
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Heterodinuclear titanium/zinc catalysis: synthesis, characterization and activity for CO2/epoxide copolymerization and cyclic ester polymerization†
Jennifer A.
Garden
 a,
Andrew J. P.
White
b and
Charlotte K.
Williams
*c
a,
Andrew J. P.
White
b and
Charlotte K.
Williams
*c
aEastCHEM School of Chemistry, Joseph Black Building, University of Edinburgh, David Brewster Road, Edinburgh EH9 3FJ, UK
bDepartment of Chemistry, Imperial College London, London, SW7 2AZ, UK
cChemistry Research Laboratory, 12 Mansfield Road, University of Oxford, Oxford, OX1 3TA, UK. E-mail: williams@chem.ox.ac.uk
First published on 3rd February 2017
Abstract
The preparation of heterodinuclear complexes, especially those comprising early-late transition metals coordinated by a simple or symmetrical ancillary ligand, represents a fundamental challenge and an opportunity to prepare catalysts benefitting from synergic properties. Here, two new mixed titanium(IV)–zinc(II) complexes, [LTi(OiPr)2ZnEt] and [LTi(OiPr)2ZnPh], both coordinated by a diphenolate tetra(amine) macrocyclic ligand (L), are prepared. The synthesis benefits from the discovery that reaction of the ligand with a single equivalent of titanium tetrakis(iso-propoxide) allows the efficient formation of a mono-Ti(IV) complex, [LTi(OiPr)2]. All new complexes are characterized by a combination of single crystal X-ray diffraction, multinuclear NMR spectroscopy and mass spectrometry techniques. The two heterobimetallic complexes, [LTi(OiPr)2ZnEt] and [LTi(OiPr)2ZnPh], feature trianionic coordination by the macrocyclic ligand and bridging alkoxide groups coordinate to both the different metal centres. The heterodinuclear catalysts are compared to the mono-titanium analogue, [LTi(OiPr)2], in various polymerization reactions. In the alternating copolymerizations of carbon dioxide and cyclohexene oxide, the mono-titanium complex is totally inactive whilst the heterodinuclear complexes show moderate activity (TOF = 3 h−1); it should be noted the activity is measured using just 1 bar pressure of carbon dioxide. In the ring opening polymerization of lactide and ε-caprolactone, the mono-Ti(IV) complex is totally inactive whilst the heterodinuclear complexes show moderate-high activities, qualified by comparison to other known titanium polymerization catalysts (L-lactide, kobs = 11 × 10−4 s−1 at 70 °C, 1 M in [lactide]) and ε-caprolactone (kobs = 5 × 10−4 s−1 at 70 °C, 0.9 M in [ε-caprolactone]).
Introduction
Synergic chemistry can take place when two metals are combined within a coordination environment either allowing through space or electronic communication so as to improve or increase the overall performance of the complex. Such heterometallic complexes have many useful applications, and have outperformed their homometallic analogues in fields such as metal-halogen exchange,1,2 CH activation3–8 and asymmetric catalysis.9,10 Despite the advances in preparing heterometallic complexes,11–14 heterometallic titanium homogeneous catalysts remain underexplored.15,16 As the second most abundant transition metal in the Earth's crust, titanium is an attractive metal for catalysis as it is sustainable, inexpensive and non-toxic.17 Furthermore, zinc is of high interest because of its low toxicity, low cost, and its lack of colour and redox chemistry. Titanium catalysts are useful in various transformations including hydroaminoalkylation reactions,18 aldol and allylic additions to ketones and aldehydes,19,20 and the epoxidation of alkenes.21 In polymerization catalysis, titanium complexes are particularly effective single site catalysts for olefin polymerization,22–24 and active titanium catalysts have been reported for oxygenated monomers including ε-caprolactone,15,25–28rac-lactide29–49 and, more recently, CO2/epoxide ring opening co-polymerization (ROCOP).27,50–57Heterodinuclear complexes have shown great promise in polymerization catalysis and have allowed greater activities and selectivities to be achieved.14,58–65 Recent breakthroughs include elegant heterodinuclear transition metal complexes based on Ti/Cr and Ti/Zr, developed by Marks and co-workers, which show higher activities in olefin polymerization than previous generations of homodinuclear analogues.14,66,67 So far, there are fewer heterodinuclear early-late transition metal polymerization catalysts, and those based on early-late first row transition metals are quite unusual, despite hints that cooperative activity enhancements can occur.16,68 In 2015, the first examples of hetero-magnesium/zinc catalysts for CO2/epoxide copolymerization were reported, which showed significantly enhanced activities (5–50 times greater) compared to their homodinuclear counterparts.69,70 The alternating copolymerization of CO2 with epoxides represents a practical and useful method of adding value to captured CO2, and there has been much recent academic and industrial interest in this field.71–78 Whilst a range of catalysts are known, those based on zinc showed particular promise and there is also recent precedent for titanium complexes showing activity.79 Therefore, it was of interest to investigate heterodinuclear titanium–zinc catalysts for the alternating copolymerization.
Heterodinuclear complex synthesis and characterization
The goal was to develop the synthesis of a hetero-Ti(IV)–Zn(II) catalyst for ROCOP. Previously, a series of di-zinc catalysts have shown very good performances,80–82 and hetero-Mg–Zn catalysts have shown even greater rates.70 As part of on-going investigations of new catalysts for alternating copolymerization, we were interested to investigate the influence of the different metal centres, therefore the same ancillary ligand (1, L) was applied; the ligand is a diphenolate tetra(amine) macrocycle. Nonetheless, a significant synthetic challenge is to develop the routes to mono-metallate such symmetrical dinucleating ligands. Previously, this was achieved using organozinc reagents and careful temperature/reaction control, so as to prepare LZn complexes. It was of interest to investigate other organometallic reagents to effect mono-metallation and in particular, to apply titanium reagents. The macrocyclic pro-ligand (1, Scheme 1) was prepared according to literature methods,81 and was subsequently reacted with one equivalent of Ti(OiPr)4 at ambient temperature, so as to form a mono-titanium complex [LTi(OiPr)2] (2). 1H NMR analysis of the reaction progress, monitored in d8-toluene, confirmed the successful double deprotonation of 1, with the loss of the phenol–OH resonances (12.10 ppm) and the disappearance of resonances associated with the Ti(OiPr)4 starting reagent (4.50 and 1.25 ppm). The 1H NMR analysis also showed the quantitative formation of complex 2, as characterized by resonances assigned to titanium coordinated iso-propoxide groups (5.13, 5.10, 1.43 and 1.27 ppm) and shifts in all the remaining ligand resonances (Fig. S1 and S2†). 1H NMR monitoring of the reaction confirmed that the relative integration of the iso-propanol and iso-propoxide resonances was 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. The complex was isolated by removal of the reaction solvent and the liberated iso-propanol. Elemental analysis and MALDI-ToF analysis of the isolated solid were fully consistent with the formation of 2. DOSY NMR analysis showed that all the product resonances possess the same diffusion coefficient (Fig. S3†), and calibration experiments suggested that the complex was monomeric in toluene (see ESI† for further details). The ligand 1 is already known to form homodinuclear complexes of Mg(II),83 Fe(III),84 Co(II/III)85 or Zn(II).81 In contrast, the addition of a second equivalent of Ti(OiPr)4 gave the same NMR spectrum as for 2, with free Ti(OiPr)4 also observed. Hexa-coordinate Fe(III) and Ti(IV) have similar ionic radii (FeIII, 69 pm; TiIV, 74 pm), but the formation of di-Ti(IV) complexes is not observed, possibly due to steric hindrance by the iso-propoxide groups, and also because charge balance in a hexa-coordinate complex is not feasible for two M(IV) centres coordinated by the macrocycle.
:1. The complex was isolated by removal of the reaction solvent and the liberated iso-propanol. Elemental analysis and MALDI-ToF analysis of the isolated solid were fully consistent with the formation of 2. DOSY NMR analysis showed that all the product resonances possess the same diffusion coefficient (Fig. S3†), and calibration experiments suggested that the complex was monomeric in toluene (see ESI† for further details). The ligand 1 is already known to form homodinuclear complexes of Mg(II),83 Fe(III),84 Co(II/III)85 or Zn(II).81 In contrast, the addition of a second equivalent of Ti(OiPr)4 gave the same NMR spectrum as for 2, with free Ti(OiPr)4 also observed. Hexa-coordinate Fe(III) and Ti(IV) have similar ionic radii (FeIII, 69 pm; TiIV, 74 pm), but the formation of di-Ti(IV) complexes is not observed, possibly due to steric hindrance by the iso-propoxide groups, and also because charge balance in a hexa-coordinate complex is not feasible for two M(IV) centres coordinated by the macrocycle.
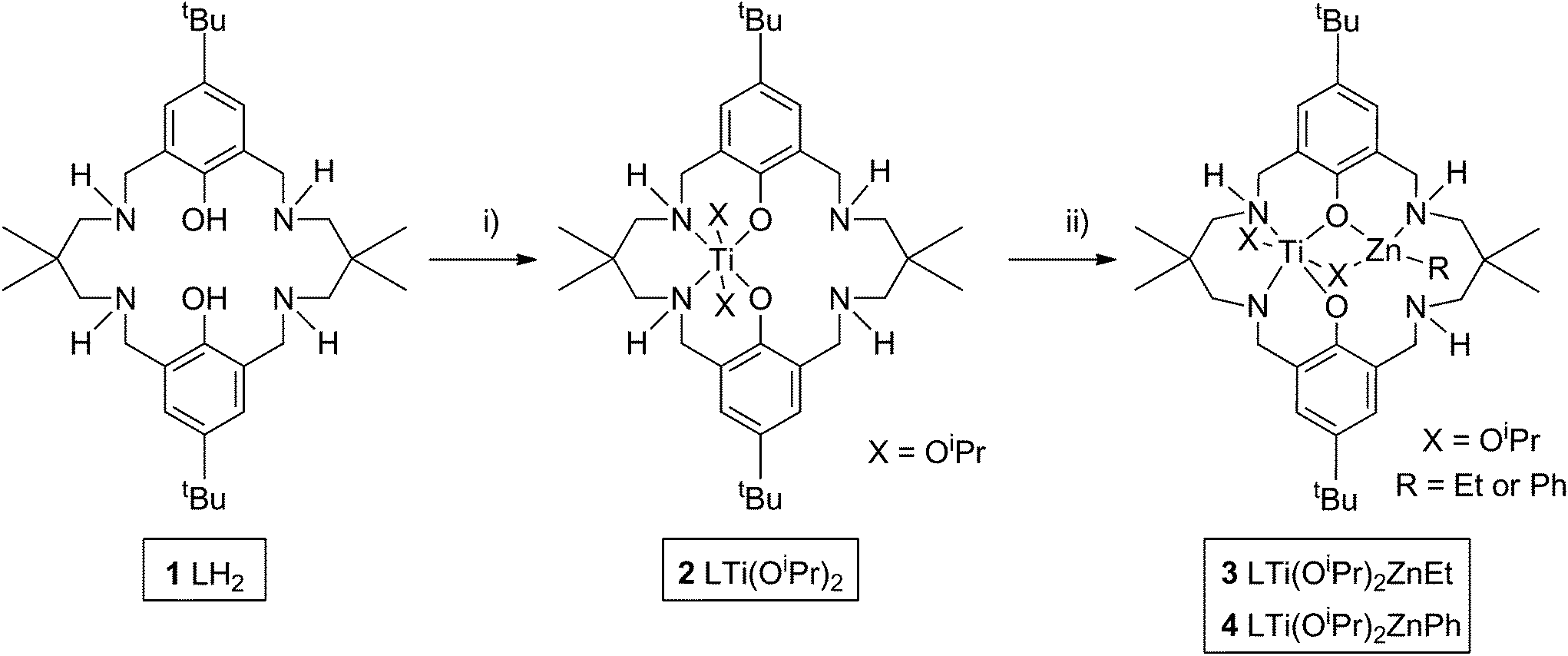 | ||
| Scheme 1 Synthesis of LTi(OiPr)2 (2), LTi(OiPr)2ZnEt (3) and LTi(OiPr)2ZnPh (4). Reagents and conditions: (i) Ti(OiPr)4, toluene, 25 °C, 1 h, 68% isolated yield (quantitative yield by 1H NMR); (ii) 3, Et2Zn, toluene, 25 °C, 10 min, 46% isolated yield (quantitative yield by 1H NMR); 4, Ph2Zn, toluene, 25 °C, 10 min, 64% isolated yield (quantitative yield by 1H NMR). | ||
The successful synthesis of a Ti(IV) complex allowed an exploration of the coordination of other metal centres in the additional coordination site of the ligand. Thus, the addition of zinc and magnesium salts, including ZnCl2 and Mg(OAc)2, to 2 was investigated.70 No reaction was observed. As an alternative method to incorporate a second metal centre, the addition of strongly reducing metals was trialled, with the aim of incorporating an oxidized magnesium or sodium centre, and with reduction of Ti(IV) to Ti(II) or Ti(III), respectively. Magnesium metal was tested, however only traces of a new product were observed by 1H NMR spectroscopy, even after 5 days at 65 °C. Sodium metal was subsequently trialled, due to its higher reduction potential. When the reaction was performed in THF solvent, or when sodium naphthalide was pre-formed as the reductant, the 1H NMR resonances of 2 were completely absent, suggesting that a reaction had occurred. The product showed resonances corresponding to LNa2 together with as yet uncharacterized titanium complexes, revealing that redistribution of the metal centres occurred, and that this route was not a viable method to cleanly insert a second metal into the ligand scaffold. In contrast, the addition of Brønsted bases NaH or Na(N(SiMe3)2) gave sharp, well-defined spectra, suggesting the presence of a new complex, and with only a little redistribution to form LNa2. The same set of new product resonances was observed using NaH or Na(N(SiMe3)2), which suggested that the major product was L3− Ti(OiPr)2Na (Fig. S4†).
As the preliminary studies suggested that the ligand could adopt a trianionic coordination mode, achieved by deprotonation of the NH, diethylzinc was selected as a reagent to prepare heterometallic complexes. The selection of zinc was driven by the excellent precedent for zinc catalysts in CO2/CHO alternating copolymerization and in lactide or ε-caprolactone polymerizations.30,86–95 The addition of Et2Zn to a yellow solution of 2, in toluene, immediately formed a vivid orange solution (Scheme 1). Orange, block crystals were deposited from toluene solvent, after 24 hours at ambient temperature. The structure was elucidated through single crystal X-ray diffraction and revealed the product to be [LTi(OiPr)2ZnEt], 3 (Fig. 1a). When Ph2Zn was used as the organo-zinc reagent, the formation of crystals of the aryl analogue, [LTi(OiPr)2ZnPh], 4 (Fig. 1b), were yielded as orange, block crystals from a hexane/THF solution at −40 °C after 2 weeks. Structurally characterized Zn/Ti complexes are rare, particularly those which are held in a symmetrical macrocyclic environment.96,97
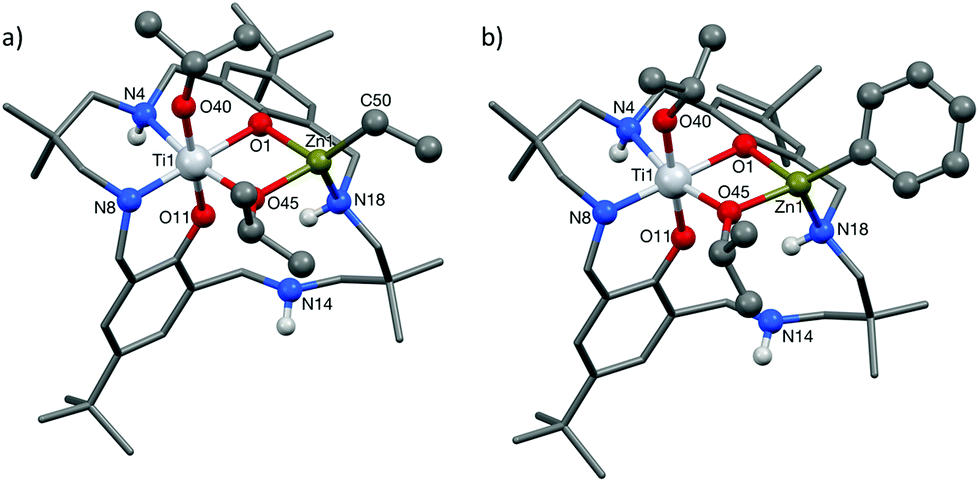 | ||
| Fig. 1 Crystal structures of (a) LTi(OiPr)2ZnEt and (b) LTi(OiPr)2ZnPh. | ||
Structural elucidation of 3 and 4 revealed that the two complexes are very similar, and the titanium centres are hexa-coordinate, connected to two phenolic oxygens, two iso-propoxide groups and two nitrogen centres, one of which is anionic (N8). The constraints of the tetra-coordinated macrocyclic ligand generates a distorted octahedral Ti geometry, with the cis angles ranging from 80.57(8)°–105.82(9)° (3), and 79.34(5)°–107.98(6)° (4), and the trans angles varying from 165.64(8)°–171.04(8)° (3), and 164.62(6)°–173.20(6)° (4). In both 3 and 4, the zinc centre has a distorted tetrahedral geometry98 and is connected to one phenolic oxygen, one iso-propoxide group and one nitrogen centre, with an additional ethyl (3) or aryl (4) ligand present to balance the charge. The large Zn1–O11 distances [3.601(2) Å, 3; 3.599(1) Å, 4] suggest that there is no coordination of the second phenolic O atom. As a result of containing both a hexa-coordinate and a tetra-coordinate metal centre, the ligand conformation is highly distorted and the two phenol rings lie almost perpendicular to each other (ring planes inclined by ca. 87°, 3; 84°, 4). This contrasts with the commonly observed “bowl” or “step”-shaped conformation of metal complexes of 1.80–82,85,99
It is also the first time that a trianionic ligand derived from 1 has been structurally characterized, demonstrating that NH deprotonation is a viable means to selectively incorporate a second metal into the complex. Only three of the four available nitrogen centres are involved in bonding to the metal atoms, and the non-coordinated nitrogen is an NH. The respective N8–C7 and N8–C9 bond lengths are 1.459(3) Å and 1.469(3) Å in 3, and 1.468(2) Å and 1.469(2) Å in 4, which rules out the presence of imine bond character. There is a clear distinction between the dative and anionic N–Ti bonds, as the anionic N–Ti bond length is significantly shorter than the N(H)–Ti bond (by 0.2 Å in 3; 0.3 Å in 4). A significant difference is also observed between the Ti–OiPr bond lengths, with the terminal iso-propoxide oxygen held 0.16 Å closer to the Ti centre than the bridging iso-propoxide oxygen, in both 3 and 4. In 3, the bridging iso-propoxide oxygen lies closer to Ti (1.975(2) Å, 3) than Zn (2.012(2) Å), whereas the iso-propoxide bridge of 4 lies almost equidistant from Ti (1.980(1) Å) and Zn (1.976(1) Å). Amine deprotonation occurs adjacent to Ti rather than Zn, which is perhaps surprising as it contrasts with reported examples of titanium zincate complexes, where it is the zinc centre which bears a formal anionic charge.96,97,100–104 It seems plausible that the amine ligand may donate electron density to the Ti centre and acidify the NH group so that it can be deprotonated by Et2Zn or Ph2Zn. Hydrogen bonding to N(14) provides further stabilization for the Zn-coordinated N18 H (N18⋯N14 2.910(3) Å, H⋯N14 2.195(8) Å, N18–H⋯N14 136.0(10)°, 3; N18⋯N14 2.892(2) Å, H⋯N14 2.178(13) Å, N18–H⋯N14 135.8(16)°, 4).
Monitoring the reaction of 2 with Et2Zn, on a 1H NMR scale in d8-toluene, revealed the immediate loss of resonances assigned to 2 and Et2Zn, accompanied by the formation of a new set of product resonances along with ethane (0.81 ppm). A distinctive sharp 1H NMR spectrum was obtained (Fig. S5 and S6†), where all signals were fully assigned to complex 3 as the sole reaction product. Consistent with a low symmetry complex, 21 individual resonances were observed for the benzylic, NH, OCH(CH3)2 and methylene protons. A distinctive doublet was observed at 5.66 ppm, attributed to one of the diastereotopic benzylic protons adjacent to the nitrogen anion, whereas the other diastereotopic proton was observed at 3.17 ppm. The benzylic and methylene protons adjacent to the metal-coordinated nitrogen atoms are diastereotopic, in contrast to those adjacent to the non-coordinated nitrogen. These resonances were fully assigned through COSY and HSQC experiments. Two individual OiPr CH resonances were observed (4.71 and 5.01 ppm), which hints that the solid-state structure is retained in solution, with one OiPr group bridging between two metal centres while the second is solely coordinated to Ti. DOSY analysis was performed, which confirmed that all product resonances possess the same diffusion coefficient. Comparison of the diffusion coefficient to standards gave a calculated molecular weight of 711 g mol−1, which is in good agreement with that of a monomeric species in d8-toluene (808 g mol−1). The 1H NMR spectra of 3 and 4 (Fig. S7 and S8†) are broadly similar, and demonstrate the generality of applying other organo-zinc reagents to prepare hetero-Ti/Zn complexes.
Applications in polymerization
Heterodinuclear 3 and 4 were tested for the alternating copolymerizations of CO2/cyclohexene oxide (CHO), using 1 mol% catalyst loading (vs. the epoxide, CHO) and 1 bar pressure of CO2 at 80 °C, the conditions were selected as being optimum for related di-zinc catalysts.71 Both complexes were moderately active, giving up to 52% conversion after 24 h (Table 1, entry 3) and exhibited high CO2 uptake, resulting in ∼94% carbonate linkages. The polymerization was moderately well-controlled, with a MW of 2190 g mol−1 and a dispersity of 1.35. The molecular weight distribution is bimodal, a feature which has been observed with many different catalysts for this copolymerization,85,105–107 and which has previously been shown to be due to chain transfer reactions between the catalyst and cyclohexanediol; the diol is itself formed by reaction between epoxide and trace water as was clearly demonstrated by Darensbourg and co-workers using detailed spectroscopic monitoring of polymerizations.108 Accordingly, the MALDI-ToF spectrum of the purified polymer showed two series of chains, which differed according to the chain end groups (Fig. S9†). One series was a α-propoxide-ω-hydroxyl end-capped polycyclohexene carbonate, whereas the second was a telechelic polymer terminated by hydroxyl groups. The mono-titanium species 2 was completely inactive for polymerizations and no polycarbonate was observed after 24 hours (entry 1). The lack of activity may provide indirect support for the notion that two metals are required for catalysis.109 Furthermore, the monometallic zinc analogue, LZn, synthesized through the reaction of ligand 1 with one equivalent of either Et2Zn or Ph2Zn, has also shown no activity towards CO2/CHO copolymerization, most likely because of the lack of an initiating co-ligand.70 Comparing catalyst 3 against other known Ti(IV) catalysts for this polymerization is rather complex as other catalysts is more difficult due to the range of different conditions used and more details are provided in the ESI regarding specific data for literature catalysts (Fig. S10 and Table S1†).56 Even given the different conditions used in testing other catalysts, catalyst 3 appears less active than the titanium diphenolate50 or titanium bis(salphen)54 complexes (Fig. S10 and Table S1†).53,56 It should however be appreciated that the other reported Ti catalysts require the addition of co-catalyst to achieve polymerizations, in the case of catalysts 3 or 4 such additives are unnecessary.| Entry | Monomer | Catalyst | Time | Temp (°C) | Conv.a (%) | M n [Đ] | M n calc. |
|---|---|---|---|---|---|---|---|
|
a Determined by 1H NMR.
b Reaction conditions: 1 mol% catalyst (vs. CHO).
c Determined by size-exclusion chromatography analysis in THF, using narrow Mn polystyrene standards as the calibrant.
d Reaction conditions: THF, [catalyst]:[L-LA] – 1:100, [L-LA] – 1.0 M.
e Determined by size-exclusion chromatography using a MALLS detector, using dn/dc values = 0.05 (PLA) and 0.078 (PCL) (refer to ESI).
f Calculated using the formula Mn = DP/[cat] = (% conversion × 144.13)/1 (assuming one chain grows per catalyst).
g
rac-Lactide was used and the Pr value was determined to be 0.68 through homodecoupled 1H NMR data.112
h Reaction conditions: THF, [catalyst]:[ε-CL] – 1:100, [ε-CL] – 0.9 M.
i Calculated using the formula Mn = DP/[cat] = (% conversion × 114.14)/1 (assuming one chain grows per catalyst).
|
|||||||
| 1b | CO2/CHO | 2 | 24 h | 80 | 0 | — | — |
| 2b | CO2/CHO | 3 | 6 h | 80 | 18 | — | — |
| 3b | CO2/CHO | 3 | 24 h | 80 | 52 | 2190 [1.35]c | — |
| 4b | CO2/CHO | 4 | 6 h | 80 | 23 | — | — |
| 5b | CO2/CHO | 4 | 24 h | 80 | 40 | 1750 [1.37]c | — |
| 6d | L-LA | 2 | 2 h | 21 | 0 | — | — |
| 7d | L-LA | 3 | 2 h | 21 | 31 | 8860 [1.15]e | 4510f |
| 8d | L-LA | 3 | 40 min | 70 | 89 | 12580 [1.13]e |
12830f |
| 9d | rac-LAg | 3 | 2 h | 50 | 92 | 11080 [1.45]e |
13260f |
| 10d | L-LA | 4 | 40 min | 70 | 75 | 6970 [1.17]e | 10750f |
| 11h | ε-CL | 2 | 24 h | 21 | 0 | — | — |
| 12h | ε-CL | 3 | 24 h | 21 | 86 | 13570 [1.26]e |
9820i |
| 13h | ε-CL | 3 | 140 min | 70 | 94 | 5360 [1.21]e | 5950i |
| 14h | ε-CL | 4 | 90 min | 70 | 94 | 5820 [1.33]e | 10700i |
Complexes 2–4 were also tested for the ring opening polymerization (ROP) of L-lactide (L-LA) ([LA] = 1 M), using 1 mol% catalyst loading (vs. L-LA) in THF solution; once again the conditions were selected as being commonly used in testing in ROP catalysis.110,111 Heterodinuclear catalysts 3 and 4 displayed moderate activities with high conversions being obtained in 40 minutes at 70 °C (Table 1, entries 8 and 10). The polymerization kinetics showed a first order dependence in monomer concentration (Fig. 2, LHS). The polymerization was well-controlled, with a linear correlation between the Mn and L-LA conversion and reasonable correlation between predicted and experimental values of MW, with dispersities <1.20 in most cases (Fig. 2, RHS). The ROP of rac-lactide was also investigated and low hetero-selectivity was observed (Pr = 0.68, Table 1, entry 9).112 The catalyst activity of 3 (kobs = 4 h−1 (= 11 × 10−4 s−1) at 70 °C, 1 mol% catalyst loading, [LA] – 1 M in THF), is competitive compared to other titanium alkoxide catalysts (refer to ESI, Fig. S11 and Table S2†).30–33 For example, 3 is 250 times faster, albeit at a higher concentration of monomer, than a related mono-titanium catalyst based on a diphenolate ligand scaffold (kobs = 16 × 10−3 h−1 at 100 °C, 1 mol% loading, [LA] – 0.5 M in benzene, Fig. S11-E†).32 In contrast to the promising activity exhibited by the heterodinuclear complexes 3 and 4, the mononuclear Ti(IV) complex 2 showed no activity at all in the ROP of L-LA (Table 1, entry 6). The heterodinuclear complex 4 which has a phenyl group attached to the zinc centre gave a slightly slower rate of propagation (6.3 × 10−4 s−1), providing some evidence that the phenyl group remains coordinated to the catalyst during the polymerization. The MALDI-ToF analysis confirms this hypothesis as it shows only polylactide chains terminated by iso-propyl (ester) and hydroxyl end groups; there was no evidence for any Zn–Et or Zn–Ph initiation of polymerizations (Fig. S12†). The reactivity enhancement observed may have a steric origin; in 3, the ligand is “tied back” to expose the zinc centre, which might facilitate lactide coordination [3, N18–Zn1–O1 84.40(8)°, N18–Zn1–O45 106.86(8)°, O1–Zn1–O45 80.38(7)°; 4, N18–Zn1–O1 86.28(6)°, N18–Zn1–O45 109.18(6)°, O1–Zn1–O45 80.66(5)°].
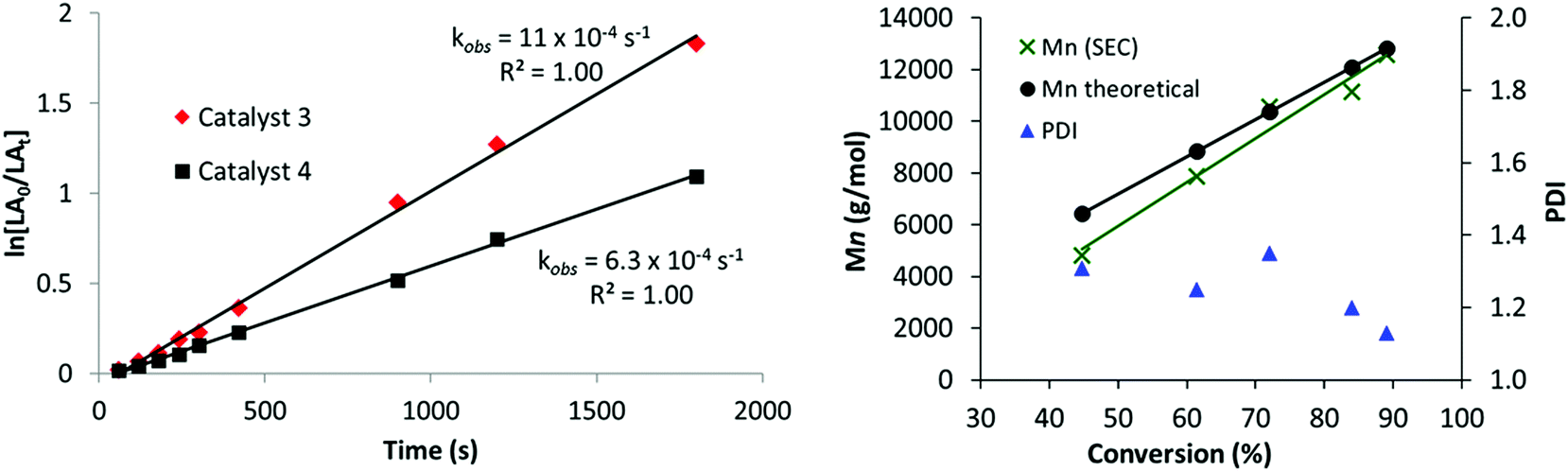 | ||
| Fig. 2 LHS: Plot of ln([L-LA0]/[L-LAt]) versus time (s) using 3 (◆) and 4 (■). RHS: Plot of the molecular weight determined by SEC (X) and calculated (●) versus the conversion for 3 (100 equiv. L-LA in THF, at 70 °C: [L-LA] – 1 M). | ||
The new catalysts were also investigated for the ROP of ε-caprolactone (ε-CL), using 1 mol% catalyst loading (vs. ε-CL), at 70 °C in THF. Once again, only the hetero-Ti(IV)–Zn complexes 3 and 4 were active with the mono-Ti(IV) complex 2 showing no activity (Table 1). Heterodinuclear 3 displayed moderate activities (refer to ESI, Fig. S13 and Table S3†)25,27 but also showed a clear induction period of approximately 45 minutes, during which very low conversions were observed. Such induction or initiation periods are common for ε-CL polymerization with other Ti and group 4 iso-propoxide catalysts,27,113,114 and are attributed to a structural rearrangement of the ligands of the initiator, allowing the monomer to access the metals coordination sphere prior to insertion.115–117 Intriguingly, no such induction period is observed with L-LA, which may suggest that the two monomers differ in their coordination mode to the heterobimetallic catalyst systems. After initiation, however, the polymerization progressed efficiently showing a linear fit to ln([ε-CL0]/[ε-CLt]) versus time data (Fig. 3, LHS), corresponding to a first-order dependence on ε-CL concentration (kobs = 5.2 × 10−4 s−1). Curiously, a much shorter induction period is observed using heterodinuclear catalyst 4, which therefore displayed much higher activity overall, in spite of the almost identical polymerization rates. Close inspection of the bond lengths and angles reveal that the Ti–OiPrterminal bond is 0.02 Å shorter and stronger in 3, which may in part account for its lower reactivity towards initiation. For both 3 and 4, MALDI-ToF analysis of the purified polycaprolactone showed two series of chains, with different end groups. While the first is an α-propoxide, ω-hydroxy end-capped PCL, the second is more unusual, with the chain ends terminated by the ligand, and a hydroxy group (Fig. S14†). This finding suggests that the anionic ligand scaffold can also act as an initiating species in catalysis, a feature which is preferably avoided to be sure of optimum performance and control.
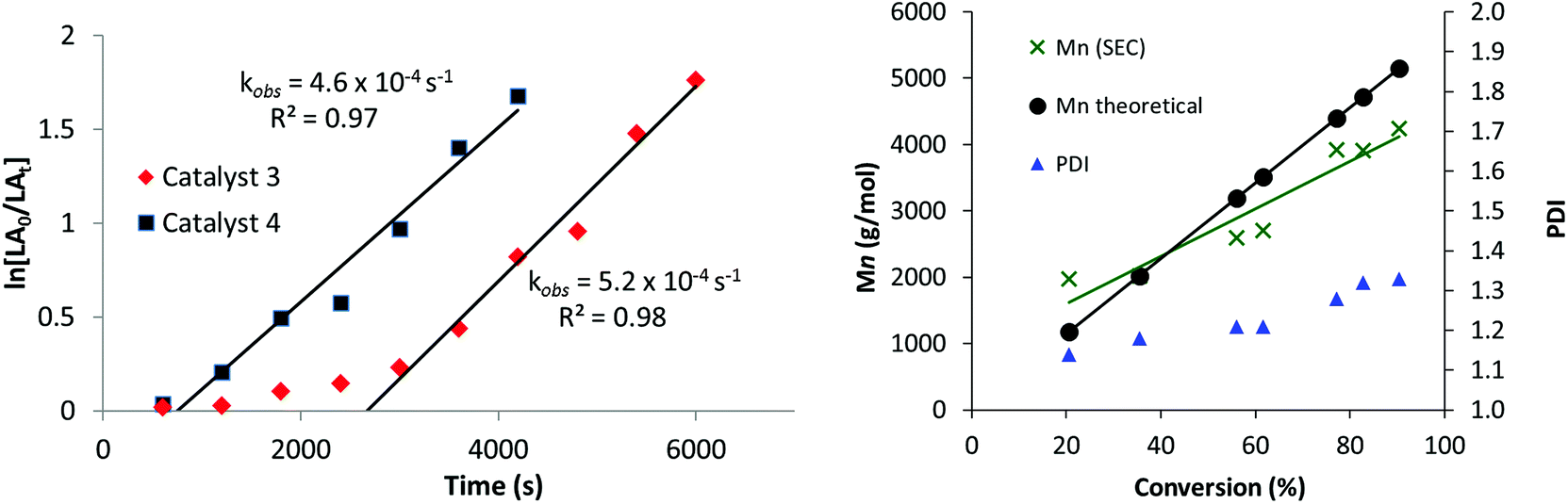 | ||
| Fig. 3 LHS: Plot of ln([ε-CL0]/[ε-CLt]) versus time (s) using 3 (100 equiv. ε-CL in THF, at 70 °C: [ε-CL] – 0.9 M). RHS: Plot of the molecular weight determined by SEC (X) and calculated (●) versus the conversion for 3 (100 equiv. ε-CL in THF, at 70 °C: [ε-CL] – 0.9 M). | ||
Conclusions
In conclusion, two new heterodinuclear Ti(IV)/Zn(II) complexes were synthesized from a symmetrical macrocyclic ligand by taking advantage of the facility to form a mono-Ti(IV) complex first and to subsequently add an organo-zinc reagent. The new complexes are examples of early-late first row transition metal heterodinuclear catalysts. They were fully characterized using X-ray crystallographic and NMR spectroscopic studies. For this particular class of macrocycles, homodinuclear complexes are well known but heterodinuclear analogues, particularly synthesized in high yields, remain very unusual. The ligand also adopts an unexpected trianionic coordination with the metals, and the complexes contain co-ligands (alkoxides) which are effective initiators for various polymerizations. The new heterodinuclear complexes show good performances in both the alternating copolymerization of epoxide and carbon dioxide and the ring opening polymerization of lactones/lactide. The mono-Ti(IV) complex is completely inactive which builds further evidence towards the importance of dinuclear polymerization catalysts and the potential for mixed metal synergic interactions. Generally, there is a growing evidence that heterodinuclear complexes are an important focus in polymerization catalysis; in future it will also be necessary to ensure that where possible earth abundant elements like titanium or zinc are applied so as to produce more sustainable and cost-effective catalysts. Further work is necessary to optimize the catalysis and could be directed towards the incorporation of different metals and co-ligands.Experimental section
All metal complexes were synthesized under anhydrous conditions, using MBraun gloveboxes and standard Schlenk techniques. Solvents and reagents were obtained from Sigma Aldrich or Strem and were used as received unless stated otherwise. THF and toluene were dried by refluxing over sodium and benzophenone and stored under nitrogen. Cyclohexene oxide (CHO) was dried over CaH2 and fractionally distilled under nitrogen. All dry solvents and reagents were stored under nitrogen and degassed by several freeze–pump–thaw cycles. A research grade CO2 cylinder supplied by BOC (100% purity), and fitted with a Drierite drying column, was used as the CO2 source for all copolymerization studies. Macrocyclic ligand 1 was synthesized following literature procedures.81 NMR spectra were recorded using a Bruker AV 400 MHz spectrometer. Correlations between proton and carbon atoms were obtained by using COSY and HSQC NMR spectroscopic methods. Elemental analysis was determined by Stephen Boyer at London Metropolitan University. SEC was performed using two Mixed Bed PSS SDV linear S columns in series, with THF as the eluent, at a flow rate of 1 mL min−1, on a Shimadzu LC-20AD instrument at 40 °C. For polycarbonate, the molecular weight (Mn) was determined by comparison against polystyrene standards. For polylactide and polycaprolactone, the MALLS detector was calibrated by polystyrene standards and the dn/dc values were measured using an external RID detector (Knauer) (refer to ESI†). The polymer samples were dissolved in SEC grade THF and filtered prior to analysis.Complex synthesis
Two sets of resonances are observed in d8-toluene solvent at 298 K which are attributed to the presence of two conformational isomers, as has been previously observed with other organometallic complexes derived from 1.70
1H NMR (d8-toluene, 400.20 MHz, 298 K): 7.38, 7.09, 7.01 and 6.82 (d, 1H, m-ArH), 7.30, 7.13, 7.04 and 6.93, (d, 0.8H, m-ArH*), 5.13 and 5.10 (sept, 1H, CH–OiPr), 5.12 and 4.55 (m, 0.8H, CH–OiPr*), 4.75 (d, J = 13.7, 3.4 Hz, 1H, benzylic–CH), 4.62 (dd, J = 12.2, 3.1 Hz, 1H, benzylic–CH), 4.40 (dd, J = 12.1, 8.1 Hz, 0.8H, benzylic–CH*), 4.32 (dd, J = 12.4 Hz, 1H, benzylic–CH), 4.08 (dd, J = 11.6 Hz, 0.8H, benzylic–CH*), 3.85 (dd, J = 11.7, 7.8 Hz, 1H, benzylic–CH), 3.64 (dd, J = 12.9, 6.3 Hz, 0.8H, benzylic–CH*), 3.35 (dd, J = 12.2, 9.9 Hz, 1H, benzylic–CH), 3.27–2.98 (m, 6H, 2× benzylic–CH, 4× benzylic–CH*, 1× methylene–CH, 1× methylene–CH*), 2.98–2.83 (m, 2.6H, 1× benzylic–CH, 1× benzylic–CH*, NH*), 2.83–2.65 (m, 2.6H, 1× methylene–CH, 2× methylene–CH*), 2.69–2.54 (m, 2.6H, 1× methylene–CH, 2× methylene–CH*), 2.58–2.35 (m, 4.8H, 1× NH, 3× methylene–CH, 1× methylene–CH*), 2.05–1.90 (m, 4.6H, 2× methylene–CH*, 2× methylene–CH, 1× NH), 1.62 (m, 1.8H, NH and NH*), 1.54 (m, 1.8H, NH and NH*), 1.43 (d, J = 6.1 Hz, 3H, CH3–iPrO), 1.37 (d, J = 6.1 Hz, 5H, CH3–iPrO*), 1.34 (s, 9H, tBu), 1.31 (s, 7H, tBu*), 1.30 (s, 7H, tBu*), 1.27 (d, 6H, CH3–iPrO), 1.25 (s, 9H, tBu), 1.19 (s, 2.4H, C(CH3)2*), 1.03 (d, 3H, CH3–iPrO), 1.02 (s, 3H, C(CH3)2), 0.98 (s, 3H, C(CH3)2), 0.86 (s, 2.4H, C(CH3)2*), 0.71 ppm (s, 3H, C(CH3)2), 0.58 (s, 2.4H, C(CH3)2*), 0.31 (d, J = 5.9 Hz, 5H, CH3–iPrO*), 0.11 (s, 3H, C(CH3)2).
13C NMR (d8-toluene, 100.63 MHz, 298 K): 128.2, 126.9, 124.3, 122.0 (m-Ar), 129.3, 127.1, 124.4, 124.2 (m-Ar*), 78.0 and 75.7 (CH–OiPr and CH–OiPr*), 62.2, 60.6, 58.0 and 57.7 (methylene–CH2), 62.7, 60.9, 58.0 and 56.9 (methylene–CH*), 57.4, 57.0, 52.2 and 51.4 (benzylic–CH2), 55.8, 53.6, 52.2 and 51.9 (benzylic–CH2*), 36.3, 35.3, 34.7, 34.1, 33.9 and 33.8 (quat. tBu, tBu*, CMe2 and CMe2*), 32.1, 32.0, 31.9 and 31.8 (tBu, tBu*, CH3–iPrO* and C(CH3)2*), 27.1 (C(CH3)2*), 26.9 (CH3–iPrO), 26.7 (C(CH3)2), 26.6 (CH3–iPrO), 26.4 (CH3–iPrO*), 26.2 (CH3–iPrO), 25.5 (C(CH3)2*), 24.1 ppm (C(CH3)2). Not all quaternary carbon resonances were not observed.
Anal. Calc. for LTi(OiPr)2:C, 67.02; H, 9.56; N, 7.82. Found: C, 66.84; H, 9.66; N, 7.71
MS (MALDI-TOF): m/z 657.8 [LTi(OiPr)]+ (100%)
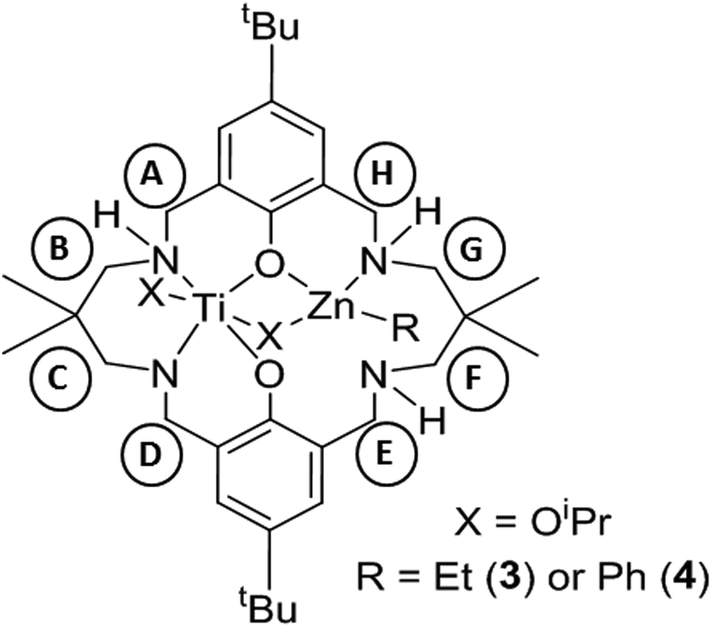
13C NMR (d8-toluene, 100.63 MHz, 298 K): 160.6 (i-Ph), 138.6 (quat. Ar–C), 127.9 (Ar–CHH), 126.6 (Ar–CHA), 125.1 (Ar–CHE) and 123.6 (Ar–CHD), 77.4 (CHC), 75.1 (CH–OiPr), 69.8 (CH–OiPr), 65.3 (CHD), 64.4 (CHB), 57.0 (CHF), 56.7 (CHG), 55.9 (CHA), 52.3 (CHH), 51.2 (CHE), 37.7, 34.6, 33.9 and 33.8 [quat. C, tBu and C(Me)2], 32.1 [C(CH3)3], 31.9 [C(CH3)3], 31.1 [C(CH3)2], 28.3 [C(CH3)2], 28.1 (CH3–iPrO), 27.3 (CH3–iPrO), 27.2 (CH3–iPrO), 27.0 (CH3–iPrO), 26.5 [C(CH3)2], 23.9 [C(CH3)2], 14.2 (Zn–CH2–CH3), −1.0 ppm (Zn–CH2–CH3). Not all quaternary aromatic carbon signals were observed.
Anal. Calc. for LTiZn(OiPr)2Et: C, 62.26; H, 8.96; N, 6.91. Found: C, 61.86; H, 8.81; N, 6.86.
Due to the air-sensitivity of LTiZn(OiPr)2Et, suitable MS data could not be collected.
1H NMR (d8-toluene, 400.20 MHz, 298 K): 7.94 (dd, J = 8.0 Hz, o-Ph, 2H), 7.40 (dd, J = 7.9, 6.9 Hz, m-Ph, 2H), 7.28 (t, J = 8 Hz, p-Ph, 1H), 7.15 and 7.05 (d, 1H, m-Ar(HA) and m-Ar(HH)), 7.00 (d, J = 2.3 Hz, 1H, m-Ar(HE)), 6.98 (d, J = 2.7 Hz, 1H, m-Ar(HD)), 5.81 (dd, J = 11.0 Hz, 1H, NHG), 5.66 (d, J = 15.5 Hz, 1H, HD), 5.24 (d, J = 12.5 Hz, 1H, HC), 5.04 (sept, J = 6.0 Hz, 1H, CH–OiPr), 4.71 (sept, J = 6.1 Hz, 1H, CH–OiPr), 4.64 (dd, J = 13.8, 10.3 Hz, 1H, HH), 4.34 (m, 4H, NHE), 4.14 (dd, J = 11.8 Hz, 1H, HA), 3.56 (d, J = 13.6 Hz, 1H, HH), 3.23 (dd, J = 12.1 Hz, 1H, HB), 3.15 (d, J = 15.7 Hz, 1H, HD), 3.05–2.88 (m, 5H, HA, HE, HG, HF), 2.69 (dd, J = 14.1 Hz, 12.1 Hz, 1H, HG), 2.52 (dd, J = 12.5 Hz, 4.1 Hz, 1H, HC), 2.43 (dd, J = 12.7 Hz, 5.6 Hz, 1H, HF), 1.99 (d, J = 11.4 Hz, 1H, HB), 1.74 (m, 1H, NHA), 1.40, 1.37, 1.33 and 1.23 (d, 3H, CH3–iPrO), 1.29 and 1.29 (s, 9H, tBu), 0.83 and 0.76 (s, 3H, C(CH3)2), 0.43 and 0.25 ppm (s, 3H, C(CH3)2).
13C NMR (d8-toluene, 100.63 MHz, 298 K): 139.7 (o-Ph), 139.1 (quat. C), 138.8 (quat. C), 127.5 (m-Ph), 127.1 and 126.7 (Ar–CHA and Ar–CHH), 126.3 and 126.1 (quat. C), 125.6 (p-Ph), 125.6 (Ar–CHE), 123.6 (Ar–CHD), 77.5 (CHC), 75.3 (CH–OiPr), 70.0 (CH–OiPr), 65.4 (CHD), 64.4 (CHB), 57.2 (CHG), 56.7 (CHF), 56.0 (CHA), 52.6 (CHH), 51.2 (CHE), 37.8, 34.6, 33.9 and 33.8 [quat. C, tBu and C(Me)2], 32.1 [C(CH3)3], 31.8 [C(CH3)3], 30.9 [C(CH3)2], 28.4 [C(CH3)2], 27.4 (CH3–iPrOH), 27.2 (CH3–iPrOH), 27.1 (CH3–iPrOH), 26.8 (CH3–iPrOH), 26.5 [C(CH3)2], 23.9 ppm [C(CH3)2]. Not all quaternary aromatic carbon signals were observed.
Anal. Calc. for LTiZn(OiPr)2Ph: C, 64.37; H, 8.46; N, 6.53. Found: C, 64.06; H, 8.32; N, 6.28.
Due to the air-sensitivity of LTiZn(OiPr)2Ph, suitable MS data could not be collected.
Acknowledgements
The authors gratefully acknowledge the EPSRC (EP/K035274/1) and Climate KIC (project EnCO2re) for funding. Dr Valentin Poirier is acknowledged for the determination of the dn/dc values for polylactide and polycaprolactone.References
- E. Hevia, J. Z. Chua, P. García-Álvarez, A. R. Kennedy and M. D. McCall, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 5294–5299 CrossRef CAS PubMed.
- T. D. Bluemke, W. Clegg, P. Garcia-Alvarez, A. R. Kennedy, K. Koszinowski, M. D. McCall, L. Russo and E. Hevia, Chem. Sci., 2014, 5, 3552–3562 RSC.
- P. C. Andrikopoulos, D. R. Armstrong, H. R. L. Barley, W. Clegg, S. H. Dale, E. Hevia, G. W. Honeyman, A. R. Kennedy and R. E. Mulvey, J. Am. Chem. Soc., 2005, 127, 6184–6185 CrossRef CAS PubMed.
- R. E. Mulvey, Organometallics, 2006, 25, 1060–1075 CrossRef CAS.
- R. E. Mulvey, F. Mongin, M. Uchiyama and Y. Kondo, Angew. Chem., Int. Ed., 2007, 46, 3802–3824 CrossRef CAS PubMed.
- A. Harrison-Marchand and F. Mongin, Chem. Rev., 2013, 113, 7470–7562 CrossRef CAS PubMed.
- F. Mongin and A. Harrison-Marchand, Chem. Rev., 2013, 113, 7563–7727 CrossRef CAS PubMed.
- A. J. Martínez-Martínez, A. R. Kennedy, R. E. Mulvey and C. T. O'Hara, Science, 2014, 346, 834–837 CrossRef PubMed.
- T. Arai, H. Sasai, K.-I. Aoe, K. Okamura, T. Date and M. Shibasaki, Angew. Chem., Int. Ed. Engl., 1996, 35, 104–106 CrossRef CAS.
- M. Shibasaki, H. Sasai and T. Arai, Angew. Chem., Int. Ed., 1997, 36, 1236–1256 CrossRef.
- M. Shibasaki and N. Yoshikawa, Chem. Rev., 2002, 102, 2187–2210 CrossRef CAS PubMed.
- R. E. Mulvey, Acc. Chem. Res., 2009, 42, 743–755 CrossRef CAS PubMed.
- B. Haag, M. Mosrin, H. Ila, V. Malakhov and P. Knochel, Angew. Chem., Int. Ed., 2011, 50, 9794–9824 CrossRef CAS PubMed.
- J. P. McInnis, M. Delferro and T. J. Marks, Acc. Chem. Res., 2014, 47, 2545–2557 CrossRef CAS PubMed.
- Y. Sarazin, R. H. Howard, D. L. Hughes, S. M. Humphrey and M. Bochmann, Dalton Trans., 2006, 340–350 RSC.
- H.-Y. Chen, M.-Y. Liu, A. K. Sutar and C.-C. Lin, Inorg. Chem., 2010, 49, 665–674 CrossRef CAS PubMed.
- S. A. Ryken and L. L. Schafer, Acc. Chem. Res., 2015, 48, 2576–2586 CrossRef CAS PubMed.
- E. Chong, P. Garcia and L. L. Schafer, Synthesis, 2014, 2884–2896 Search PubMed.
- S. E. Denmark and J. Fu, Chem. Rev., 2003, 103, 2763–2794 CrossRef CAS PubMed.
- B. Schetter, B. Ziemer, G. Schnakenburg and R. Mahrwald, J. Org. Chem., 2008, 73, 813–819 CrossRef CAS PubMed.
- E. P. Talsi, D. G. Samsonenko and K. P. Bryliakov, Chem. – Eur. J., 2014, 20, 14329–14335 CrossRef CAS PubMed.
- H. H. Brintzinger, D. Fischer, R. Mulhaupt, B. Rieger and R. M. Waymouth, Angew. Chem., Int. Ed. Engl., 1995, 34, 1143–1170 CrossRef CAS.
- M. Bochmann, J. Chem. Soc., Dalton Trans., 1996, 255–270 RSC.
- E. Y. X. Chen and T. J. Marks, Chem. Rev., 2000, 100, 1391–1434 CrossRef CAS PubMed.
- A. D. Schwarz, A. L. Thompson and P. Mountford, Inorg. Chem., 2009, 48, 10442–10454 CrossRef CAS PubMed.
- A. D. Schwarz, K. R. Herbert, C. Paniagua and P. Mountford, Organometallics, 2010, 29, 4171–4188 CrossRef CAS.
- L. C. Liang, S. T. Lin and C. C. Chien, Inorg. Chem., 2013, 52, 1780–1786 CrossRef CAS PubMed.
- R. L. Webster, N. Noroozi, S. G. Hatzikiriakos, J. A. Thomson and L. L. Schafer, Chem. Commun., 2013, 49, 57–59 RSC.
- F. Zhang, H. Song and G. Zi, J. Organomet. Chem., 2010, 695, 1993–1999 CrossRef CAS.
- C. Bakewell, G. Fateh-Iravani, D. W. Beh, D. Myers, S. Tabthong, P. Hormnirun, A. J. P. White, N. Long and C. K. Williams, Dalton Trans., 2015, 44, 12326–12337 RSC.
- T. K. Saha, B. Rajashekhar and D. Chakraborty, RSC Adv., 2012, 2, 307–318 RSC.
- J.-C. Buffet, A. N. Martin, M. Kol and J. Okuda, Polym. Chem., 2011, 2, 2378–2384 RSC.
- H. C. Tseng, H. Y. Chen, Y. T. Huang, W. Y. Lu, Y. L. Chang, M. Y. Chiang, Y. C. Lai and H. Y. Chen, Inorg. Chem., 2016, 55, 1642–1650 CrossRef CAS PubMed.
- J. Ejfler, M. Kobylka, L. B. Jerzykiewicz and P. Sobota, J. Mol. Catal. A: Chem., 2006, 257, 105–111 CrossRef CAS.
- B. Gao, Q. Duan, Y. H. Li, D. N. Li, L. Q. Zhang, Y. Cui, N. H. Hu and X. Pang, RSC Adv., 2015, 5, 13437–13442 RSC.
- D. J. Gilmour, R. L. Webster, M. R. Perry and L. L. Schafer, Dalton Trans., 2015, 44, 12411–12419 RSC.
- S. L. Hancock, M. F. Mahon and M. D. Jones, Dalton Trans., 2011, 40, 2033–2037 RSC.
- S. L. Hancock, M. F. Mahon and M. D. Jones, Chem. Cent. J., 2013, 7, 135–143 CrossRef PubMed.
- Y. J. Kim and J. G. Verkade, Macromol. Rapid Commun., 2002, 23, 917–921 CrossRef CAS.
- A. J. Nielson and J. M. Waters, Polyhedron, 2010, 29, 1715–1726 CrossRef CAS.
- N. Petzetakis, M. Pitsikalis and N. Hadjichristidis, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1092–1103 CrossRef CAS.
- B. Rajashekhar, S. K. Roymuhury, D. Chakraborty and V. Ramkumar, Dalton Trans., 2015, 44, 16280–16293 RSC.
- C. Romain, L. Brelot, S. Bellemin-Laponnaz and S. Dagorne, Organometallics, 2010, 29, 1191–1198 CrossRef CAS.
- C. Romain, S. Choua, J. P. Collin, M. Heinrich, C. Bailly, L. Karmazin-Brelot, S. Bellemin-Laponnaz and S. Dagorne, Inorg. Chem., 2014, 53, 7371–7376 CrossRef CAS PubMed.
- A. Sauer, A. Kapelski, C. Fliedel, S. Dagorne, M. Kol and J. Okuda, Dalton Trans., 2013, 42, 9007–9023 RSC.
- E. Sergeeva, J. Kopilov, I. Goldberg and M. Kol, Inorg. Chem., 2010, 49, 3977–3979 CrossRef CAS PubMed.
- C. K. Su, H. J. Chuang, C. Y. Li, C. Y. Yu, B. T. Ko, J. D. Chen and M. J. Chen, Organometallics, 2014, 33, 7091–7100 CrossRef CAS.
- E. L. Whitelaw, M. D. Jones, M. F. Mahon and G. Kociok-Kohn, Dalton Trans., 2009, 9020–9025 RSC.
- A. L. Zelikoff, J. Kopilov, I. Goldberg, G. W. Coates and M. Kol, Chem. Commun., 2009, 6804–6806 RSC.
- K. Nakano, K. Kobayashi and K. Nozaki, J. Am. Chem. Soc., 2011, 133, 10720–10723 CrossRef CAS PubMed.
- Y. Wang, Y. Qin, X. Wang and F. Wang, Catal.: Sci. Technol., 2014, 4, 3964–3972 CAS.
- Y. Wang, Y. Qin, X. Wang and F. Wang, ACS Catal., 2015, 5, 393–396 CrossRef CAS.
- C. C. Quadri and E. Le Roux, Dalton Trans., 2014, 43, 4242–4246 RSC.
- M. Mandal and D. Chakraborty, J. Polym. Sci., Part A: Polym. Chem., 2015, 54, 809–824 CrossRef.
- C.-K. Su, H.-J. Chuang, C.-Y. Li, C.-Y. Yu, B.-T. Ko, J.-D. Chen and M.-J. Chen, Organometallics, 2014, 33, 7091–7100 CrossRef CAS.
- J. Hessevik, R. Lalrempuia, H. Nsiri, K. W. Tornroos, V. R. Jensen and E. Le Roux, Dalton Trans., 2016, 45, 14734–14744 RSC.
- C. Y. Li, C. J. Yu and B. T. Ko, Organometallics, 2013, 32, 172–180 CrossRef CAS.
- J. Wang, H. Li, N. Guo, L. Li, C. L. Stern and T. J. Marks, Organometallics, 2004, 23, 5112–5114 CrossRef CAS.
- H. Li and T. J. Marks, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15295–15302 CrossRef CAS PubMed.
- S. K. Mandal and H. W. Roesky, Acc. Chem. Res., 2010, 43, 248–259 CrossRef CAS PubMed.
- F. Hild, P. Haquette, L. Brelot and S. Dagorne, Dalton Trans., 2010, 39, 533–540 RSC.
- M. Delferro and T. J. Marks, Chem. Rev., 2011, 111, 2450–2485 CrossRef CAS PubMed.
- M. Normand, E. Kirillov, T. Roisnel and J.-F. Carpentier, Organometallics, 2012, 31, 1448–1457 CrossRef CAS.
- B. Gao, R. Duan, X. Pang, X. Li, Z. Qu, H. Shao, X. Wang and X. Chen, Dalton Trans., 2013, 42, 16334–16342 RSC.
- C. Boulho, H. S. Zijlstra and S. Harder, Eur. J. Inorg. Chem., 2015, 2015, 2132–2138 CrossRef CAS.
- S. Liu, A. Motta, M. Delferro and T. J. Marks, J. Am. Chem. Soc., 2013, 135, 8830–8833 CrossRef CAS PubMed.
- S. Liu, A. Motta, A. R. Mouat, M. Delferro and T. J. Marks, J. Am. Chem. Soc., 2014, 136, 10460–10469 CrossRef CAS PubMed.
- J. Kuwabara, D. Takeuchi and K. Osakada, Chem. Commun., 2006, 3815–3817 RSC.
- P. K. Saini, C. Romain and C. K. Williams, Chem. Commun., 2014, 50, 4164–4167 RSC.
- J. A. Garden, P. K. Saini and C. K. Williams, J. Am. Chem. Soc., 2015, 137, 15078–15081 CrossRef CAS PubMed.
- M. I. Childers, J. M. Longo, N. J. Van Zee, A. M. LaPointe and G. W. Coates, Chem. Rev., 2014, 114, 8129–8152 CrossRef CAS PubMed.
- N. Takeda and S. Inoue, Makromol. Chem., 1978, 179, 1377–1381 CrossRef CAS.
- D. R. Moore, M. Cheng, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2003, 125, 11911–11924 CrossRef CAS PubMed.
- K. Nakano, S. Hashimoto and K. Nozaki, Chem. Sci., 2010, 1, 369–373 RSC.
- M. R. Kember, A. Buchard and C. K. Williams, Chem. Commun., 2011, 47, 141–163 RSC.
- G.-P. Wu, D. J. Darensbourg and X.-B. Lu, J. Am. Chem. Soc., 2012, 134, 17739–17745 CrossRef CAS PubMed.
- M. W. Lehenmeier, S. Kissling, P. T. Altenbuchner, C. Bruckmeier, P. Deglmann, A.-K. Brym and B. Rieger, Angew. Chem., Int. Ed., 2013, 52, 9821–9826 CrossRef CAS PubMed.
- Y. Liu, W.-M. Ren, J. Liu and X.-B. Lu, Angew. Chem., Int. Ed., 2013, 52, 11594–11598 CrossRef CAS PubMed.
- F. Al-Qaisi, E. Streng, A. Tsarev, M. Nieger and T. Repo, Eur. J. Inorg. Chem., 2015, 2015, 5363–5367 CrossRef CAS.
- A. Buchard, F. Jutz, M. R. Kember, A. J. P. White, H. S. Rzepa and C. K. Williams, Macromolecules, 2012, 45, 6781–6795 CrossRef CAS.
- M. R. Kember, P. D. Knight, P. T. R. Reung and C. K. Williams, Angew. Chem., Int. Ed., 2009, 48, 931–933 CrossRef CAS PubMed.
- M. R. Kember, A. J. P. White and C. K. Williams, Inorg. Chem., 2009, 48, 9535–9542 CrossRef CAS PubMed.
- M. R. Kember and C. K. Williams, J. Am. Chem. Soc., 2012, 134, 15676–15679 CrossRef CAS PubMed.
- A. Buchard, M. R. Kember, K. G. Sandeman and C. K. Williams, Chem. Commun., 2011, 47, 212–214 RSC.
- M. R. Kember, A. J. P. White and C. K. Williams, Macromolecules, 2010, 43, 2291–2298 CrossRef CAS.
- G. A. Luinstra, Polym. Rev., 2008, 48, 192–219 CrossRef CAS.
- S. I. Vagin, R. Reichardt, S. Klaus and B. Rieger, J. Am. Chem. Soc., 2010, 132, 14367–14369 CrossRef CAS PubMed.
- S. Klaus, M. W. Lehenmeier, E. Herdtweck, P. Deglmann, A. K. Ott and B. Rieger, J. Am. Chem. Soc., 2011, 133, 13151–13161 CrossRef CAS PubMed.
- W. C. Ellis, Y. Jung, M. Mulzer, R. Di Girolamo, E. B. Lobkovsky and G. W. Coates, Chem. Sci., 2014, 5, 4004–4011 RSC.
- Y. Liu, W.-M. Ren, K.-K. He and X.-B. Lu, Nat. Commun., 2014, 5, 5687–5693 CrossRef PubMed.
- S. Kissling, P. T. Altenbuchner, M. W. Lehenmeier, E. Herdtweck, P. Deglmann, U. B. Seemann and B. Rieger, Chem. – Eur. J., 2015, 21, 8148–8157 CrossRef CAS PubMed.
- C. K. Williams, L. E. Breyfogle, S. K. Choi, W. Nam, V. G. Young, M. A. Hillmyer and W. B. Tolman, J. Am. Chem. Soc., 2003, 125, 11350–11359 CrossRef CAS PubMed.
- C. K. Williams, N. R. Brooks, M. A. Hillmyer and W. B. Tolman, Chem. Commun., 2002, 2132–2133 RSC.
- C. Romain and C. K. Williams, Angew. Chem., Int. Ed., 2014, 53, 1607–1610 CrossRef CAS PubMed.
- B. M. Chamberlain, M. Cheng, D. R. Moore, T. M. Ovitt, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2001, 123, 3229–3238 CrossRef CAS PubMed.
- J. Streuff, M. Feurer, G. Frey, A. Steffani, S. Kacprzak, J. Weweler, L. H. Leijendekker, D. Kratzert and D. A. Plattner, J. Am. Chem. Soc., 2015, 137, 14396–14405 CrossRef CAS PubMed.
- C. G. Vonk, J. Cryst. Mol. Struct., 1973, 3, 201–207 CrossRef CAS.
- L. Yang, D. R. Powell and R. P. Houser, Dalton Trans., 2007, 955–964 RSC.
- M. R. Kember, F. Jutz, A. Buchard, A. J. P. White and C. K. Williams, Chem. Sci., 2012, 3, 1245–1255 RSC.
- D. G. Sekutowski and G. D. Stucky, Inorg. Chem., 1975, 14, 2192–2199 CrossRef CAS.
- K. Folting, J. C. Huffman, R. L. Bansemer and K. G. Caulton, Inorg. Chem., 1984, 23, 3289–3292 CrossRef CAS.
- P. A. Seewald, G. S. White and D. W. Stephan, Can. J. Chem., 1988, 66, 1147–1152 CrossRef CAS.
- P. G. Cozzi, T. Carofiglio, C. Floriani, A. Chiesi-Villa and C. Rizzoli, Organometallics, 1993, 12, 2845–2848 CrossRef CAS.
- A. Gansäuer, I. Winkler, D. Worgull, D. Franke, T. Lauterbach, A. Okkel and M. Nieger, Organometallics, 2008, 27, 5699–5707 CrossRef.
- H. Sugimoto and K. Kuroda, Macromolecules, 2008, 41, 312–317 CrossRef CAS.
- W. J. van Meerendonk, R. Duchateau, C. E. Koning and G.-J. M. Gruter, Macromolecules, 2005, 38, 7306–7313 CrossRef CAS.
- Y. Xiao, Z. Wang and K. Ding, Chem. – Eur. J., 2005, 11, 3668–3678 CrossRef CAS PubMed.
- G.-P. Wu and D. J. Darensbourg, Macromolecules, 2016, 49, 807–814 CrossRef CAS.
- C. Romain, A. Thevenon, P. K. Saini and C. K. Williams, in Carbon Dioxide and Organometallics, ed. X.-B. Lu, Springer International Publishing, Cham, 2015, pp. 101–141 Search PubMed.
- P. J. Dijkstra, H. Du and J. Feijen, Polym. Chem., 2011, 2, 520–527 RSC.
- R. H. Platel, L. M. Hodgson and C. K. Williams, Polym. Rev., 2008, 48, 11–63 CrossRef CAS.
- T. M. Ovitt and G. W. Coates, J. Am. Chem. Soc., 2002, 124, 1316–1326 CrossRef CAS PubMed.
- Y. A. Piskun, I. V. Vasilenko, S. V. Kostjuk, K. V. Zaitsev, G. S. Zaitseva and S. S. Karlov, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1230–1240 CrossRef CAS.
- A. J. Chmura, M. G. Davidson, M. D. Jones, M. D. Lunn, M. F. Mahon, A. F. Johnson, P. Khunkamchoo, S. L. Roberts and S. S. F. Wong, Macromolecules, 2006, 39, 7250–7257 CrossRef CAS.
- P. Dubois, N. Ropson, R. Jérôme and P. Teyssié, Macromolecules, 1996, 29, 1965–1975 CrossRef CAS.
- Z. Zhong, P. J. Dijkstra, C. Birg, M. Westerhausen and J. Feijen, Macromolecules, 2001, 34, 3863–3868 CrossRef CAS.
- A. Duda and S. Penczek, Macromolecules, 1995, 28, 5981–5992 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1507078 and 1507079. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt04193k |
| This journal is © The Royal Society of Chemistry 2017 |