
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Towards broad spectrum activity-based glycosidase probes: synthesis and evaluation of deoxygenated cyclophellitol aziridines†
Sybrin P.
Schröder
 a,
Jasper W.
van de Sande
a,
Wouter W.
Kallemeijn
b,
Chi-Lin
Kuo
b,
Marta
Artola
a,
Eva J.
van Rooden
a,
Jianbing
Jiang
a,
Thomas J. M.
Beenakker
a,
Bogdan I.
Florea
a,
Wendy A.
Offen
c,
Gideon J.
Davies
c,
Adriaan J.
Minnaard
d,
Johannes M. F. G.
Aerts
b,
Jeroen D. C.
Codée
a,
Gijsbert A.
van der Marel
a and
Herman S.
Overkleeft
*a
a,
Jasper W.
van de Sande
a,
Wouter W.
Kallemeijn
b,
Chi-Lin
Kuo
b,
Marta
Artola
a,
Eva J.
van Rooden
a,
Jianbing
Jiang
a,
Thomas J. M.
Beenakker
a,
Bogdan I.
Florea
a,
Wendy A.
Offen
c,
Gideon J.
Davies
c,
Adriaan J.
Minnaard
d,
Johannes M. F. G.
Aerts
b,
Jeroen D. C.
Codée
a,
Gijsbert A.
van der Marel
a and
Herman S.
Overkleeft
*a
aDepartment of Bioorganic Synthesis, Leiden Institute of Chemistry, Leiden University, Einsteinweg 55, 2333 CC Leiden, The Netherlands. E-mail: h.s.overkleeft@chem.leidenuniv.nl
bDepartment of Medical Biochemistry, Leiden Institute of Chemistry, The Netherlands
cDepartment of Chemistry, York Structural Biology Laboratory, The University of York, Heslington, York, YO10 5DD, UK
dStratingh Institute for Chemistry, University of Groningen, Nijenborgh 7, 9747 AG Groningen, The Netherlands
First published on 3rd November 2017
Abstract
Activity-based protein profiling has emerged as a powerful tool for visualizing glycosidases in complex biological samples. Several configurational cyclophellitol isomers have been shown to display high selectivity as probes for glycosidases processing substrates featuring the same configuration. Here, a set of deoxygenated cyclophellitols are presented which enable inter-class profiling of β-glucosidases and β-galactosidases.
Glycosidases are hydrolytic enzymes that catalyze the hydrolysis of oligosaccharides and glycoconjugates.1 Glycosidases are involved in a broad array of human pathologies and are therefore promising therapeutic targets.2 Activity-based protein profiling (ABPP) provides an effective tool for the analysis of enzyme composition, expression and activity, and can assist in the discovery of enzyme inhibitors.3 ABPP relies on the addition of a mechanism-based inhibitor to the sample of interest, which reacts with the catalytic machinery of the target enzyme in a covalent and irreversible manner. The inhibitor carries a reporter tag and is termed an activity-based probe (ABP). As the first example on the development of ABPP, Cravatt and co-workers have described irreversible fluorophosphonate based ABPs which display high reactivity towards a wide range of serine hydrolases (SH).4 These ABPs were used to reveal active SH composition and expression level in various tissues, and as well in the development of highly specific and potent inhibitors against, for instance, monoacylglycerol lipase in a competitive ABPP setting.5 Cyclophellitol (1, Fig. 1), first isolated from Phellinus sp. is a covalent irreversible retaining β-glucosidase inhibitor.6 The configurational isomers, galacto-cyclophellitol (2)7 and manno-cyclophellitol (3)8 are inactivators for β-galactosidases and β-mannosidases, respectively, and their selectivity arises from their absolute configurations. We have previously reported several cyclophellitol based ABPs for selective glycosidase profiling.9–11 For example, treatment of mouse tissue lysates with acyl ABP 4 resulted in tissue-specific fluorescent labelling of the three retaining β-glucocerebrosidases (GBA1, 2 and 3) and lactase-phlorizin hydrolase (LPH) without non-specific cross-labelling. Later studies revealed that alkyl ABP 5 also effectively labels these retaining β-glucosidases.12 While the selectivity of these probes is an attractive value, it also impedes the study of multiple enzyme classes with a single probe, as for example can be done by the broad-spectrum SH ABPs developed by Cravatt and co-workers.4 Therefore, the development of broad spectrum glycosidase ABPs would be of interest. Since the substrate specificity of glucosidases, mannosidases and galactosidases is dependent on the carbohydrate hydroxyl configuration at C2 and C4 (axial or equatorial), removal of these alcohols may abolish active site preference. We therefore synthesized four deoxygenated ABPs 6–9 and we set out to establish whether these compounds would be effective ABPs indiscriminate towards glucosidases and galactosidases (and for 8–9, mannosidases).
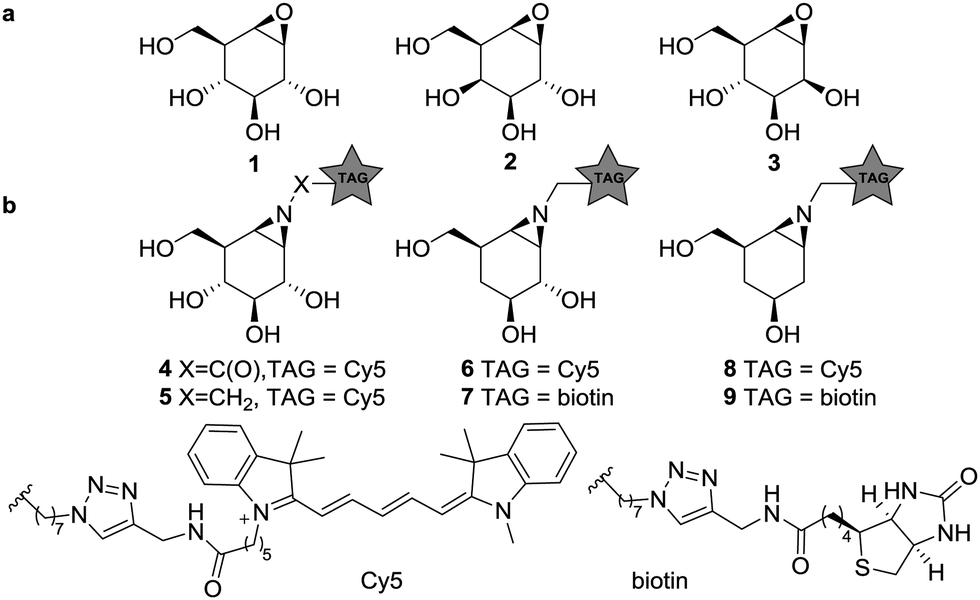 | ||
| Fig. 1 (a) Structures of cyclophellitols 1–3, selective covalent and irreversible retaining glycosidase inhibitors. (b) Selective β-glucosidase ABPs 4–5, 4-deoxy ABPs 6–7 and 2,4-deoxy ABPs 8–9. | ||
The synthesis of compounds 6–9 was achieved from key-intermediates 12 and 15, which were prepared as follows. The primary alcohol in diol 10 (available by chemistry developed by Madsen and co-workers, Scheme 1a)13 was protected as the p-methoxybenzyl ether (PMB) after which tosylation of the secondary alcohol afforded intermediate 11. Treatment of 11 with lithium aluminum hydride followed by deprotection afforded 12. For cyclohexene 15 (Scheme 1b), chiral aldehyde 13 (available by chemistry developed by Helmchen and co-workers)14 was asymmetrically allylated using Brown's reagent and subsequently subjected to ring-closing metathesis using the Grubbs 2nd generation catalyst to give 14. Benzoylation and detritylation afforded cyclohexene 15. Both cyclohexenes 12 and 15 could be reacted with trichloroacetonitrile to afford the corresponding primary imidates (Scheme 1c), which upon treatment with N-iodosuccinimide (NIS) cyclized to afford the cyclic imidates 16 and 19 stereospecifically. Acidolysis and base treatment resulted in formation of aziridines 17 and 20, which were alkylated with an azidoalkyl spacer and deprotected to give alkyl aziridines 18 and 21, and then ligated with a reporter tag using click chemistry to afford deoxygenated ABPs 6–9 (see ESI†).
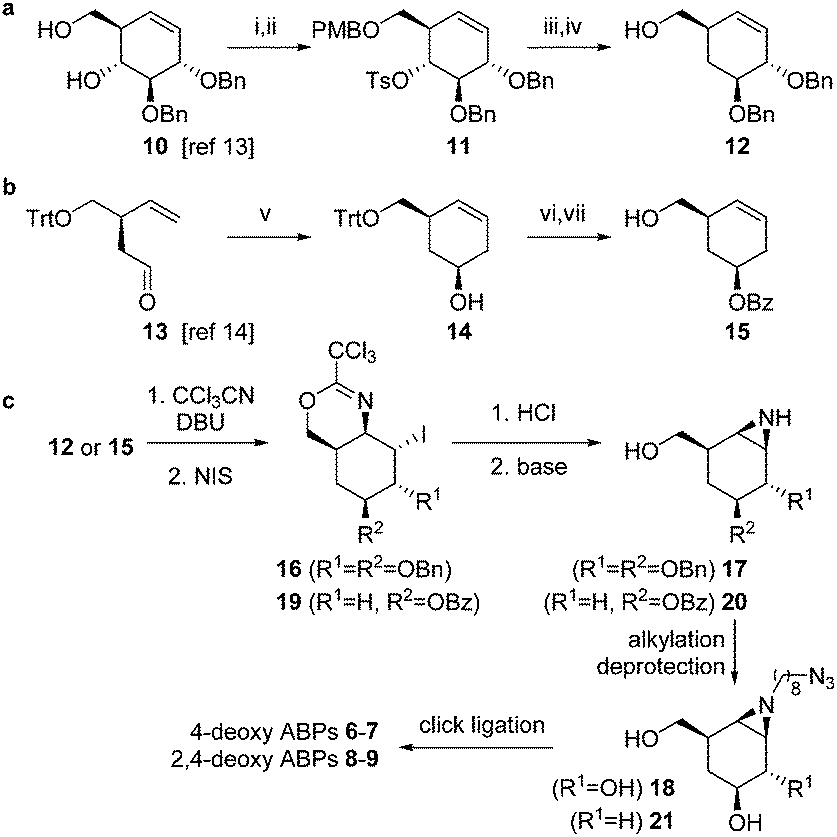 | ||
Scheme 1 The synthesis of deoxygenated ABPs required two different synthetic strategies (a and b) to afford key intermediates 12 and 15, which were further functionalized to ABPs 6–9 following similar synthetic transformations (c). Reagents and conditions (i) PMBCl, 2-aminoethyl diphenylborinate (10 mol%), K2CO3, KI, MeCN, 60 °C, 95%; (ii) TsCl, pyridine, 60 °C, 90%; (iii) LiAlH4, THF, reflux, 86%; (iv) HCl (10%), DCM/HFIP, 86%; (v) 1. (+)-Ipc2B(allyl)borane, THF, −90 °C; 2. Grubbs II (5%), DCM, reflux, 77%, dr 86![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :14; (vi) BzCl, Et3N, DMAP, DCM, 84%; (vii) CSA, MeOH, DCM, 94%. :14; (vi) BzCl, Et3N, DMAP, DCM, 84%; (vii) CSA, MeOH, DCM, 94%. | ||
The labelling efficiency and specificity towards recombinant β-glucosidases, β-galactosidases and β-mannosidases was investigated for fluorescent ABPs 6 and 8, and compared to that of β-glucosidase selective probe 5. After enzyme incubation with a range of ABP concentrations (30 minutes, 37 °C, optimal enzyme pH), recombinant Homo sapiens GBA1 is highly efficiently labelled by ABP 5, with protein bands visualized at probe concentrations as low as 10 nM (Fig. 2a). Additionally, 4-deoxy ABP 6 also labelled GBA1 efficiently, although the potency was slightly reduced, likely due to lacking hydrogen-bonding interactions within the active site residue(s) as the result of the absence of OH-4. The labelling potency of 2,4-deoxy ABP 8 proved to be drastically reduced. All bands could be competed with selective β-glucosidase inactivator 1. Also, denatured enzyme (1% SDS, 100 °C, 5 min) is not labelled by any of the probes indicating that all labelling is activity-based. As expected, binding kinetics studies revealed that 5 is a far more potent GBA1 inactivator (kinact/KI = 27.51 ± 0.85 μM−1 min−1) than 6 (kinact/KI = 0.14 ± 0.08 μM−1 min−1). Kinetic parameters for 8 could not be determined under our assay conditions reflecting its low potency. Labelling of Cellvibrio japonicus β-galactosidase from glycoside hydrolase family 35 (CjGH35A)15 could be detected at micromolar concentrations of 5 and 8. Interestingly, 4-deoxy ABP 6 was significantly more potent and labelled the enzyme at concentrations down to 100 nM, similar to GBA1 labelling; which is surprising given the extensive interactions of O-4, from a galacto-cyclophellitol aziridine, with the enzyme (see ESI†). Indeed, kinetic parameters for 6 in CjGH35A (kinact/KI = 0.16 ± 0.07 μM−1 min−1) are comparable to that in GBA1 (kinetic parameters of 5 and 8 could not be determined under our assay conditions, due to low potency). Competition experiments indicated the labelling was activity-based, although full competition was not achieved which could indicate a fraction of non-specific binding. Lastly, we found that none of the ABPs showed significant labelling of GH2 β-mannosidase from Helix pomatia (see ESI†). Indeed, it is known that OH-2 is highly important for coordination to the glycosidase catalytic nucleophile and stabilizing the transition state (TS).16 Moreover, probes 5, 6 and 8 mimic preferentially adopt a 4H3 configuration, which is characteristic for β-glucosidase and β-galactosidase transition states, whereas β-mannosidases often favour a B2,5 TS.17 Next, the glycosidase profiling by probes 6 and 8 in lysates from mouse (C57bl/6j, Jackson's laboratories) kidney and liver tissue, and human wild-type fibroblast cells (Lonza) was investigated, and compared to that of β-glucosidase selective probe 5. The samples were incubated with the ABPs under identical conditions (1 μM ABP, pH 5.0, 37 °C, 30 min). In mouse liver lysates, 5 labels a distinct band at ∼65 kDa (Fig. 3a), corresponding to the molecular weight of GBA1 (58–66 kDa18), and this band is fully abolished when the sample was pre-incubated with GBA1 specific inhibitor 1.19 Similarly, incubation with 6 resulted in a band at the same height. This band could only be partially competed with 1, and pre-incubation by specific β-galactosidase inactivator 2 resulted in partial competition as well. Ultimately, full competition was achieved by pre-incubation with both cyclophellitols 1 and 2. Thus, in contrast to labelling with 5, it was concluded that incubation with 6 resulted in the concomitant labelling of GBA1 and a β-galactosidase, presumably acid β-galactosidase (GLB1, ∼63 kDa).20 To identify the bands labelled on gel, the proteins were enriched by incubation of the lysate with biotin probe 7 and subsequent pull-down with streptavidin beads. After tryptic digestion, the peptide fragments were identified by LC-MS/MS and the abundance of the protein hits was quantified as previously described,21 in unsupervised mode using the default settings of the PLGS (Waters) and IsoQuant software. Indeed, incubation with 7 resulted in enrichment of GBA1 and GLB1, corresponding to the mixed band at ∼65 kDa on gel. In contrast, labelling with ABP 8 did not result in labelling of GBA1 or GLB1. Multiple bands were visible on gel, but no competition was observed except for one band at ∼115 kDa of an unknown protein. We therefore conclude that the other bands were a result of non-specific binding. Regardless, the lysate was incubated with 9 and the labelled proteins were identified by proteomic analysis. Whereas no enrichment of GBA1 could be detected, the probe did show increased levels of GLB1 labelling. Additionally, LC-MS/MS analysis detected enrichment of β-galactosylceramidase (GALC), an 80 kDa protein which is processed in the lysosomes into two catalytically active subunits of ∼30 and ∼50 kDa,22 and is responsible for the hydrolysis of galactosylceramide. Interestingly, fluorescent labelling with 8 did not identify GALC on gel. However, it is known that expression levels of GALC in the liver are low.23
 | ||
| Fig. 2 Fluorescent labelling of recombinant H. sapiens GBA1 and C. japonicus β-galactosidase with ABPs 5, 6 and 8 (left panel), and competition experiments with selective cyclophellitol inhibitors 1 or 2 (right panel). (a) GBA1 is potently labelled by 5 and 6, but not 8. All labelling was activity-based, indicated by competition experiments with GBA1 selective inhibitor 1. (b) β-Galactosidase is potently labelled by 5, in an activity-based fashion. | ||
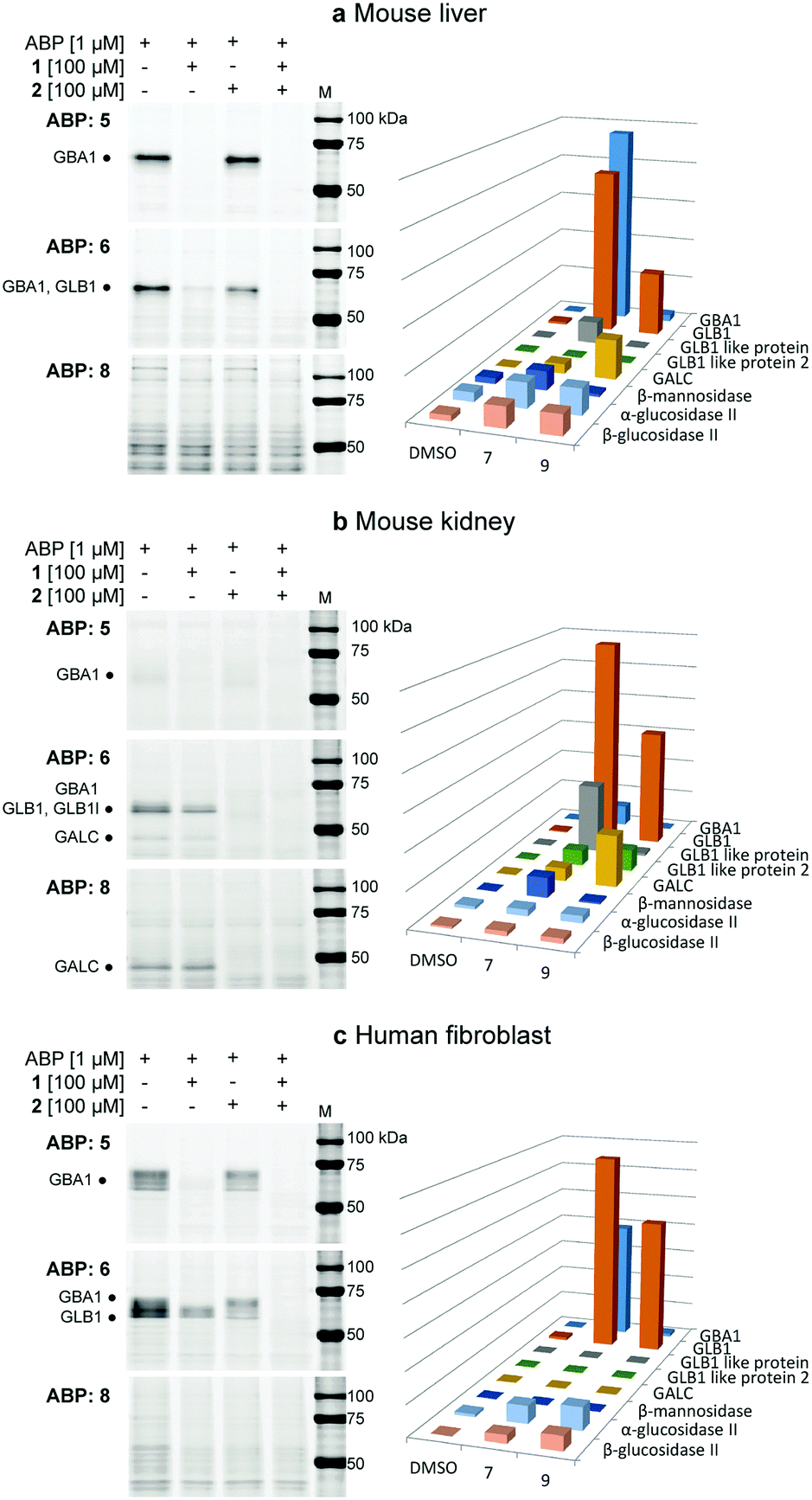 | ||
| Fig. 3 Fluorescent labelling of mouse liver (a), kidney (b) and human fibroblast (c) lysates with probes 5, 6 and 8, complemented with competition experiments with β-glucosidase specific covalent inhibitor 1 and β-galactosidase specific 2. The bar charts display the LC-MS/MS pull-down analyses with the biotinylated ABP analogues corresponding to the gels on the left, compared to the negative control (DMSO). (a) Probe 5 only labels GBA1, while 6 labels GBA1 and GLB1, and 8 only shows non-specific labelling of proteins in gel, although pull-down analysis shows enrichment of GLB1 and GALC with 9. (b) Only low amounts of GBA1 could be detected by specific probe 5. 4-Deoxy probe 6 labels GBA1, GLB1 (like) protein and GALC. ABP 8 shows specific fluorescent labelling of GALC on gel. (c) ABP 5 labels GBA1 only, while 6 labels both GBA1 and GLB1. ABP 8 does not significantly label proteins, however pull-down analysis shows enrichment of GLB1. | ||
In mouse kidney lysates (Fig. 3b), 5 only labels GBA1 minimally. In contrast, bands at ∼65 kDa and ∼45 kDa were observed with probe 6 which could be fully competed out by pre-incubation with 2, but not 1. This indicated the labelling of mainly β-galactosidases in this sample, presumably GLB1 and GALC, which was subsequently confirmed by proteomics experiments as described above. Enrichment of GBA1 was also detected but less pronounced, in line with the fluorescent assay. Labelling with probe 6 was also observed for GLB1-like protein 1 (GLB1l, ∼68 kDa) and 2 (GLB1l2, processed mass unknown), which both possess the GLB1 active site motif.24 While 2,4-deoxy ABP 8 did not significantly label GBA1 or GLB1, proteomics identified GLB1 and GLB1l2 by pull-down with 9. Interestingly, a strong band at ∼45 kDa was labelled, which could be competed with 2. Indeed, proteomics revealed increased labelling of GALC by this probe and it was therefore concluded that 8 is a GALC specific ABP.
Lastly, the labelling of human fibroblast lysates was investigated (Fig. 3c). A pattern of bands between 55–70 kDa is labelled by 5 which can subsequently be fully competed for with 1, indicating the labelling of different isoforms18 of GBA1 in this lysate. Similarly, 6 labels bands between 55–70 kDa, however with increased signal intensity. Approximately 50% competition was achieved by pre-incubation with either 1 or 2, whereas pre-incubation with both inhibitors completely abrogated labelling. Indeed, pull-down analysis identified both GBA1 and GLB1 enrichment by probe 7. Finally, 8 did not show significant labelling of proteins in the sample, whereas GLB1 was enriched by ABP 9 according to LC-MS/MS analysis.
In summary, we have developed syntheses towards a set of deoxygenated probes which were modelled on cyclophellitol aziridine 5. None of the probes display activity towards β-mannosidases. Deoxygenation at C2 and C4 (ABPs 8–9) enables labelling of purified β-glucosidases and β-galactosidases, albeit with low potency. Nevertheless, it was found that 8 displayed unexpected specificity towards β-galactosylceramidase in mouse kidney lysate where the enzyme is abundant.23 Deoxygenation at C4 (ABPs 6–7) affects the binding kinetics for GBA1, however in vitro labelling proceeded with high efficiency and moreover these probes now identify retaining β-galactosidases as well. Based on the results presented herein we conclude that, indeed, probes designed for simultaneously profiling retaining glycosidases processing differently configured glycosides are within reach. We had earlier observed that probes designed for a specific class of retaining glycosidases may possess cross-reactivity towards other glycosidases,10 however these results could not be predicted. Here, we show that by substitution of the hydroxyl group that distinguishes glucose from galactose for hydrogen on an otherwise unaltered cyclophellitol aziridine yields probes labelling both glucosidases and galactosidases. The strategy is however not general, since stripping off OH-2 yields a largely inactive probe. Future research on differently configured, OH-deleted cyclophellitol aziridines is required to establish if and to what extent this strategy is viable to deliver a set of broad-spectrum glycosidase ABPs, for use in combination with the specific exo-glycosidase probes already in hand for dissecting large numbers of retaining glycosidases from various species.
We thank The Netherlands Organization for Scientific Research (NWO-CW, ChemThem grant to JMFGA and HSO), the European Research Council (ERC-2011-AdG-290836 “Chembiosphing” to HSO, and ERC-2012-AdG-322942 “Glycopoise”, to GJD and WAO), Sanofi Genzyme (research grant to JMFGA and HSO and postdoctoral contract to MA), ChemAxon for the Instant JChem software and Diamond Light Source for access to beamline I02 (proposal number mx-9948) that contributed to the results presented here. GJD is a Royal Society Ken Murray Research Professor.
Conflicts of interest
There are no conflicts to declare.Notes and references
- G. Davies and B. Henrissat, Structure, 1995, 3, 853–859 CrossRef CAS PubMed.
- K. Ohtsubo and J. D. Marth, Cell, 2006, 126, 855–867 CrossRef CAS PubMed.
- M. J. Evans and B. F. Cravatt, Chem. Rev., 2006, 106, 3279 CrossRef CAS PubMed.
- Y. Liu, M. P. Patricelli and B. F. Cravatt, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 14694–14699 CrossRef CAS.
- J. Z. Long, W. Li, L. Booker, J. J. Burston, S. G. Kinsey, J. E. Schlosburg, F. J. Pavón, A. M. Serrano, D. E. Selley, L. H. Parsons, A. H. Lichtman and B. F. Cravatt, Nat. Chem. Biol., 2008, 5, 37–44 CrossRef PubMed.
- S. Atsumi, K. Umezawa, H. Iinuma, H. Naganawa, H. Nakamura, Y. Iitaka and T. Takeuchi, J. Antibiot., 1989, 43, 49–53 CrossRef.
- L. I. Willems, T. J. M. Beenakker, B. Murray, B. Gagestein, H. Van Den Elst, E. R. Van Rijssel, J. D. C. Codée, W. W. Kallemeijn, J. M. F. G. Aerts, G. A. Van Der Marel and H. S. Overkleeft, Eur. J. Org. Chem., 2014, 6044 CrossRef CAS.
- V. W. F. T. Tony and K. M. Shing, J. Chem. Soc., Perkin Trans. 1, 1994, 2017–2025 Search PubMed.
- W. W. Kallemeijn, K. Y. Li, M. D. Witte, A. R. A. Marques, J. Aten, S. Scheij, J. Jiang, L. I. Willems, T. M. Voorn-Brouwer, C. P. A. A. Van Roomen, R. Ottenhoff, R. G. Boot, H. Van Den Elst, M. T. C. Walvoort, B. I. Florea, J. D. C. Codée, G. A. Van Der Marel, J. M. F. G. Aerts and H. S. Overkleeft, Angew. Chem., Int. Ed., 2012, 51, 12529–12533 CrossRef CAS PubMed.
- J. Jiang, C. L. Kuo, L. Wu, C. Franke, W. W. Kallemeijn, B. I. Florea, E. Van Meel, G. A. Van Der Marel, J. D. C. Codée, R. G. Boot, G. J. Davies, H. S. Overkleeft and J. M. F. G. Aerts, ACS Cent. Sci., 2016, 2, 351–358 CrossRef CAS PubMed.
- L. I. Willems, T. J. M. Beenakker, B. Murray, S. Scheij, W. W. Kallemeijn, R. G. Boot, M. Verhoek, W. E. Donker-Koopman, M. J. Ferraz, E. R. Van Rijssel, B. I. Florea, J. D. C. Codée, G. A. Van Der Marel, J. M. F. G. Aerts and H. S. Overkleeft, J. Am. Chem. Soc., 2014, 136, 11622–11625 CrossRef CAS PubMed.
- J. Jiang, T. J. M. Beenakker, W. W. Kallemeijn, G. A. Van Der Marel, H. Van Den Elst, J. D. C. Codée, J. M. F. G. Aerts and H. S. Overkleeft, Chem. – Eur. J., 2015, 21, 10861 CrossRef CAS PubMed.
- F. G. Hansen, E. Bundgaard and R. Madsen, J. Org. Chem., 2005, 70, 10139–10142 CrossRef CAS PubMed.
- G. Franck, K. Brödner and G. Helmchen, Org. Lett., 2010, 12, 3886–3889 CrossRef CAS PubMed.
- J. Larsbrink, A. J. Thompson, M. Lundqvist, J. G. Gardner, G. J. Davies and H. Brumer, Mol. Microbiol., 2014, 94, 418 CrossRef CAS PubMed.
- S. G. Withers, Carbohydr. Polym., 2001, 44, 325–337 CrossRef CAS.
- G. J. Davies, A. Planas and C. Rovira, Acc. Chem. Res., 2012, 45, 308–316 CrossRef CAS PubMed.
- S. van Weely, J. M. F. G. Aerts, M. B. van Leeuwen, J. C. Heikoop, W. E. Donker-Koopman, J. A. Barranger, J. M. Tager and A. W. Schram, Eur. J. Biochem., 1990, 191, 669–677 CrossRef CAS PubMed.
- M. D. Witte, W. W. Kallemeijn, J. Aten, K.-Y. Li, A. Strijland, W. E. Donker-Koopman, A. M. C. H. van den Nieuwendijk, B. Bleijlevens, G. Kramer, B. I. Florea, B. Hooibrink, C. E. M. Hollak, R. Ottenhoff, R. G. Boot, G. A. van der Marel, H. S. Overkleeft and J. M. F. G. Aerts, Nat. Chem. Biol., 2010, 6, 907–913 CrossRef CAS PubMed.
- S. Tomino and M. Meisler, J. Biol. Chem., 1975, 250, 7752 CAS.
- J. Kuharev, P. Navarro, U. Distler, O. Jahn and S. Tenzer, Proteomics, 2015, 15, 3140–3151 CrossRef CAS PubMed.
- S. Nagano, T. Yamada, N. Shinnoh, H. Furuya, T. Taniwaki and J. I. Kira, Clin. Chim. Acta, 1998, 276, 53–61 CrossRef CAS.
- P. Luzi, M. A. Rafi, M. Zaka, M. Curtis, M. T. Vanier and D. A. Wenger, Mol. Genet. Metab., 2001, 73, 211–223 CrossRef CAS PubMed.
- J. Le Carré, D. F. Schorderet and S. Cottet, Mol. Vision, 2011, 17, 1287–1297 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and compound characterization, SDS-page images, proteomics and kinetics data. See DOI: 10.1039/c7cc07730k |
| This journal is © The Royal Society of Chemistry 2017 |