyl hydrazone ligands.14 They reported the transformation of catechol into o-benzoquinone in presence of catalyst and O2, which would correspond to an oxidase activity (oxidation without incorporation of oxygen atom in the substrate). Similar results were obtained a few years later with tetradentate ligands containing imidazole, phenolate and/or pyridine moieties.15 At the same time, the manganese(II) complex of another tetradentate ligand, N,N′-bis(2-pyridylmethyl)-1,2-ethanediamine (bispicen), managed to oxidise the 4-tert-butylcatechol into its quinone equivalent mixed with extradiol intermediates.16 Reaction conditions turned out to have an important effect on the resulting products.
We have developed a bioinspired approach for design of the metal supported catalysts based on the model of metalloproteins.17–21 This approach was recently applied using the manganese dependent dioxygenase (MndD) as a model, the N,N′-bis[(pyridin-2-yl)methyl]prop-2-yn-1-amine (L) as a clickable ligand for grafting and [Mn2L2(Cl)2(μ-Cl)2] as a precursor metal complex (Scheme 1).18 This ligand has the particular advantage to mimic the facial triad present in the active site of the MndD and allows an efficient grafting in the silica support.22–25 Indeed, the alkyne side function allows grafting of the ligand in an azido-functionalised support using “click chemistry”.26–28 In addition, the two pyridine and the tertiary amine functions present in the ligand can coordinate to the Mn(II) ion and thus mimic the metal environment of the MndD.
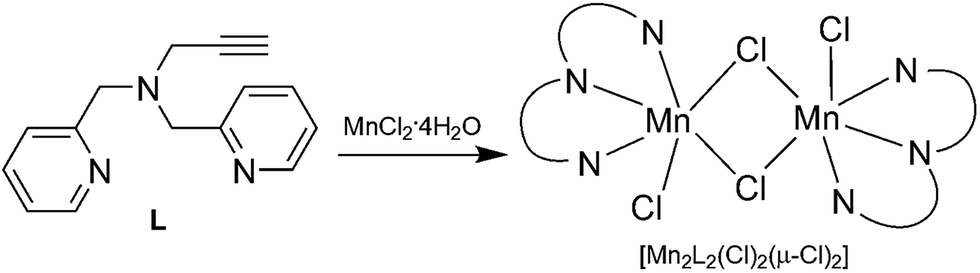 | ||
| Scheme 1 Synthesis of complex [Mn2L2(Cl)2(μ-Cl)2]. | ||
In the present work we are going further in providing a comprehensive investigation of the [Mn2L2(Cl)2(μ-Cl)2] grafting, knowing that this dimer is in equilibrium with the monomer in solution.18 We will also compare two different local environments of the metal complex in the solid to test the influence of a pyridine side function meant here to mimic the histidine function neighbouring the metal active site in MndD. Both homogeneous and heterogeneous systems are tested for the first time on the oxidation of catechol derivatives.
Results and discussion
Incorporation of the MnII complex in the mesoporous silica: synthesis strategy to isolate the metal sites with control of their close environment
The alkyne function present in the ligand L (Scheme 1) was used to covalently graft the manganese complex into the pores of a mesoporous silica of the LUS type, which presents a 2D hexagonal structure just like the classic MCM-41 silica.29 The LUS silica was synthesised using a ultra-fast microwave assisted hydrothermal synthesis that affords a high quality mesostructured porous silica.30 Due to the yield and selectivity advantages of the copper(I)-catalysed Huisgen azide–alkyne cycloaddition (also known as “click chemistry”), this reaction was chosen for the grafting process.26–28 We had the choice to design our ligands either with an azide or an alkyne arm, the latter being preferred for two reasons. First, the incorporation of an azide function in the ligand could have been more problematic for the synthesis of the complex than the incorporation of an alkyne arm because of the coordinating potential of the azide moiety. Secondly, the synthesis of the azide tethering silane was easier than the alkyne tether, the azidopropyltriethoxysilane precursor being then synthesised starting from chloropropyltriethoxysilane upon nucleophilic substitution of the chlorine atom by an azide moiety.31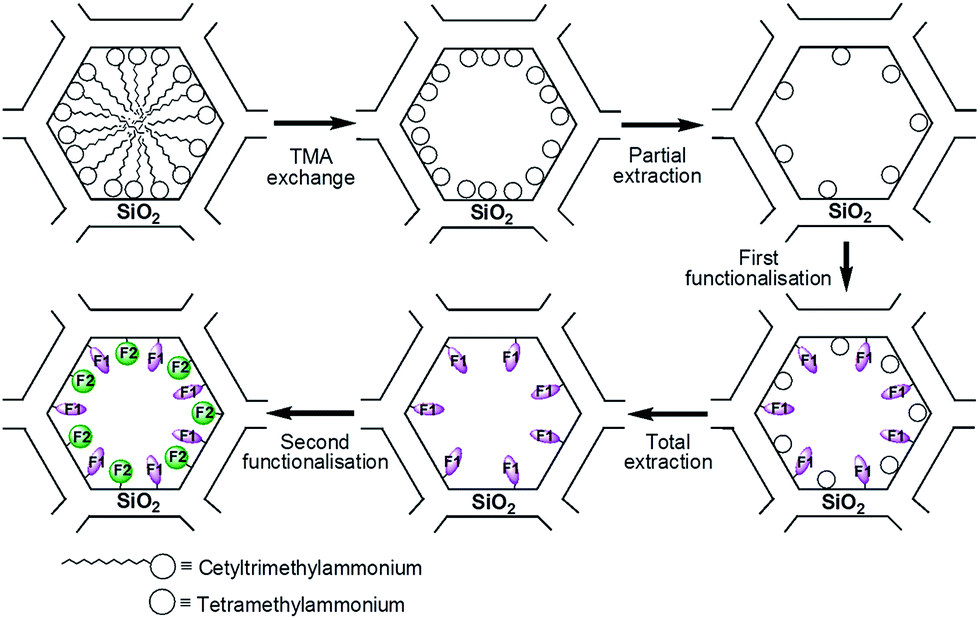 | ||
| Scheme 2 Molecular stencil patterning strategy. TMA = tetramethylammonium. | ||
The molecular stencil patterning (MSP) technique was used to incorporate the complexes into the solid (Scheme 2).32–34 This strategy takes advantage of the cationic nature of the surfactant (cetyltrimetylammonium, CTA+) present in the mesoporous silica. The general procedure is a four step strategy: (i) the template is partially removed with hydrochloric acid; due to electrostatic repulsion, the remaining surfactant rearranges thus making some silica surface available, (ii) a first organosilane (F1) is grafted onto this freed surface, (iii) all of the remaining surfactant is removed, freeing silica surface once again, and (iv) the second function (F2) is grafted, providing a double functionalised material. In the present work, however, a slightly different protocol was applied where CTA+ was exchanged with tetramethylammonium (TMA+) prior to the first partial extraction (Scheme 2).34 This modification allowed a better diffusion of the F1 grafting function since there was more available pore space. If the grafted functions are modelled as spheres arranged in 2D hexagonal packing on the pore surface, isolating a function F2 will result in having it surrounded by 6 functions F1. Every F1 will thus be adjacent to 3 functions F2, which corresponds to a ratio F1/F2 of 2.32
The nomenclature used for the synthesised solids is the following. LUS stands for the as-made support containing CTA+ molecules inside the pores, while the material exchanged with TMA+ is denoted as LUS-TMA. Partial extraction is represented by an – Ex suffix where x is the percentage of extracted TMA+. For instance, a material where 15% of TMA+ was extracted is named LUS-TMA-E15. Totally extracted materials are simply noted LUS-E. Likewise, when the remaining TMA+ is removed from a LUS-TMA-Ex material, the resulting one is denoted as LUS-Ex. Grafting reactions are symbolised by prefixes. For instance, silica functionalised with azide (N3) and trimethylsilyl (TMS) groups will be named N3–TMS@LUS. In the case of double functionalisation, the last function incorporated is placed at the beginning of the name: a material functionalised with F1 and then with F2 will thus be called F2–F1@LUS.
In the present work two materials containing a manganese(II) complex were studied using [Mn2L2(Cl)2(μ-Cl)2]18 as metal precursor: a new solid {Mn}-Py@LUS-E15 (S1), where pyridine (Py) functions are in the vicinity of the metal complexes, and the already reported {Mn}-TMS@LUS-E (S2),18 where the manganese complexes are surrounded by hydrophobic trimethylsilyl (TMS) functions (Scheme 3). In this nomenclature, the {Mn} function is the product of the reaction between [Mn2L2(Cl)2(μ-Cl)2] and the azide (N3) tether present in both host materials. The TMS function was chosen to mimic some hydrophobic residues present in the enzymatic pocket of the metalloprotein, whereas Py function was selected to mimic the His200 residue present in the second coordination sphere of the Mn active site, which is known to help in the activation of molecular oxygen during the catalytic cycle of MndD.7,35,36
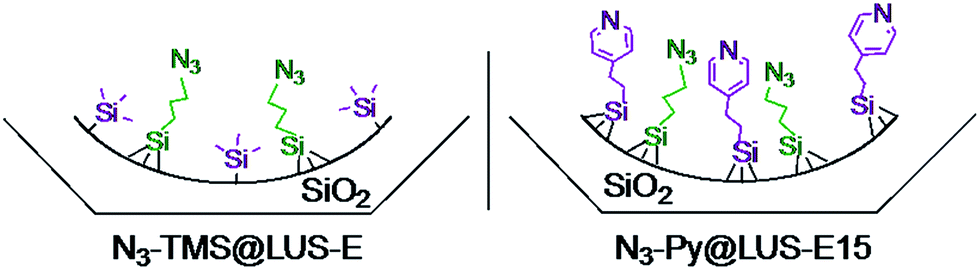 | ||
| Scheme 3 Host materials for the grafting of the Mn(II) complex: N3–TMS@LUS-E and N3–Py@LUS-E15. | ||
Optimisation of the “click chemistry” grafting of the Mn complex
Even though click chemistry is known to be efficient, one must be careful about the conditions implied in those reactions. Indeed, the copper-catalysed Huisgen cycloaddition has proven to be sensitive to coordinating molecules when the classical copper catalyst is used. In principle, copper(II) sulphate or acetate is directly used and reduced to copper(I) in situ, most of the time by treatment with sodium ascorbate. For instance, reaction with L and benzyl azide has been reported and performed under these conditions.37 Instead of sodium ascorbate, methanol can also be used as reductant.38 Some preliminary tests between the ligand L and azidopropyltrimethoxysilane were performed in order to optimise the conditions for the click reaction on the solid support. Under these conditions, coordination of copper to the ligand L was observed in all cases, even when copper(I) bromide was used. A tris(triphenylphosphine)copper(I) bromide complex was required as catalyst to avoid this problem. This change of catalyst was inspired by organic syntheses of triethoxysilanes using the microwave-assisted Huisgen copper-catalysed reaction reported in the literature.39 Given that triethoxysilanes are particularly sensitive to water and to many chemical functions such as alcohols or carboxylic acids, this kind of synthesis was quite a challenge. However, using microwave irradiation and CuBr(PPh3)3 as catalyst, the desired organosilane was obtained in a yield typical for click chemistry (above 90%).Then, several tests were performed to click the manganese(II) complex [Mn2L2(Cl)2(μ-Cl)2] in the azide-functionalised silica N3@LUS-E. After an overnight reaction with CuBr(PPh3)3, the IR spectrum of the resulting material still exhibited a band around 2100 cm−1, which is characteristic of the presence of azide moieties. After a one-week reaction, no absorption occurred in this region, proving that all the azide moieties had reacted. This long reaction time can be explained by the porous nature of the material. All the reactive sites at the silica surface are not as accessible as in solution, and diffusion inside the porous material becomes a non-negligible parameter. Removal of the copper catalyst was performed by washing with methanol. This technique was efficient enough to remove the catalyst, as confirmed by EDX microscopy, EPR and elemental analysis: no trace of copper could be found beyond the detectable threshold. Therefore, this technique was applied to incorporate the manganese complex into the host materials N3–Py@LUS-E15 and N3–TMS@LUS-E, affording solids {Mn}-Py@LUS-E15 (S1), {Mn}-TMS@LUS-E (S2), respectively.
Characterisation of the hybrid materials
The porous structure of the host materials was checked using powder X-ray diffraction (XRD) during the different steps of the synthesis. The original 2D hexagonal porous structure was maintained during the functionalisation of the silica and the incorporation of the manganese complex (Fig. 1).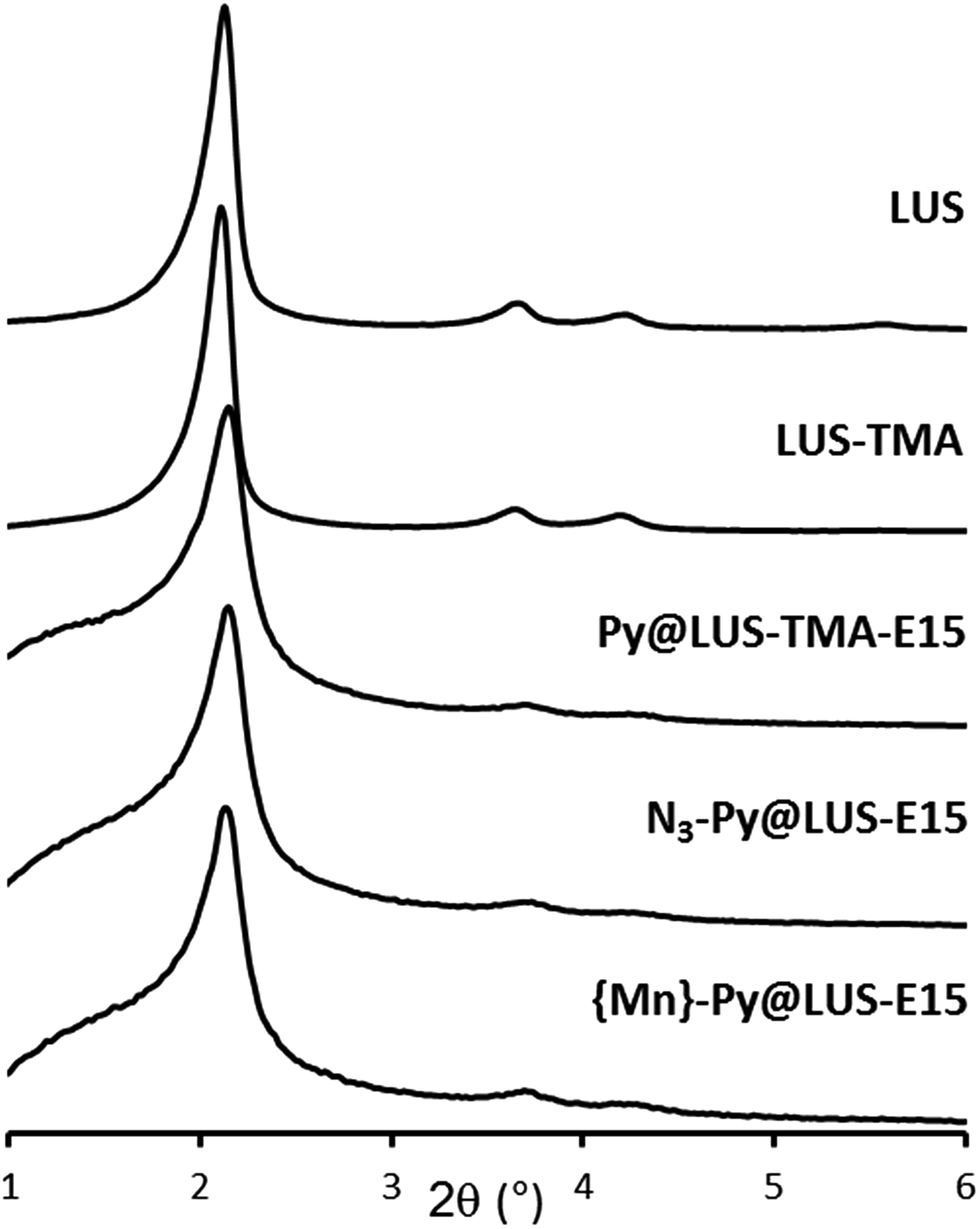 | ||
| Fig. 1 Powder XRD patterns of the support LUS-E and the materials LUS-TMA, Py@LUS-TMA-E15, N3–Py@LUS-E15 and {Mn}-Py@LUS-E15 (S1). | ||
Thermogravimetric analyses (TGA) indicate that after exchange of CTA+ surfactant molecules present in the as-made silica by TMA+, almost 2/3 of the internal surface of the silica was covered by TMA+ cations, which corresponds to 2.1 mmol of TMA+ per gram of SiO2. Starting from this solid, two intermediate materials were synthesised: TMS@LUS-TMA and Py@LUS-TMA-E15. In order to obtain a F1/F2 molar ratio of 2, TMS functionalisation was performed without any partial extraction. Indeed, TMS is grafted to the support with only one covalent bond and the ammonia generated by the aperture of the azasilane used for the functionalisation displaces a small part of the TMA+. However, in the case of pyridine, which possesses a triethoxysilane tether function, 15% of TMA+ was extracted before the functionalisation reaction in order to favour the grafting of the function. In other words, TMS functionalisation was performed on LUS-TMA material while pyridine functionalisation was carried out with LUS-TMA-E15 material. The F1/F2 molar ratio was calculated from TGA, elemental analyses, and quantitative solid 29Si-NMR (Table 1 and Fig. S1†). Py/N3 and TMS/N3 molar ratios of 1.9 and 1.8 can be deduced, and these are close to the desired value of 2.0 required to insure site isolation. Note that the TMS amount cannot be precisely deduced neither from TGA – the mass loss is very low and occurs in a broad temperature range – nor from elemental analyses due to the presence of carbon atoms in TMA+. Integration of M and Q massifs from solid 29Si-NMR could however give a precise value of the quantity of grafted TMS functions per gram of SiO2 (Fig. 2).
| Material | Vpb (cm3 g−1) | Mn (wt%) | F1/Sic | F2/Sid | Cl/Mne |
|---|---|---|---|---|---|
| a {Mn}-Py@LUS-E15 = S1; {Mn}-TMS@LUS-E = S2.b Porous volume deduced from sorption N2 isotherms using t-plot, ±0.01 cm3 g−1.c Molar ratio, F1 = TMS or Py.d Molar ratio, F2 = N3.e Molar ratio, deduced from chemical analyses. | |||||
| LUS-E | 0.86 | — | — | — | — |
| N3–TMS@LUS-E | 0.26 | — | 0.046 | 0.025 | — |
| N3–Py@LUS-E15 | 0.28 | — | 0.064 | 0.034 | — |
| S118 | 0.31 | 1.7 | — | — | 0.7 |
| S2 | 0.22 | 2.4 | — | — | 0.5 |
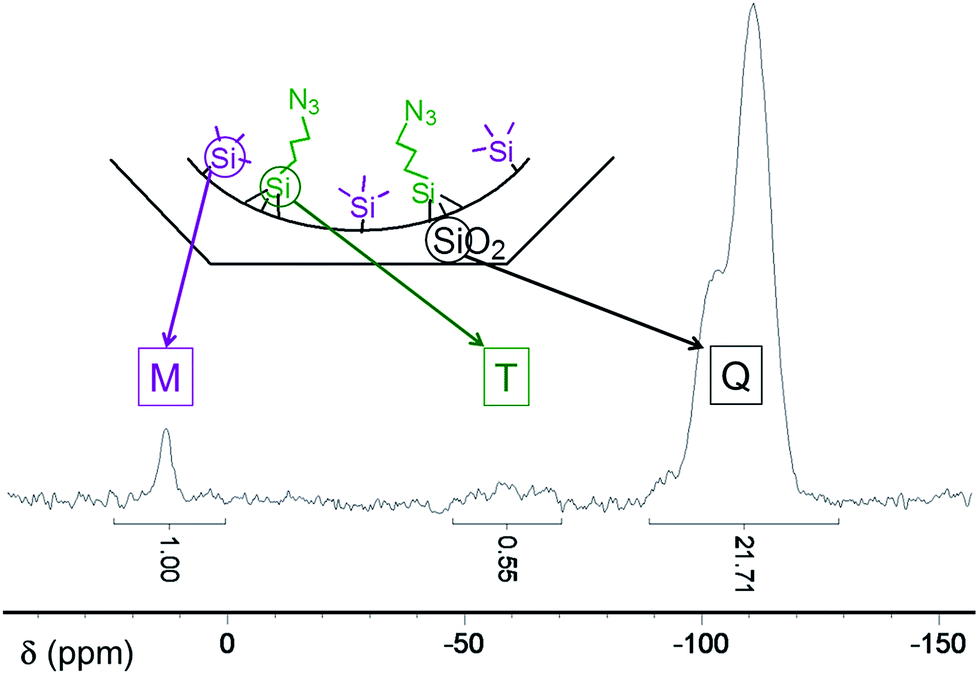 | ||
| Fig. 2 Solid 29Si HPDEC NMR spectrum of N3–TMS@LUS-E. | ||
The two hybrid materials {Mn}-Py@LUS-E and {Mn}-TMS@LUS-E present a manganese weight loading of about 2%, 1.7 and 2.4%, respectively (Table 1). The chloride counter-anions present in the initial manganese complex are only partially retained in the final materials. The Cl/Mn molar ratio in the grafted materials (Cl/Mn = 0.7 and 0.5 for {Mn}-Py@LUS-E and {Mn}-TMS@LUS-E, respectively) is approximately 25% of the original chloride anions present in the dinuclear precursor (Cl/Mn = 2). This diminution tends to prove that the chloride bridges are partially lost during the grafting process questioning about the presence of mononuclear species.
Nitrogen sorption isotherms were performed to measure the remaining porous volume of the final materials (Table 1 and Fig. 3). The pore volume, measured using the t-plot method, was reduced from 0.86 cm3 g−1 in LUS-E down to 0.2–0.3 cm3 g−1 in the final materials, indicating a partial filling of the pores after the incorporation of the complex. However, there was still enough pore volume to allow the substrate diffusion and its access to the manganese(II) centres for catalytic applications.
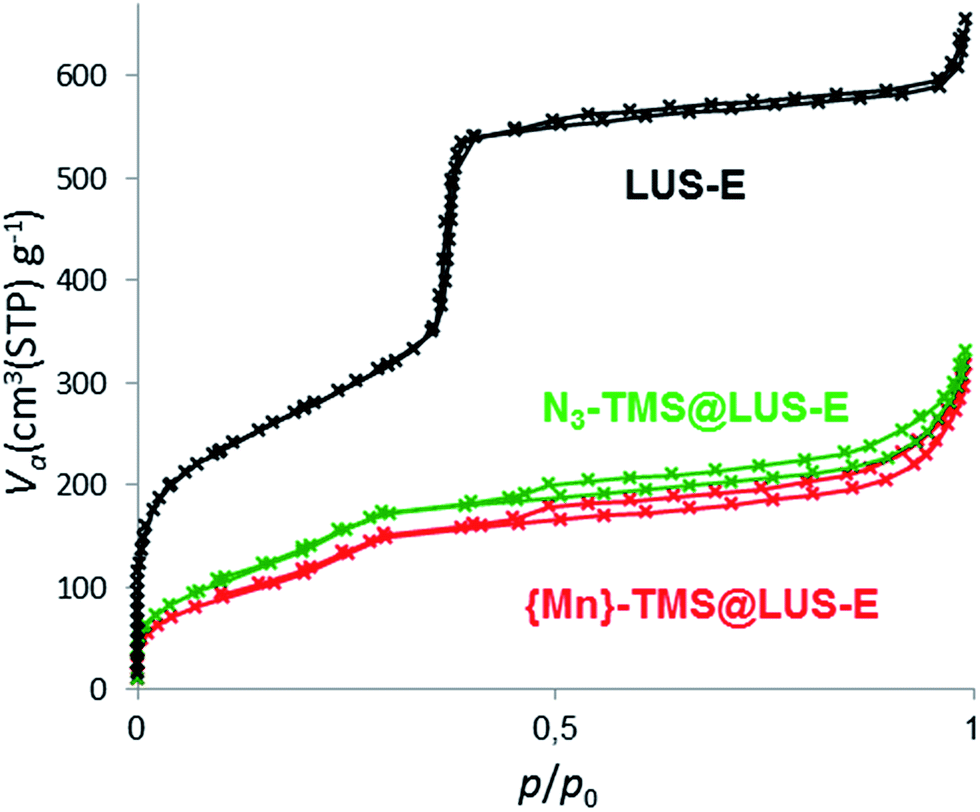 | ||
| Fig. 3 Nitrogen sorption isotherms at 77 K of LUS-E, N3–TMS@LUS-E, {Mn}-TMS@LUS-E (S2). | ||
Nature of the metal complexes inside the solid
In order to determine the relative amount of mono- and dinuclear species, magnetic and EPR measurements were performed for both solids {Mn}-Py@LUS-E15 (S1), {Mn}-TMS@LUS-E (S2).The magnetic properties of S1 and S2 are shown in Fig. 4 and S2† in the form of the χMT vs. T plot (χM being the molar magnetic susceptibility per MnII ion and T the absolute temperature). They are qualitatively similar to those of the dinuclear precursor [Mn2L2(Cl)2(μ-Cl)2].18 χMT remains constant from room temperature down to ca. 100 K and it abruptly decreases upon further cooling, suggesting the occurrence of weak antiferromagnetic interactions between the MnII ions within the grafted metal complex. The least-squares fits of the experimental data through the appropriate expression derived from the spin Hamiltonian for an antiferromagnetically coupled dinuclear manganese(II) complex (H = −JS1S2),18 assuming the presence of a variable amount of mononuclear manganese(II) species (α), gave J = −1.5 cm−1 and g = 2.0 for both S1 and S2 with α = 0.25 (S1) and 0.38 (S2) (solid lines in Fig. 4 and S2†). The calculated α values indicate the presence of a relatively larger amount of mononuclear MnII species resulting from the loss of the two bridging chlorides in S2, in agreement with the slightly smaller value of the Cl/Mn ratio, as discussed above.
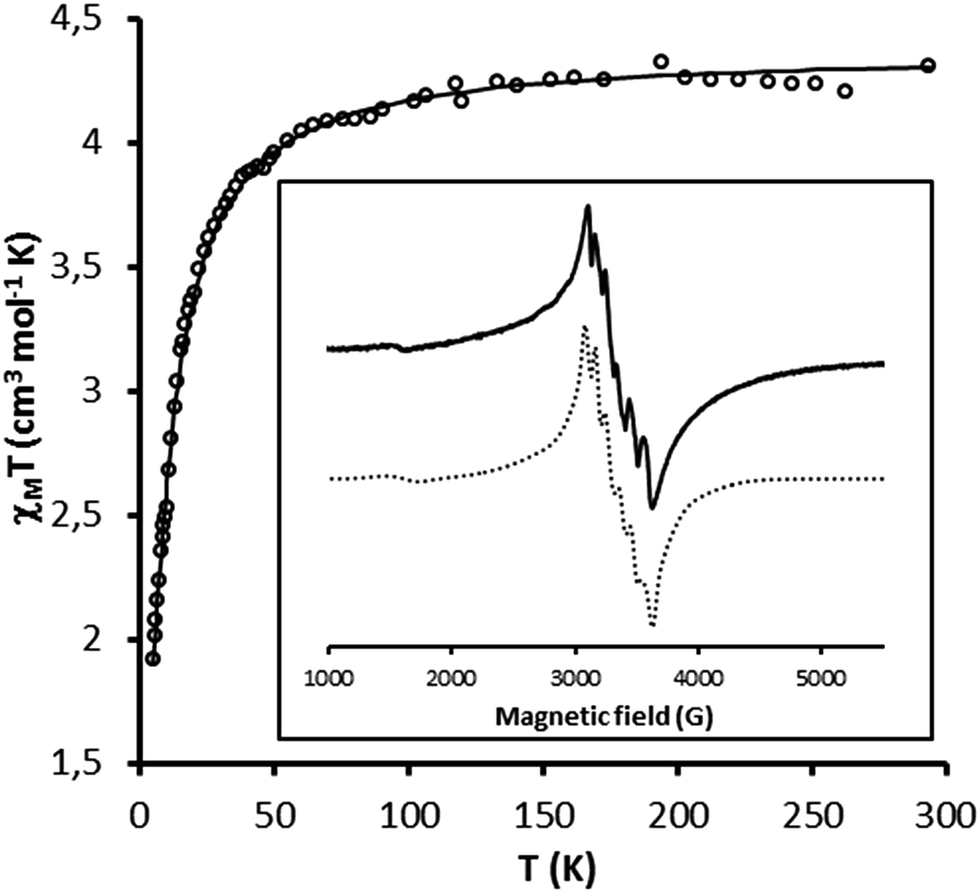 | ||
| Fig. 4 Temperature dependence of χMT for S1. The solid line represents the best fit discuss in the text. Insert: solid-state EPR spectra of S1 at 120 K and its simulation (dotted line). | ||
The calculated −J value in S1 and S2 materials are twenty five-fold larger than that found for the dinuclear precursor (J = −0.059 cm−1).18 The stronger antiferromagnetic coupling for the grafted metal complex could be explained by the bending of the Mn–Cl–Mn angle (θ) within the double-bridged Mn2(μ-Cl)2 diamond core with respect to that found in the dinuclear precursor (θ = 97.5°).18 This situation agrees with the general trend found in related double-bridged di-μ-chlorodimanganese(II) complexes reported in the literature, which exhibit weak ferro- (J = 0.66–1.1 cm−1 with θ = 93.5–96.4°)40–42 to moderate antiferromagnetic couplings (−J = 4.8–5.2 cm−1 with θ = 90.2–90.7°).43,44 Overall, these results suggest that the molecular structure of the dinuclear precursor was likely modified upon grafting within the pore walls of the mesoporous silica.
EPR spectra of S1 and S2 solids confirmed the presence of both monomeric and dimeric Mn species. In both cases the spectra were simulated assuming the monomeric ratio α deduced from the magnetic measurements (Fig. 4 and S2†). The shape of the simulated signals matched well that of the experimental signals assuming one type of monomer and two types of dimers, a dimer 1 with D = 0.0333 cm−1 and a dimer 2 with D = 0.0733 cm−1. The calculated parameters for the different species were: for S1, 62.5% of dimer 1 with g = [1.990 1.990 2.120], 12.5% of dimer 2 with g = [2.000 2.000 2.150], 25% of monomer with g = [2.055 2.018 1.940], and for S2, 28% of dimer 1 with g = [1.990 2.000 2.130], 34% of dimer 2 with g = [1.990 2.100 2.200], 38% of monomer with g = [2.080 2.010 1.940] (see ESI† for additional parameters). The presence of two different dimers revealed by EPR is not surprising given the various possibilities of interaction with the functionalised material or the triazole cycle generated during the Huisgen cycloaddition. The Cl/Mn molar ratio deduced from the elemental analysis for S1 and S2 (0.7 and 0.5, respectively) led us to suppose that the solid likely contains a dimer 1 with two chloride bridges, a dimer 2 with one chloride bridge and a monomer without any chloride ion in the first coordination sphere (Scheme 4 and Fig. S2†). Considering this hypothesis, the calculated Cl/Mn molar ratios would be 0.68 and 0.45 for S1 and S2, respectively, which are within the margin of error of elemental analysis. In addition, the difference of g values for dimer 2 in both solids could be explained either by the presence of a second bridge provided by the different supports or by a different geometry around the metal ion induced by the solid matrix.
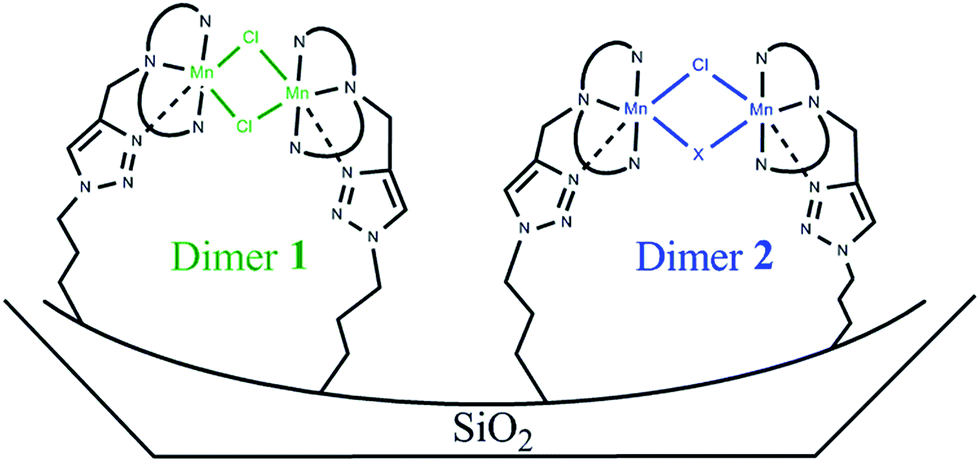 | ||
| Scheme 4 Representation of dimers 1 and 2. X represents a possible second bridge for dimer 2. Dashed bonds are possible bonds created after the click reaction. | ||
Catalytic properties
We first tested the oxidation of 3,5-di-t-butylcatechol (3,5-DTBC), known to be easier to study if compared to catechol, as it leads to more stable intermediates and thus to fewer by-products, due to the presence of 3- and 5-t-butyl blocked positions.45 In order to appreciate the effect of the manganese complex anchoring into the mesoporous silica, we systematically compared the heterogeneous catalyst and its homogeneous analogue. We mainly identified one product, named A, which is the quinone equivalent of 3,5-DTBC (Scheme 5) and confirmed the oxidase activity of our catalysts (Table 2).14| Entry | Catalyst | Base (%) | Conversion (%) | Selectivityb (%) |
|---|---|---|---|---|
| a [Mn2L2(Cl)2(μ-Cl)2] = [Mn]; {Mn}-Py@LUS-E15 = S1; {Mn}-TMS@LUS-E = S2. Standard reaction conditions: 50 mL MeOH, 0.25 mmol 3,5-DTBC, 10% catalyst, O2, room temperature, 5 h; base = 2,2,6,6-tetramethylpiperidine.b Selectivity for product A.c Presence of unknown products.d T = 65 °C. | ||||
| 0 | — | — | 0 | 0 |
| 1 | — | 200 | ∼20c | 0 |
| 2 | MnCl2 | 0 | 100 | 100 |
| 3 | MnCl2 | 10 | 100 | 100 |
| 4 | [Mn] | 10 | 100 | 100 |
| 5 | [Mn]d | 10 | 100 | 100 |
| 6 | S1 | 0 | 100 | 100 |
| 7 | S2 | 0 | 100 | 100 |
Without any catalyst and in the presence of a base (Table 2, entry 1), a mixture of unidentified products and the starting material was obtained. In the absence of base, no reaction occurred, without or with MnCl2 as catalyst (entries 0 and 2, respectively). In homogeneous conditions, with a catalytic amount of base (10%), complete conversion was observed and product A was isolated exclusively whatever the catalyst used, i.e. MnCl2 or [Mn2L2(Cl)2(μ-Cl)2] (entries 3 and 4). Working under reflux did not change the reactivity (entry 5).
Heterogeneous catalysts led also to product A even in the absence of an additional base (entries 6 and 7 for {Mn}-Py@LUS-E15 and {Mn}-TMS@LUS-E, respectively). It is not surprising since it was already observed that pyridine has a negligible effect on this reaction contrary to triethylamine.46 Nevertheless, our heterogeneous catalysts turned out to be as efficient as the homogeneous counterpart in the case of 3,5-DTBC, with the advantage of being easier to separate and potentially recyclable.
We then turned our attention to a more complex substrate, the 4-t-butyl catechol (4-TBC), for which we mainly identified two products, named B16 and C (Scheme 6).47,48 Without catalyst and in the presence of base, a total conversion of 4-TBC to a mixture of unidentified products was obtained (Table 3, entry 1), whereas without base but in presence of [Mn2L2(Cl)2(μ-Cl)2] no reaction occurred (entry 2). With both catalyst and base, a mixture of products B and C were isolated with a 100% total conversion (entry 3).
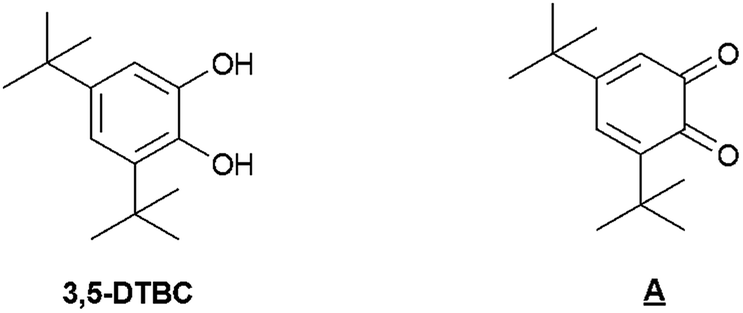 | ||
| Scheme 5 3,5-DTBC and its quinone equivalent A. | ||
| Entry | Catalyst | Base (%) | Oxidant | Conversion (%) | Selectivity (%) | |
|---|---|---|---|---|---|---|
| B | C | |||||
| a [Mn2L2(Cl)2(μ-Cl)2] = [Mn]; {Mn}-Py@LUS-E15 = S1; {Mn}-TMS@LUS-E = S2. Standard reaction conditions: 50 mL MeOH, 0.25 mmol 4-TBC, 10% catalyst, O2, room temperature, 5 h; base = 2,2,6,6-tetramethylpiperidine.b Mixture of unidentified products.c T = 65 °C.d 3 h. | ||||||
| 1 | — | 200 | O2 | 100b | 0 | 0 |
| 2 | [Mn] | 0 | O2 | 0 | 0 | 0 |
| 3 | [Mn] | 200 | O2 | 100 | 60 | 40 |
| 4 | [Mn] | 200 | Air | 100 | 74 | 26 |
| 5 | [Mn] | 200 | Airc | 100 | 66 | 34 |
| 6 | [Mn] | 10 | Air | 48 | 100 | 0 |
| 7 | [Mn] | 200 | Aird | 100 | 38 | 62 |
| 8 | Mn(NO3)2 | 10 | O2 | 0 | 0 | 0 |
| 9 | MnCl2 | 10 | O2 | 0 | 0 | 0 |
| 10 | [Mn] | 10 | O2 | 70 | 100 | 0 |
| 11 | S1 | 0 | O2 | <5 | <5 | 0 |
| 12 | S2 | 0 | O2 | <5 | <5 | 0 |
| 13 | S1 | 200 | O2 | 100 | 100 | 0 |
 | ||
Scheme 6 4-TBC and the two main oxidation products ![[B with combining low line]](https://www.rsc.org/images/entities/char_0042_0332.gif) and and ![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) . . | ||
Changing the nature of oxidant (air instead of oxygen) led to a slight modification of the B/C ratio in favour of B product (entries 3 and 4), whereas heating under reflux had almost no significant effect on this ratio (entries 4 and 5). Reducing the reaction time from 5 h (entry 4) to 3 h (entry 7) diminished the amount of B in favour of C. We thus assume that there is equilibrium between B and C depending on the reaction conditions. Finally, using a catalytic amount of base considerably diminished the activity of the catalyst (entries 4 and 6). Concerning the nature of the catalyst, with either Mn(NO3)2 (entry 8) or MnCl2 (entry 9) no reaction occurred, which proves that the ligand L is required for the activity of the catalyst (entry 10). In the absence of base, the heterogeneous catalysts had almost no activity in the case of 4-TBC (entries 11 and 12), whereas it showed oxidase activity with 3,5-DTBC. In the former case, an addition of stoichiometric amount of base was required to go to completion, leading selectively to only one compound, product B (entry 13). Indeed 3,5-DTBC is easier to oxidise since the intermediates are more stable.49,50 It is noteworthy that in all cases no notable changes were observed in the N2 sorption isotherms, before and after catalytic tests, leading us to believe that the material do not undergo major modifications throughout the whole catalytic process (Fig. S3†). Nonetheless it remains to study the recyclability of these bio-inspired catalysts.
Experimental
Chemicals
All chemicals were used without further purification. Solvents were dried using molecular sieves if needed. Propargylamine, 2-picolyl chloride hydrochloride and 3-chloropropyltriethoxysilane were purchased from Sigma-Aldrich, tetrabutylammonium iodide and 2,2,6,6-tetramethylpiperidine were purchased from Acros Organics, tetramethylammonium bromide, hexamethyldisilazane were purchased from Alfa Aesar, 2-(4-pyridylethyl)triethoxysilane was provided by ABCR and bromotris(triphenylphosphine)copper bromide was purchased from Santa Cruz Biotechnology.Synthesis of the ligand and the azide tether
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C) cm−1.
C) cm−1.Synthesis of the grafted complexes
[Mn2L2(Cl)2(μ-Cl)2] was used to synthesise the hybrid materials.18 The complex was incorporated in a LUS mesoporous silica where azide functions were previously grafted using the so-called molecular stencil pattering technique.34 A Berghof speedwave oven was used for the synthesis of the mesoporous silica LUS using the procedure described elsewhere.18,30 It provides a microwave irradiation at 2450 MHz with a power of 1450 W. The experimental conditions used were: T = 180 °C, ramp = 1 min, plateau = 9 min (total heating time = 10 min).30Synthesis of {Mn}-Py@LUS-E15 (S1)
Synthesis of {Mn}-TMS@LUS-E (S2)18
General procedure for the catalytic tests
The substrate (0.25 mmol) and the base (2,2,6,6-tetramethylpiperidine) were dissolved in MeOH (50 mL) in a single-necked round-bottom flask. To this solution was added the catalyst and the gas was passed through the reaction mixture for 5 h with magnetic stirring.Physico-chemical characterisation
Infrared spectra were recorded from KBr pellets using a Mattson 3000 IRTF spectrometer. Nitrogen sorption isotherms at 77 K were determined with a volume device Belsorp Max on solids that were dried at 80 °C under vacuum overnight. Low angle X-ray powder diffraction (XRD) experiments were carried out using a Brucker (Siemens) D5005 diffractometer using Cu K monochromatic radiation. Liquid NMR spectra were recorded on a Bruker AC 200 spectrometer. Solid NMR spectra were measured on an Advance III Bruker 500 MHz Wide Bore spectrometer, with a CP/MAS DVT (double resonance, variable temperature) probe 4 mm. The delay time between pulses (pulse width 5 μs) in HPDEC experiments was set to 500 s. Spinning speed was set to about 5 kHz and the number of scans was 5000. Frequency 13C = 125.76 MHz, 29Si = 99.36 MHz. EPR spectra were recorded on a Brucker Elexsys e500 X-band (9.4 GHz) spectrometer with a 4102ST cavity (modulation: 2 G, power: 4 mW). The simulated spectra were calculated using the Easyspin toolbox from Matlab. Variable-temperature magnetic susceptibility measurements were made using a Quantum Design MPMS-XL-5 SQUID susceptometer, using an applied field of 1000 G.Conclusions
The N,N′-bis[(pyridin-2-yl)methyl]prop-2-yn-1-amine (L) ligand is a versatile molecule allowing both coordination to a metal ion and grafting to a solid support using click chemistry. The functionalisation method herein reported is not restricted to this particular complex and can be applied to any metal complex provided that the ligand contains a free alkyne group. The site isolation is ensured by the molecular stencil patterning strategy that allows homogeneous grafting of two different functions in a precise ratio. The azide anchor function is surrounded by pyridyl or TMS functions. Once again, the method is not limited to these particular groups and can be extended to other isolating functions. We have demonstrated that the use of CuBr(PPh3)3 as catalyst for the Huisgen cycloaddition avoids copper complexation to L allowing therefore a better yield for the click reaction. The Mn hybrid materials here presented have been tested in the catalytic oxidation of two catechol derivatives (3,5-DTBC and 4-TBC). Their activity has been compared with that of the molecular analogue. With both catechol derivatives tested, oxidation could be achieved using molecular dioxygen as oxidant. This reaction can be compared to the natural oxidase enzymes. Nevertheless, the heterogeneous catalysts were more advantageous as no base was necessary in the case of 3,5-DTBC and only one product was obtained in the case of 4-TBC. Therefore, once grafted in a confined and controlled environment, the metal complex does exhibit a different and more advantageous behaviour than the free complex. The broad range of isolating functions available offers numerous options to control the activity of any inserted complex, which is not possible for complexes in solution. Controlling the environment at a larger range than the sole ligand might allow tuning the catalyst activity, in a similar fashion to the natural enzymes.Acknowledgements
J. C. thanks the French “Ministère de l'Enseignement, de la Recherche et de la Technologie” (MERT) for a PhD fellowship, and the C-MIRA program of Rhône-Alpes region in France for financial support to the collaboration between the Universitat de València (Spain) and the Ecole Normale Supérieure de Lyon (France). The Spanish work was supported by the MICINN (Spain) (Project CTQ2010-15364) and the Generalitat Valenciana (Spain) (Projects PROMETEO/2009/108 and ISIC/2012/002).Notes and references
- B. C. Gates, Catalytic Chemistry, Wiley, 1992 Search PubMed.
- W. Kaim and B. Schwederski, Bioinorganic chemistry: Inorganic Elements in the Chemistry of Life, John Wiley & Sons, 1st edn, 1994 Search PubMed.
- E. A. Lewis and W. B. Tolman, Chem. Rev., 2004, 104, 1047–1076 CrossRef CAS PubMed.
- L. M. Mirica, X. Ottenwaelder and T. D. P. Stack, Chem. Rev., 2004, 104, 1013–1045 CrossRef CAS PubMed.
- E. I. Solomon, U. M. Sundaram and T. E. Machonkin, Chem. Rev., 1996, 96, 2563–2605 CrossRef CAS PubMed.
- P. C. A. Bruijnincx, G. van Koten and R. J. M. K. Gebbink, Chem. Soc. Rev., 2008, 37, 2716–2744 RSC.
- J. P. Emerson, M. L. Wagner, M. F. Reynolds, L. Que, M. J. Sadowsky and L. P. Wackett, J. Biol. Inorg Chem., 2005, 10, 751–760 CrossRef CAS PubMed.
- T. D. H. Bugg and S. Ramaswamy, Curr. Opin. Chem. Biol., 2008, 12, 134–140 CrossRef CAS PubMed.
- L. Que and M. F. Reynolds, Met. Ions Biol. Syst., 2000, 37, 505–525 CAS.
- V. Georgiev, T. Borowski, M. R. A. Blomberg and P. E. M. Siegbahn, J. Biol. Inorg Chem., 2008, 13, 929–940 CrossRef CAS PubMed.
- J. P. Emerson, E. G. Kovaleva, E. R. Farquhar, J. D. Lipscomb and L. Que, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 7347–7352 CrossRef CAS PubMed.
- A. K. Whiting, Y. R. Boldt, M. P. Hendrich, L. P. Wackett and L. Que, Biochemistry, 1996, 35, 160–170 CrossRef CAS PubMed.
- H. Sakurai, S. Shimomura and T. Yoshimura, Biochem. Biophys. Res. Commun., 1983, 115, 618–624 CrossRef CAS PubMed.
- A. Ramadan and I. M. El-Mehasseb, Transition Met. Chem., 1998, 23, 183–189 CrossRef.
- M. U. Triller, D. Pursche, W. Y. Hsieh, V. L. Pecoraro, A. Rompel and B. Krebs, Inorg. Chem., 2003, 42, 6274–6283 CrossRef CAS PubMed.
- N. Shaikh, A. Panja, P. Banerjee and M. Ali, Transition Met. Chem., 2003, 28, 871–880 CrossRef CAS.
- S. Calmettes, B. Albela, O. Hamelin, S. Menage, F. Miomandre and L. Bonneviot, New J. Chem., 2008, 32, 727–737 RSC.
- J. Chaignon, S. E. Stiriba, F. Lloret, C. Yuste, G. Pilet, L. Bonneviot, B. Albela and I. Castro, Dalton Trans., 2014, 43, 9704–9713 RSC.
- S. Abry, A. Thibon, B. Albela, P. Delichère, F. Banse and L. Bonneviot, New J. Chem., 2009, 33, 484–496 RSC.
- V. Jollet, B. Albela, K. Senechal-David, P. Jegou, E. Kolodziej, J. Sainton, L. Bonneviot and F. Banse, Dalton Trans., 2013, 42, 11607–11613 RSC.
- W.-J. Zhou, B. Albela, M.-Y. He and L. Bonneviot, Polyhedron, 2013, 64, 371–376 CrossRef CAS.
- C. T. Kresge, M. E. Leonowicz, W. J. Roth, J. C. Vartuli and J. S. Beck, Nature, 1992, 359, 710–712 CrossRef CAS.
- D. Brunel, N. Bellocq, P. Sutra, A. Cauvel, M. Lasperas, P. Moreau, F. Di Renzo, A. Galarneau and F. Fajula, Coord. Chem. Rev., 1998, 178, 1085–1108 CrossRef.
- K. Moller and T. Bein, Chem. Mater., 1998, 10, 2950–2963 CrossRef CAS.
- D. E. De Vos, M. Dams, B. F. Sels and P. A. Jacobs, Chem. Rev., 2002, 102, 3615–3640 CrossRef CAS PubMed.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS PubMed.
- H. C. Kolb and K. B. Sharpless, Drug Discovery Today, 2003, 8, 1128–1137 CrossRef CAS PubMed.
- J. E. Moses and A. D. Moorhouse, Chem. Soc. Rev., 2007, 36, 1249–1262 RSC.
- L. Bonneviot, M. Morin and A. Badiei, Patent, WO 01/55 031A031, 2001.
- J. Chaignon, Y. Bouizi, L. Davin, N. Calin, B. Albela and L. Bonneviot, Green Chem., 2015, 17, 3130–3140 RSC.
- M. Ortega-Munoz, J. Lopez-Jaramillo, F. Hernandez-Mateo and F. Santoyo-Gonzalez, Adv. Synth. Catal., 2006, 348, 2410–2420 CrossRef CAS.
- S. Abry, B. Albela and L. Bonneviot, C. R. Chim., 2005, 8, 741–752 CrossRef CAS.
- A. Badiei, L. Bonneviot, N. Crowther and G. M. Ziarani, J. Organomet. Chem., 2006, 691, 5911–5919 CrossRef CAS.
- K. Zhang, B. Albela, M. Y. He, Y. M. Wang and L. Bonneviot, Phys. Chem. Chem. Phys., 2009, 11, 2912–2921 RSC.
- M. W. Vetting, L. P. Wackett, L. Que, J. D. Lipscomb and D. H. Ohlendorf, J. Bacteriol., 2004, 186, 1945–1958 CrossRef CAS PubMed.
- V. Georgiev, T. Borowski and P. E. M. Siegbahn, J. Biol. Inorg Chem., 2006, 11, 571–585 CrossRef CAS PubMed.
- S. Huang, R. J. Clark and L. Zhu, Org. Lett., 2007, 9, 4999–5002 CrossRef CAS PubMed.
- W. S. Brotherton, H. A. Michaels, J. T. Simmons, R. J. Clark, N. S. Dalal and L. Zhu, Org. Lett., 2009, 11, 4954–4957 CrossRef CAS PubMed.
- K. Buerglova, N. Moitra, J. Hodacova, X. Cattoen and M. W. C. Man, J. Org. Chem., 2011, 76, 7326–7333 CrossRef PubMed.
- I. Romero, M. N. Collomb, A. Deronzier, A. Llobet, E. Perret, J. Pecaut, L. Le Pape and J. M. Latour, Eur. J. Inorg. Chem., 2001, 69–72 CrossRef CAS.
- J. Z. Wu, E. Bouwman, A. M. Mills, A. L. Spek and J. Reedijk, Inorg. Chim. Acta, 2004, 357, 2694–2702 CrossRef CAS.
- G. A. van Albada, A. Mohamadou, W. L. Driessen, R. de Gelder, S. Tanase and J. Reedijk, Polyhedron, 2004, 23, 2387–2391 CrossRef CAS.
- F. H. Kohler, N. Hebendanz, G. Muller, U. Thewalt, B. Kanellakopulos and R. Klenze, Organometallics, 1987, 6, 115–125 CrossRef.
- B. Brauer, D. Schaarschmidt, C. Flohrer, T. Ruffer, S. Tripke, A. Hildebrandt, B. Walfort, L. Sorace and H. Lang, Inorg. Chim. Acta, 2011, 365, 277–281 CrossRef.
- K. S. Banu, T. Chattopadhyay, A. Banerjee, M. Mukherjee, S. Bhattacharya, G. K. Patra, E. Zangrando and D. Das, Dalton Trans., 2009, 8755–8764 RSC.
- I. C. Szigyarto, L. I. Simandi, L. Parkanyi, L. Korecz and G. Schlosser, Inorg. Chem., 2006, 45, 7480–7487 CrossRef CAS PubMed.
- T. R. Demmin and M. M. Rogic, J. Org. Chem., 1980, 45, 4210–4214 CrossRef CAS.
- M. Lanfranchi, L. Prati, M. Rossi and A. Tiripicchio, J. Mol. Catal. A: Chem., 1995, 101, 75–80 CrossRef CAS.
- J. Mukherjee and R. Mukherjee, Inorg. Chim. Acta, 2002, 337, 429–438 CrossRef CAS.
- J. Reim and B. Krebs, J. Chem. Soc., Dalton Trans., 1997, 3793–3804 RSC.
- J. Rosenthal and S. J. Lippard, J. Am. Chem. Soc., 2010, 132, 5536–5537 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: TGA profiles of materials, magnetic measurements and N2 sorption isotherms. See DOI: 10.1039/c7ra00514h |
| This journal is © The Royal Society of Chemistry 2017 |