
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIntegrative self-sorting of coordination cages based on ‘naked’ metal ions
Witold M.
Bloch
 and
Guido H.
Clever
*
and
Guido H.
Clever
*
Department of Chemistry and Chemical Biology, TU Dortmund University, Otto-Hahn-Straße 6, 44227 Dortmund, Germany. E-mail: guido.clever@tu-dortmund.de
First published on 29th June 2017
Abstract
Coordination-driven self-assembly of metal ions and organic ligands has been extensively utilised over the past four decades to access a variety of nano-sized cage assemblies, with functions ranging from sensing and catalysis to drug delivery. Many of the reported examples, however, are highly symmetric architectures that contain one type of organic ligand carrying not more than a single functionality. This contrasts significantly with the level of structural and functional complexity encountered in biological macromolecular hosts, which are able to bind and chemically convert smaller molecules in their highly-decorated internal cavities. To address this disparity, rational approaches that facilitate heteroleptic assembly by regulating integrative self-sorting of metal ions and multiple ligand components have emerged. Among these, routes to access coordination cages from ‘naked’ metal cations that offer more than two coordination sites are still in early development, as the complexity of the self-sorted products in terms of composition and stereochemistry presents an entropic challenge. This feature article highlights recent progress in controlling integrative self-sorting of multi-component cage systems with a focus on structures composed of ‘naked’ metal cations and two different ligands. Once heteroleptic self-assembly strategies find a wider implementation in supramolecular design, the resultant interplay between tailored combinations of precisely positioned substituents promises enhanced functionality in nanoscale structures.
 Guido H. Clever (left) and Witold M. Bloch (right) | Witold M. Bloch obtained his PhD at the University of Adelaide in 2014 under the supervision of Christopher Sumby and Christian Doonan. In 2015, he received an Alexander von Humboldt fellowship which he commenced in Göttingen and is now continuing at TU Dortmund in the group Guido Clever. His research interests include porous nanomaterials and the rational design of multi-component cage structures. |
Guido H. Clever is a Professor of Bioinorganic Chemistry at Technical University Dortmund. He studied chemistry in Heidelberg and received his PhD from LMU Munich under the supervision of Thomas Carell. From 2007 to 2010 he was an AvH/JSPS postdoctoral researcher and Assistant Professor in the group of Mitsuhiko Shionoya at the University of Tokyo. In 2010 he became a Junior Professor at the University of Göttingen, where he was appointed on a W2 tenured position in 2013. In 2015 he accepted a call on a Full Professorship at TU Dortmund. He was awarded the ADUC prize 2012 for young investigators and the Dozentenpreis of the FCI in 2015. His research on stimuli-responsive, multi-functional coordination cages is currently supported by an ERC consolidator grant. His further interests include metal-mediated DNA architectures. |
1. Introduction
In nature, the assembly of multi-component macromolecules such as ribosomes, microtubules and virus capsids is achieved through integrative self-sorting – a highly-optimised recognition process in which combinations of multiple molecular entities are arranged in a precise and controlled manner. Driven by complementary supramolecular effects such as π-stacking, hydrogen-bonding, charge, dipole and dispersive interactions, as well as steric repulsion, integrative self-sorting in nature continues to inspire chemists that seek to achieve increased functionality in artificial supramolecular systems.Coordination-driven self-assembly has been one of the most successful approaches to rationally design a large assortment of discrete 2D and 3D nano-architectures with control over their size and shape.1–6 In particular, hollow 3D assemblies, also known as coordination cages, have received significant interest due to the diverse functions that arise from an internal cavity accessible to guest inclusion.7–12
In recent years, the focus of the field of coordination cages has been gradually shifting from structure toward function, resulting in the emergence of coordination cages tailor-designed for applications such as catalysis,13–15 controlled drug release,16–18 molecular separation,19–21 sensing22–24 and stabilization of reactive intermediates.25,26 A common limitation of these examples, however, is that they are often composed of only one type of ligand – the key component from which the assembly draws its overall function. In order to expand the degree of functionality, current efforts in the field have returned to progressing structural design, with a particular focus on understanding the phenomenon of integrative self-sorting27–29 and thus developing approaches that facilitate the assembly of heteroleptic cages composed of multiple different ligands.
To date, most of the efforts directed at accessing heteroleptic supramolecular structures have focused on minimizing the degree of self-sorting in the system by utilizing cis-protected metal centres as building blocks (e.g. [M(en)]; M = PdII or PtII, en = ethylenediamine) that restrict the number of bound bridging ligands (Fig. 1a).2,3,30 In combination with this, the utilization of different donor combinations contributed by bis-, tris- or tetrakis-monodentate bridging ligands has been a particularly effective approach in constructing multi-component assemblies with accessible cavities. For example, Stang, Zheng and others have constructed heteroleptic prisms through the charge-separation approach between adjacent carboxylate/pyridine donors.30,31 Similar multi-component architectures based on organometallic half-sandwich units have been prepared by Jin, Therrien and others.32,33 Mukherjee has exploited combinations of imidazole and pyridyl ligands to assemble several cage architectures possessing versatile guest-binding properties.34–36 Fujita,37,38 Kobayashi and others39,40 have employed steric repulsion to access a variety of 2D and 3D heteroleptic structures based on ‘side chain-directed’ control. Whilst using cis-protected metal ions is an effective approach to access heteroleptic structures, the number of different ligands that can coordinate to a square-planar metal centre is limited to two.41 On the other hand, a greater degree of complexity of cage structures is possible from unprotected (‘naked’) metal ions such as PdII offering all of its four coordination sites (Fig. 1b). While strategies for a rational synthesis of the schematically depicted tetra-functional [M2LALBLCLD] cage are still elusive, we review herein approaches to control integrative self-sorting of such systems based on two different ligands.
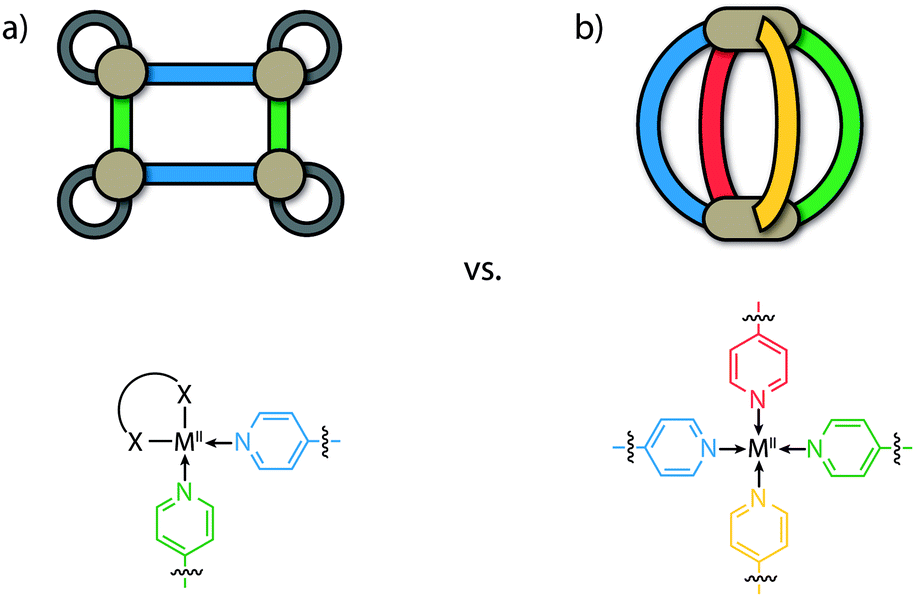 | ||
| Fig. 1 Comparison of heteroleptic self-assemblies based on square-planar metal cations. (a) Supramolecular rectangle [{M(en)}4LA2LB2] formed from cis-protected cations with two different ligands, (b) [M2LALBLCLD] cage formed from ‘naked’ cations and four different ligands. | ||
The construction of hollow heteroleptic cages based on naked metal ions and more than one type of ligand is certainly not a straightforward task; the tendency to form entropically driven mixtures can often hinder this aim. It is worth noting that because naked metal ions avail all of their coordination sites for ligand binding, there is a stronger entropic contribution in self-sorting processes compared to analogues systems composed of cis-protected metal ions.
Using the archetypal [M2L4] framework as an example, the outcomes of self-sorting in a fully-dynamic two-ligand-system can be grouped into three scenarios: (1) [Pd2LAnLB4−n] (n = 0–4) statistical mixtures – often encountered in the absence of complementary interactions between the two entities LA and LB; (2) [Pd2LA4] + [Pd2LB4] narcissistic self-sorting – occurs when there is a significant energetic penalty arising from the combination of ligands LA and LB (commonly due to a difference in ligand size and/or shape); (3) integrative self-sorting to give a single heteroleptic product, e.g. cis-[Pd2LA2LB2] – often driven by significant enthalpic factors that originate from the combination of adjacent LA and LB components in the assembly (Fig. 2). Often, this situation can be achieved through the utilization of secondary interactions or through geometric principles. It is important to note that the above-mentioned scenarios are not limited to assembly reactions where metal precursors are combined with a mix of ligand components. When kinetically allowed, mixtures of pre-assembled homoleptic cages can often undergo ligand shuffling (scenario 1 or 3; an example is discussed below) but may as well remain stable in one another's presence (scenario 2).
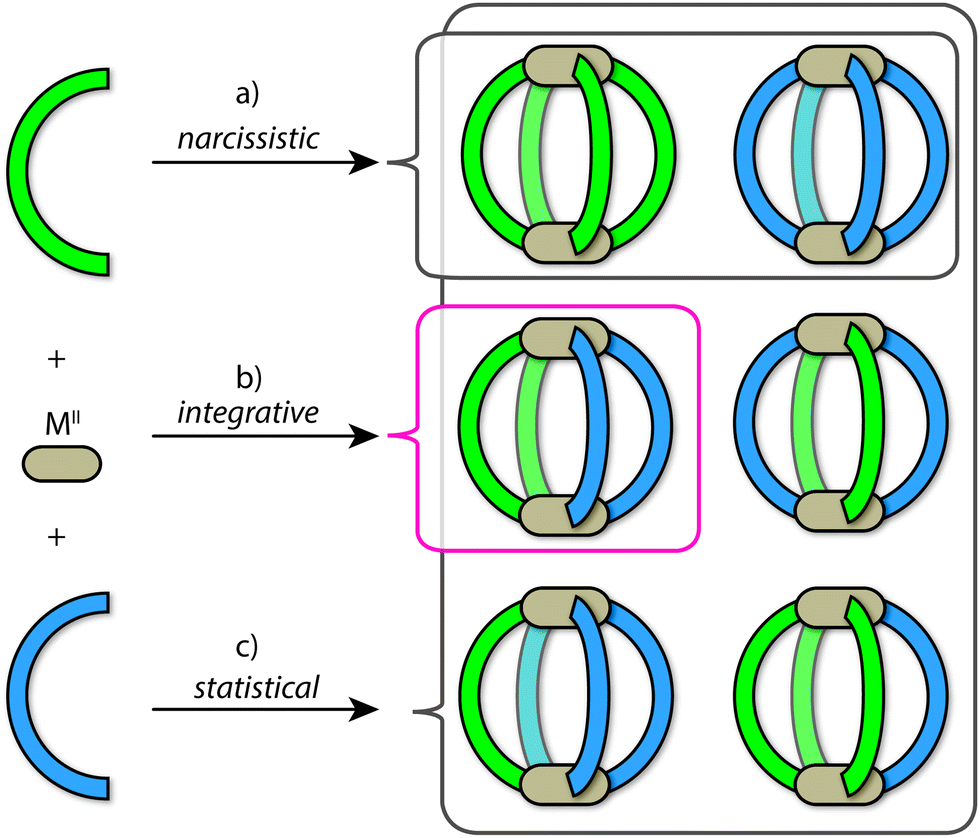 | ||
| Fig. 2 The three different self-sorting outcomes in coordination-driven self-assembly; (a) a narcissistic self-sorted mixture; (b) integrative self-sorting that gives rise to a single heteroleptic product of choice (here the cis-[Pd2LA2LB2] species); (c) statistical mixture according to a Boltzmann distribution. | ||
Herein we use the term ‘integrative self-sorting’ to describe the process by which a non-statistical speciation of heteroleptic cage compounds is achieved from a mixture of metal ions and two different ligands or their respective homoleptic cages. Whilst narcissistic self-sorting is not the focus of this review, it is important to place its importance in the overall context of self-sorting: narcissistically self-sorted systems allow for the interplay of functions between well-defined, independent molecular species present in a mixture, while integrative self-sorting allows for the construction of cage assemblies of increased complexity and function.
In this feature article, we focus on recent reports of integrative self-sorting of hollow, homometallic coordination cages based on ‘naked’ metal cations, i.e. without cis-protecting ligands at the metal centre. We note that examples of heterometallic cages as well as multi-component assemblies from cis-protected metal ions have been the subject of recent reviews.8,30,42–44 Additionally, self-sorting phenomena have also been studied in the context of dynamic covalent organic cages and examples include imine-based systems by Mastalerz,45 boronic ester cages by Beuerle46 and Severin,47 as well as amino acid-bridged cages by Wessjohann and Rivera.48
The examples of self-sorting discussed herein will be divided into two categories: (1) assembly-dependent integrative self-sorting; (2) coordination-dependent integrative self sorting. In the former route, the selective combination of different building blocks is governed by the entire structure, for example by a complementary shape of the components or by interactions between ligand backbones and guest templates. On the other hand, the latter route relies upon structural features of the proximate heteroleptic coordination environment (e.g. steric and/or electronic match of the donor groups around the metal centre). The different approaches (Fig. 3) and underlying principles which facilitate integrative self-sorting will be discussed in order to obtain a general overview of how to build-up structural complexity in coordination cages.
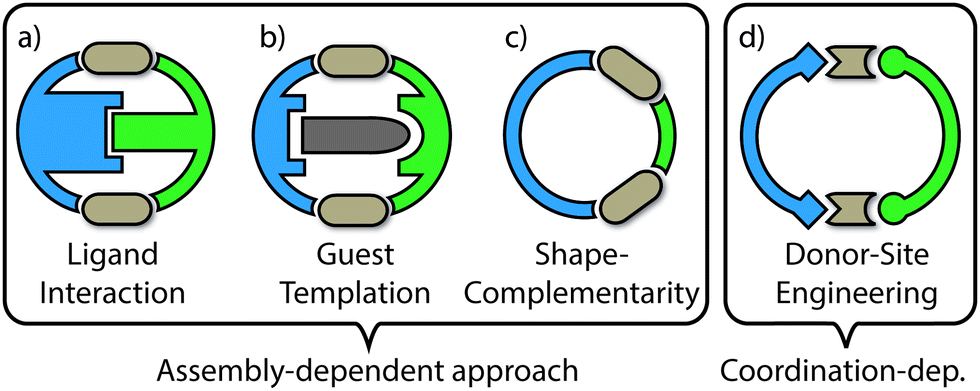 | ||
| Fig. 3 Schematic comparison of selected strategies for achieving rationally assembled heteroleptic structures: (a) specific stabilizing interactions between two different ligands; (b) the same, but mediated by a templating guest; (c) geometric design based on shape-complementarity between the different ligands; (d) specific combination of donors around the metal centres guiding the heteroleptic assembly. Approaches (a–c) depend on the shape and functionality of the overall assembly, i.e. the ligand backbones. Strategy (d) is controlled by the immediate environment of the coordinated metal centres. In (a) and (b) the cavity is usually sacrificed but (c) and (d) allow for post-assembly guest uptake. | ||
2. Assembly-dependent integrative self-sorting
Integrative self-sorting is favoured when there is an associated energetic benefit arising from the combination of two or more complementary ligand components in the cage structure. For example, a heteroleptic structure can bring together intra-assembly supramolecular interactions (e.g. hydrogen bond donor/acceptor combinations) that would otherwise be absent in a narcissistic mixture of complexes. If such an enthalpic benefit provides a stabilizing effect that is more prominent than the entropic penalty associated with the formation of a heteroleptic product, the formation of statistical mixtures can be overcome. The following paragraphs describe different variations of major enthalpic effects with the help of selected examples from the recent literature.2.1. Ligand interaction
Utilizing the approach of sub-component self-assembly,49 Nitschke and co-workers investigated the role of π-stacking interactions in the assembly and self-sorting of tetrahedral-shaped cages composed of electron-rich/poor subunits and divalent metal cations (FeII, CoII and ZnII).50 Whilst sub-component assembly of NDI subunit S1 gave the expected [M416]8+ tetrahedron, reacting bis(3-aminophenyl)pyrene sub-unit S2 with either FeII, CoII or ZnII and formylpyridine yielded a close-packed [M426]8+ pseudo-tetrahedron. X-ray analysis of the latter cage revealed prominent π-stacking interactions between neighbouring pyrene backbones, giving rise to an essentially ‘close-packed’ structure. Furthermore, reacting S2 with formylpyridine and MII(NTf2)2 in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1 ratio produced a different, lower nuclearity homoleptic assembly, [M222(CH3CN)4]4+, which was found to incorporate electron-deficient aromatic guests in its electron-rich cavity. Prompted by this observation, self-sorting experiments were carried out with electron-poor NDI sub-unit S1 and various electron-rich pyrene isomers. One particular combination led to a striking outcome; combining S1 and S2 in a 1:1 ratio with formylpyridine and MII(NTf2)2 led to a [M41224]8+ heteroleptic assembly (Fig. 4). X-ray analysis of this sample revealed an unusual triple-decker pyrene–pyrene–NDI stack, contrasting with the more commonly observed donor–acceptor alternating stacks. Furthermore, the [M41224]8+ structure could be accessed through a cage-to-cage transformation of [M416]8+ and [M426]8+ or a ligand-induced transformation of [M222(CH3CN)4]4+ with S1 in the presence of 2-formylpyridine.
:2:1 ratio produced a different, lower nuclearity homoleptic assembly, [M222(CH3CN)4]4+, which was found to incorporate electron-deficient aromatic guests in its electron-rich cavity. Prompted by this observation, self-sorting experiments were carried out with electron-poor NDI sub-unit S1 and various electron-rich pyrene isomers. One particular combination led to a striking outcome; combining S1 and S2 in a 1:1 ratio with formylpyridine and MII(NTf2)2 led to a [M41224]8+ heteroleptic assembly (Fig. 4). X-ray analysis of this sample revealed an unusual triple-decker pyrene–pyrene–NDI stack, contrasting with the more commonly observed donor–acceptor alternating stacks. Furthermore, the [M41224]8+ structure could be accessed through a cage-to-cage transformation of [M416]8+ and [M426]8+ or a ligand-induced transformation of [M222(CH3CN)4]4+ with S1 in the presence of 2-formylpyridine.
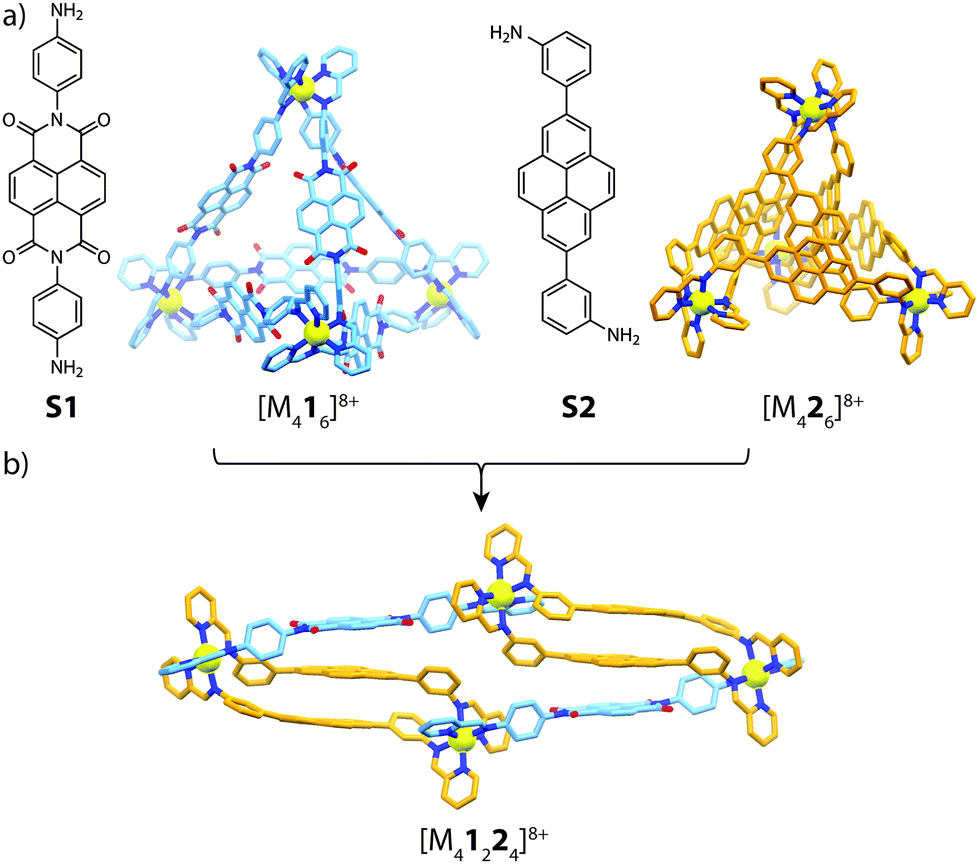 | ||
| Fig. 4 (a) Electron-poor (S1) and electron-rich (S2) sub-components and their respective homoleptic assemblies; (b) the heteroleptic [M41224]8+ product (M = FeII, CoII, ZnII). | ||
In 2011, Hooley and co-workers examined the formation of heteroleptic [Pd2LA2LB2]4+ cages by introducing endohedral steric bulk into bis-monodentate ligands with identical geometries and donor types.51 By modification of the ligand backbone, ligands with different endohedral functionality were prepared: R = NH2 (4); R′ = CF3 (5) or NHPh (6) (Fig. 5). The least sterically demanding ligands (3 and 4) were observed to smoothly form homoleptic [Pd2L4]4+ assemblies with PdII. On the other hand, sterically bulkier ligands 5 and 6 formed complex mixtures containing a number of different species. Pd-mediated assembly of ligands 3 and 4 gave a mixture of [Pd23n44−n]4+ (n = 0–4) complexes with a bias towards [Pd23341]4+ as indicated by NMR and ESI-MS analysis. Further studies revealed that combining 3 with the medium-sized NHCOCF3 ligand 5 in a 3:1 ratio gave a well resolved mixture consisting of [Pd23351]4+ and [Pd234]4+ in a ratio of 3:1 respectively. This experiment demonstrated that indeed, control of heteroleptic cage formation can be achieved by ‘filling’ the empty space of the cavity with endohedral steric bulk.
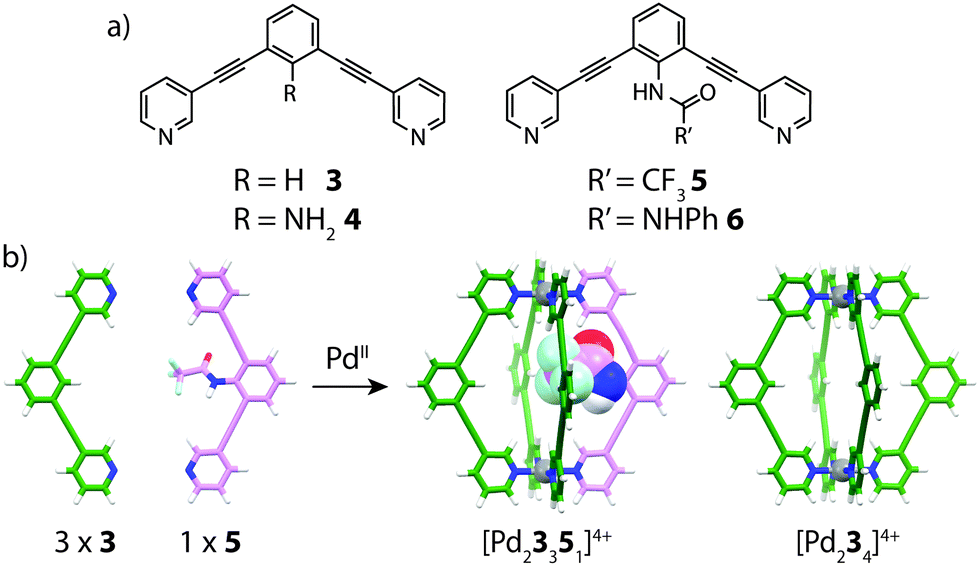 | ||
| Fig. 5 (a) Endohedrally functionalized ligands 3–6 bearing varying degrees of steric bulk; (b) integrative self-sorting of ligands 3 and 5 to give a 3:1 mixture of [Pd23351]4+ and [Pd234]4+. The NHCOCF3 endohedral substituent in the heteroleptic assembly is shown as a space-filling representation. | ||
For both of the systems discussed above, it is important to note that whilst exploiting complementary interactions between adjacent ligands is a successful approach for enabling the assembly of heteroleptic architectures, the basis of the structural control relies upon components interacting in close proximity (or within a cavity). Consequently, the possibility for an accessible cavity that would be able to bind further guest molecules is eliminated in these systems.
2.2. Templating effects
When the formation of heteroleptic cage assemblies relies on two ligands with similar dimensions and/or donor groups, the fine energetic balance between the possible products can be controlled by the incorporation of a guest template; often resulting in selective stabilization of a particular structure.An early example of this was reported in 1999 by Albrecht and co-workers, who examined the self-sorting of different alkyl-bridged catechol ligands in TiIII helicates.52 In this study, the internal cavity of the anionic helicates was lined with multiple catechol oxygen atoms that could facilitate the binding of hard metal cations (according to the HSAB principle). Interestingly, the binding of different alkali cation guests inside the cavity of the cage could regulate the outcome of self-sorting. For example, addition of an Li+ guest drove the system from a narcissistic mixture to a mixture containing both homoleptic and heteroleptic species. A similar effect has recently been reported in a dynamic library of orthoester cryptands which could rearrange to hetero- or homo-component host–guest complexes in the presence of different templating alkali metal ions.53
In 2015, the group of Yoshizawa examined the self-sorting of anthracene-functionalized [Pd2LA2LB2]4+ cages using a fullerene guest as template.54 In accordance with previous studies,55 the short and long bis-monodentate ligands (7 and 8) were both observed to form Pd-mediated homoleptic coordination cages, [Pd274]4+ and [Pd284]4+. A comparison of their X-ray structures revealed that the cavity of the latter assembly is larger by 90 Å3, which was exploited for the binding C70 and functionalized C60 guests. In the absence of a guest template, a 1:1 mixture of the two homoleptic cages was found to spontaneously rearrange into a statistical mixture of cages: [Pd27n84−n]4+ (n = 0–4). However, addition of C60 to this mixture resulted in guest encapsulation and simultaneous re-organization to a single [C60@Pd27282]4+ heteroleptic product (Fig. 6). This was confirmed by 2D NMR and ESI-MS which also revealed the absence of other heteroleptic host–guest complexes, suggesting that the C60 template provides a large energetic contribution that stabilizes this particular structure. Force-field calculations revealed the cis-isomer to be the most energetically favoured which was attributed to optimal host–guest interactions with the C60 guest.
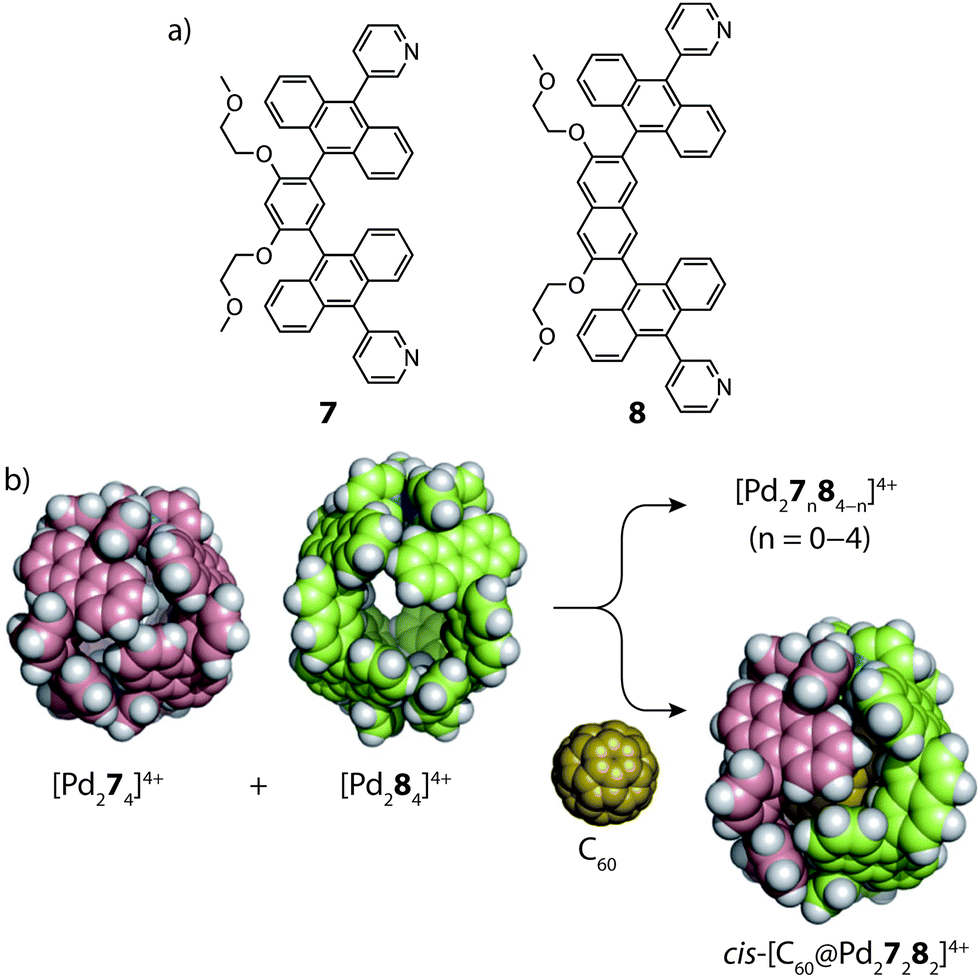 | ||
| Fig. 6 (a) Short (7) and long (8) anthracene-functionalized ligands; (b) C60-templated integrative self-sorting of cages [Pd274]4+ and [Pd284]4+ to give the heteroleptic host–guest complex cis-[C60@Pd27282]4+. Reproduced with permission from ref. 54. | ||
Whilst these examples clearly demonstrate that templating effects can steer self-sorting toward a heteroleptic product, the overall approach – which relies upon an already occupied cavity – may not facilitate the exchange of other guests without perturbing the self-sorted assembly.
2.3. Shape-complementarity
Using geometric principles to design cage assemblies of desired shapes and sizes is a well-established approach in metallosupramolecular chemistry.2 This design strategy, which has been also termed ‘edge-directed self-assembly’,56 often involves utilizing the encoded geometric features of two-component systems in a complementary manner. In most of the reported examples that make use this approach, however, usually one of the organic components is irreversibly attached to a metal centre in the form of a tightly joined organometallic fragment, resulting in a non-dynamic system with straight-forward design criteria.57 A more complicated scenario arises in a heteroleptic system formed from naked metal ions where the ligand components are able to exchange dynamically. Therefore, whilst the concepts of geometric design are indeed powerful, controlling integrative self-sorting in fully-dynamic three-component systems comprises a more complicated task due to the propensity of ligand exchange in energetically similar structures.In 2010, Li and Zhou reported a ligand-substitution strategy that enables access to heteroleptic cages composed of angular dicarboxylate ligands and CuII2 paddle-wheel nodes (Fig. 7).58 In this work, the authors prepared a number of different homoleptic architectures which could be interconverted or disassembled by the addition of a competing ligand. In two particular examples, partial ligand substitution of a cuboctahedral homoleptic cage precursor was observed, which led to novel heteroleptic architectures. Reacting [(Cu2)12924] with ligand 10, which has a 60° bend angle, resulted in a structural transformation to a lower nuclearity, mixed-ligand structure, [(Cu2)696106]. The same reaction with ligand 11, which has a 120° bend angle, resulted in the selective substitution of 12 of the available 24 vertices with preservation of the overall topology to give [(Cu2)129121112]. Interestingly, this structure is composed of two ligands of equal geometries but unequal lengths. X-ray analysis revealed that the parent [(Cu2)393] triangular fragments were conserved in each transformation, suggesting that the [(Cu2)393] motif may be energetically favoured in the dynamic self-assembly process. Whilst it is evident that geometric recognition between ligands of distinct angles and lengths plays a role in this example, the assembly and characterization of these cages appears to be dependent on crystallization methods. It is therefore unknown whether solid-state packing effects contribute to the stabilization of the heteroleptic structure.
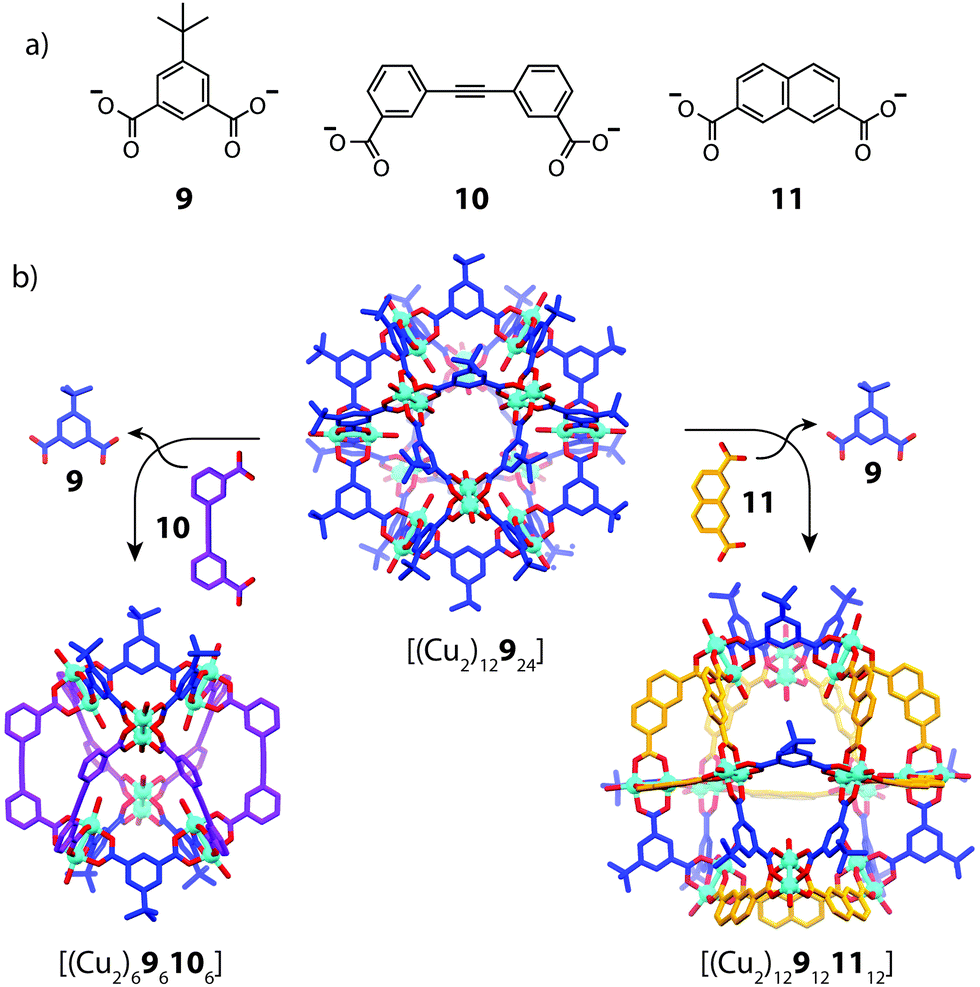 | ||
| Fig. 7 (a) Bent dicarboxylate ligands 9–11 bearing angles of 120°, 60° and 120° respectively; (b) transformation of the homoleptic [(Cu2)12924] cage to heteroleptic species [(Cu2)696106] (left) and [(Cu2)129121112] (right) by ligand substitution. | ||
Fujita and co-workers reported a related heteroleptic cuboctahedral system composed from PdII cations and bis-monodentate pyridyl ligands. Homoleptic Pd-mediated self-assembly of ligands 12 and 13, which bear similar bend angles (120°) but different overall lengths, gave cuboctahedral [Pd121224]24+ and [Pd121324]24+, respectively, in accordance with previously established geometric criteria.59 Combining these ligands with PdII in a 1:1:1 ratio, however, led to clean integrative self-sorting to give a novel [Pd1212121312]24+ heteroleptic architecture (Fig. 8).60 The mixed-ligand structure was supported by ESI-MS, DOSY NMR as well as X-ray crystallography, which revealed a C3v pseudocantellated tetrahedral topology. Further self-sorting studies demonstrated that the ligand-length ratio (derived from the difference in length between the two ligand components) is crucial in achieving integrative self-sorting; when two ligands of similar lengths were combined with PdII, a statistical mixture of [Pd12LAnLB24−n]24+ (n = 0–24) species was obtained. It is worth noting that this system represents a high degree of structural control: random mapping of two ligands in a [Pd1212n1324−n]24+ structure can give rise to 7 × 105 possible combinations.
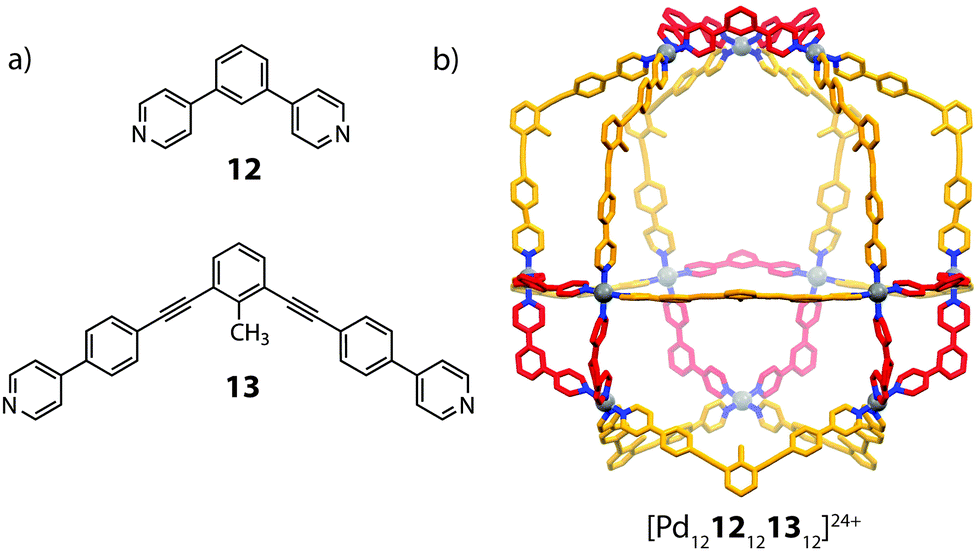 | ||
| Fig. 8 (a) The short (12) and long (13) angular ligands (b) a heteroleptic [Pd1212121312]24+ cantellated tetrahedron. | ||
Clever and co-workers explored integrative self-sorting of [Pd2LA2LB2]4+ coordination cages utilising geometric design, rather than ligand length, to steer the formation of the heteroleptic product.61 Therefore, combinations of banana-shaped ligands 14–16 with different bend angles and donor sites were systematically examined (Fig. 9). First, two obviously shape-complementary ligands were studied: a 60° inward-bent acridone ligand (14) and an 120° outward-bent phenanthrene ligand (16). Preliminary DFT modelling of the possible [Pd2LAnLB4−n] (n = 0–4) structures revealed that the cis-[Pd2142162] assembly is by far the most energetically favoured due to a shape-complementary arrangement of ligands with respect to the PdII centres.
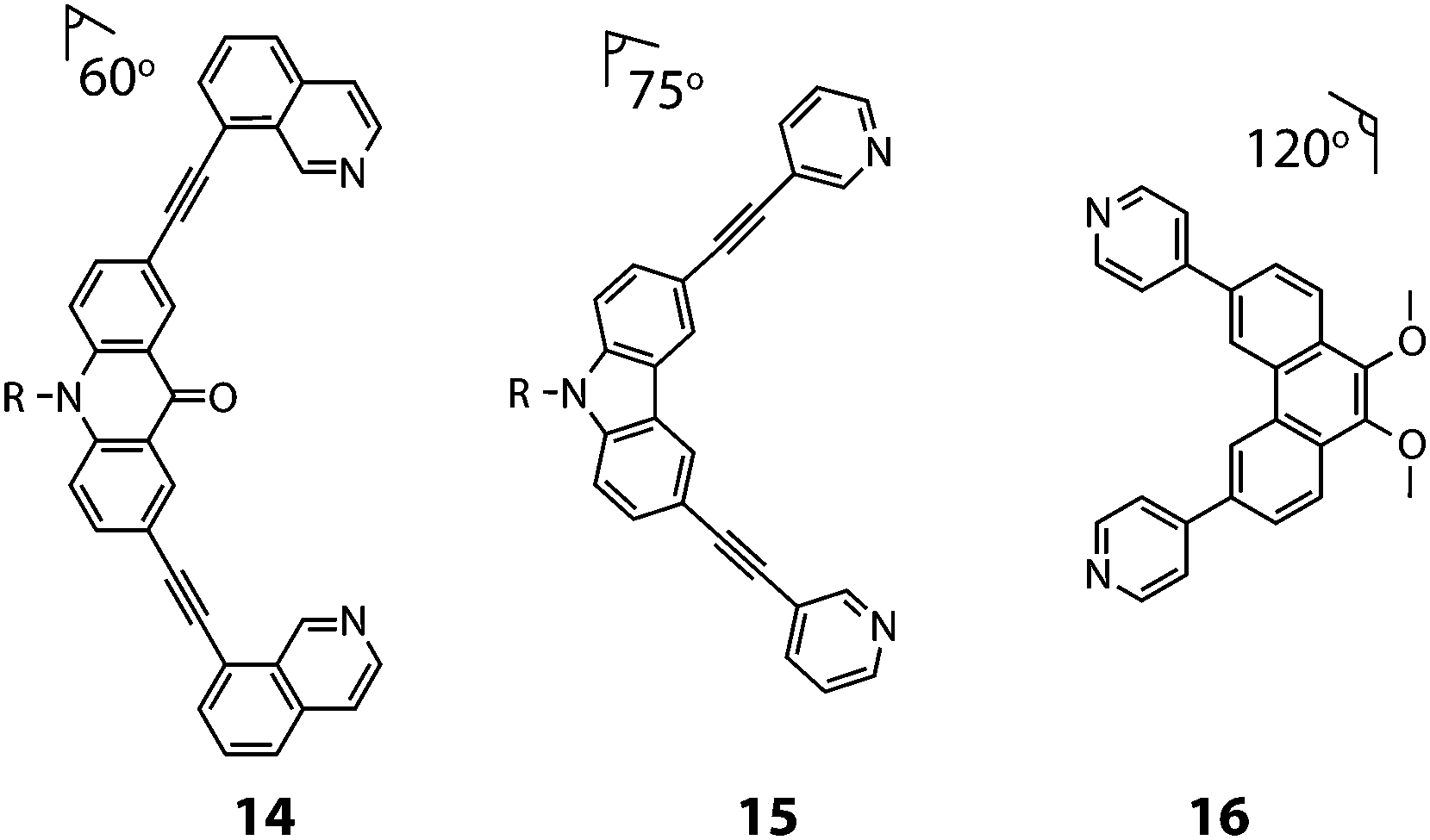 | ||
| Fig. 9 The angularly distinct bis-monodentate ligands (14–16) used to assemble [Pd2LA2LB2]4+ heteroleptic cages (R = C6H13). | ||
Studies examining the homoleptic structures revealed that a helical [Pd2144]4+ cage is formed from 14 and PdII cations in which the isoquinoline ligands are forced to adopt a twisted conformation. In contrast, the Pd-mediated self-assembly of 16 led to a [Pd4168]8+D4h-symmetric box structure (Fig. 10a). The combination of 14 and 16 with Pd(II) in a 1:1:1 ratio led to the clean formation of a new species with a distinct NMR spectrum. 2D NMR as well as ESI-MS experiments supported a heteroleptic assembly obeying a formula of [Pd2142162]4+. Furthermore, 1H–1H NOESY analysis revealed several evident inter-ligand contacts consistent with the DFT-calculated structure of cis-[Pd2142162]4+. The heteroleptic cage could also be accessed via a cage-to-cage transformation of [Pd2144]4+ and [Pd4168]8+ which confirmed that cis-[Pd2142162]4+ is the thermodynamic minimum of the system. Additionally, the authors successfully probed the unusually-shaped cavity of the bent heteroleptic structure through guest-binding experiments with isomeric naphthalene di-sulfonates. NMR titrations revealed that the anisotropic cage cavity exhibits stronger binding toward the guest isomer possessing a complementary ‘bent’ geometry (Fig. 10b). The shape-dependent guest binding specificity was thus found to be opposite to the one of a previously reported ‘straight’ [Pd2L4]4+ cage which exhibited preference for binding the linear di-sulfonate guest.62
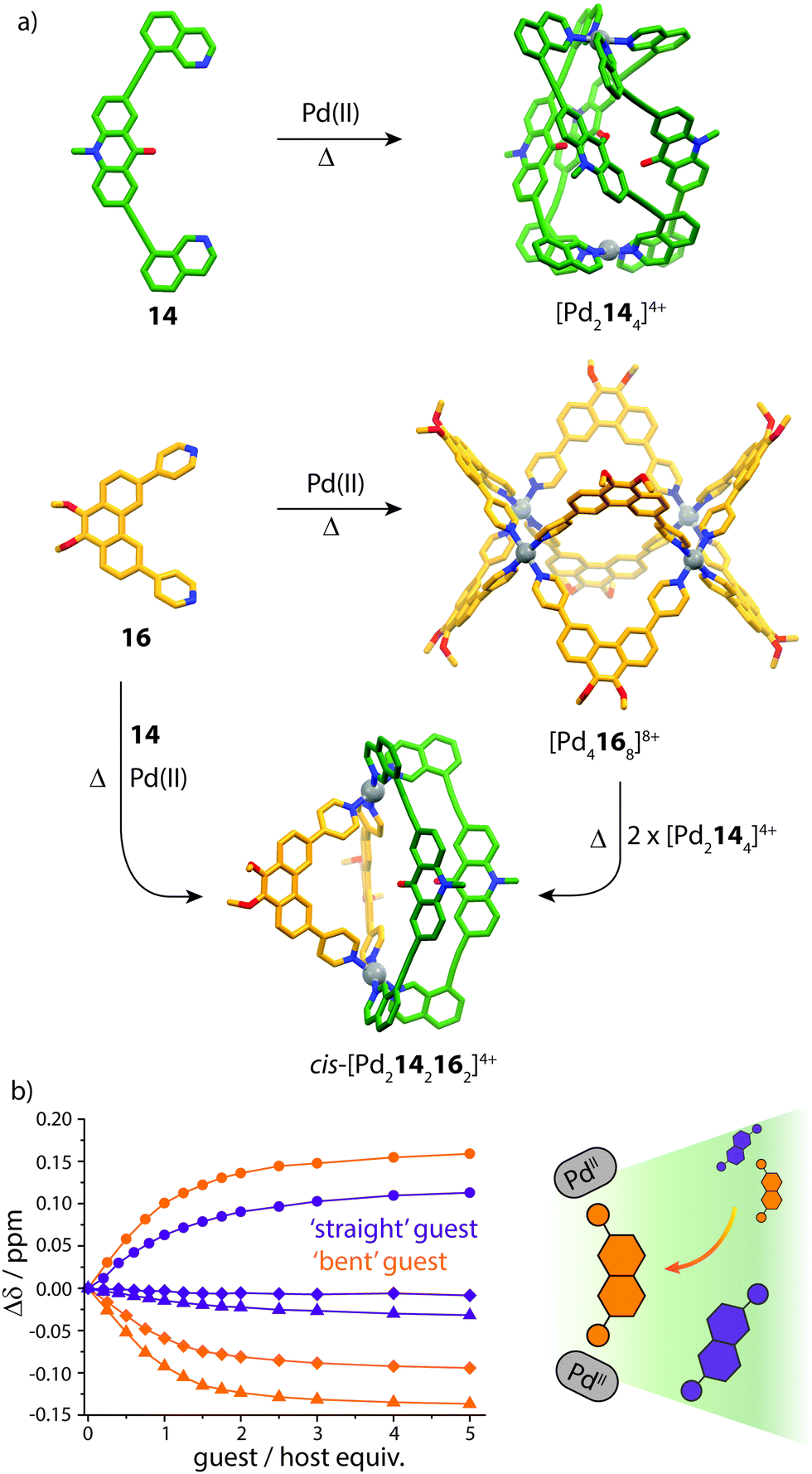 | ||
| Fig. 10 (a) Self-assembly of homoleptic and heteroleptic cages from shape-complementary ligands 14 and 16. Combining the two ligands with PdII or mixing their homoleptic cages leads to clean integrative self-sorting to give a cis-heteroleptic cage product; (b) 1H NMR titrations of the heteroleptic cage with ‘straight’ and ‘bent’-shaped di-sulfonate guests. Reproduced with permission from ref. 61. | ||
Subsequently, Clever and co-workers undertook further studies to examine the interplay of complementary ligand geometries in [Pd2LA2LB2]4+ cages. The Pd-mediated self-sorting of carbazole ligand (15) was examined with the two angular components (14 and 16) established in their first system. Ligand 15 bears a 75° bend-angle (Fig. 9) and has been previously studied in the context of mono- and double-cage assembly.63 Evidence for a [Pd2152162]4+ heteroleptic assembly was obtained by 1H NMR and ESI-MS analysis of a 1:1:1 mixture of 15, 16 and PdII. A similar result was obtained from heating a 2:1 mixture of the homoleptic assemblies [Pd2154]4+ and [Pd4168]8+ (Fig. 11). Single-crystal X-ray analysis confirmed the heteroleptic [Pd2152162]4+ cage structure in which each pair of ligands simultaneously bridges two PdII centres in a cis-conformation. The [Pd2152162]4+ cage structure represents the first heteroleptic cage exclusively formed from simple pyridine donors, demonstrating that geometric factors are more dominant in this system than specific donor combinations. On the other hand, combining ligands 14 and 15 with PdII in a 1:1:1 ratio (or mixing their corresponding homoleptic cages) led to a topologically-novel heteroleptic structure. Analysis of the sample by ESI-MS revealed prominent peaks corresponding to a cage with the formula [Pd2142152]4+, while NMR analysis revealed a complicated spectrum with each ligand losing its inherent two-fold symmetry. 1H–1H NOESY analysis revealed several intra-ligand contacts which suggested that ligand 14 is in a locked anti-conformation rather than the normally encountered syn-conformation. Indeed, X-ray analysis revealed an unprecedented structural motif for metallosupramolecular structures; a trans-[Pd2(anti-14)2152]4+ cage structure resembling a “doubly-bridged figure-eight” topology.64 Furthermore, the authors were able to show a degree of morphological control by transforming each heteroleptic assembly to the cis-[Pd2142162]4+ cage by the addition of a competing ligand (Fig. 11). This heteroleptic-to-heteroleptic process was driven by the ideal shape of ligands 14 and 16, representing a high degree of structural control in the system.
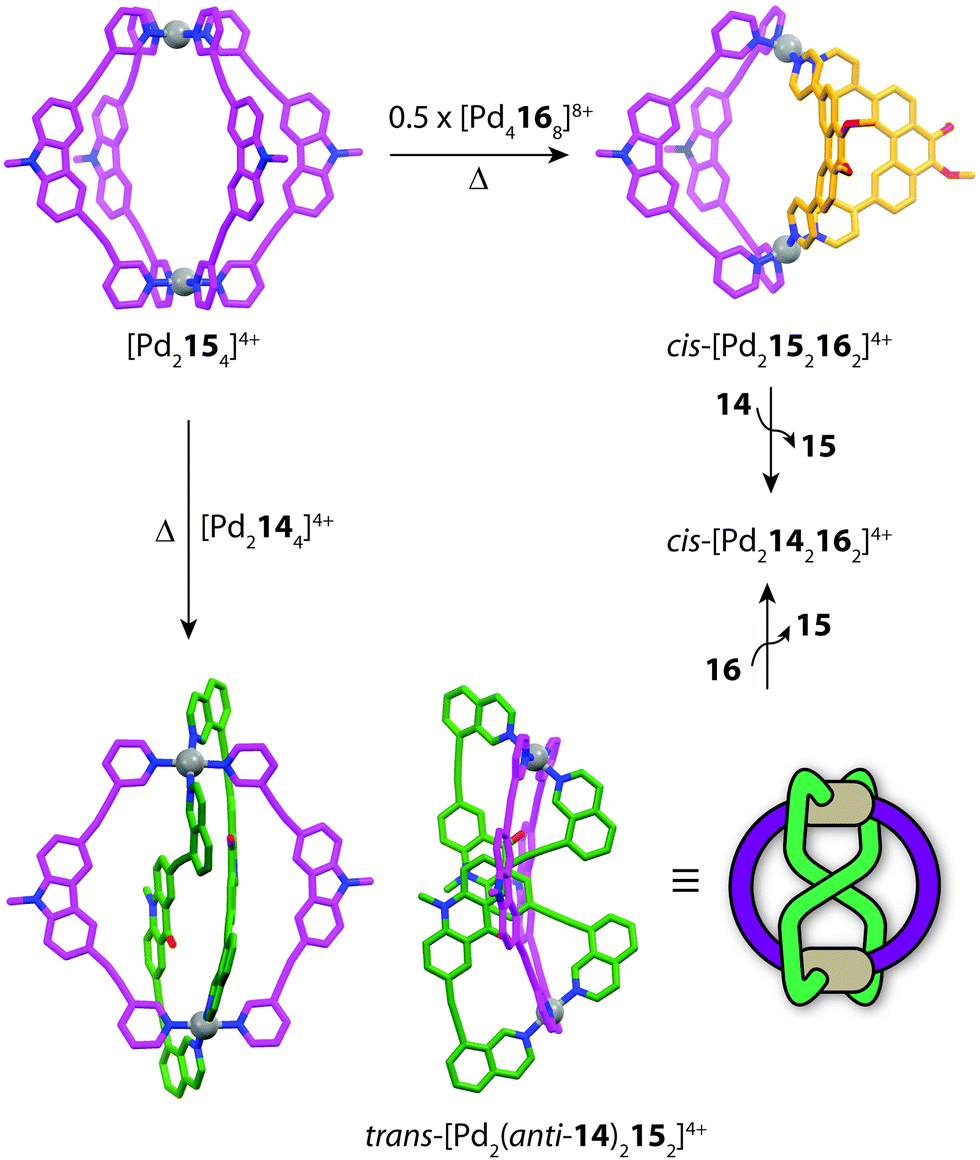 | ||
| Fig. 11 Cage-to-cage integrative transformations of [Pd2154]4+ to form either cis or trans heteroleptic cages. Each heteroleptic cage can be transformed to cis-[Pd2142162]4+ by addition a competing ligand (14 or 16). | ||
2.4. Other effects
Nitschke and co-workers reported the synthesis and self-sorting of two related tetrahedral cages, [Zn416]8+ and [Zn4176]8+, prepared by sub-component self-assembly of ZnII and NDI (S1) or porphyrin-derived (S17) subunits.65 A study of the host–guest properties revealed that each cage can respond to a different stimulus; the electron-poor NDI units of [Zn416]8+ were found to thread up to two electron-rich crown-ether macrocycles, while [Zn4176]8+ was found to accommodate a C70 guest in its cavity (Fig. 12a and b). Importantly, the host–guest properties of each cage were exclusive with respect to one another. This was supported by multiple experiments, including ESI-MS for [Zn416+Cn]8+ (C = catenated crown ether), which revealed direct catenation for n = 1 and 2. Mixing the two cages in a 1:1 ratio resulted in a near-to statistical mixture of seven constitutionally distinct heteroleptic [Zn41n176−n]8+ cages. This mixture was observed to reshuffle upon addition of either the crown-ether or C70, favouring assemblies which can interact with the respective guest stimulus. For example, addition of C70 to the mixture of cages resulted in narcissistic self-sorting into [C70@Zn416]8+ and [Zn4176]8+, while addition of the crown-ether resulted in a defined self-sorted mixture of heteroleptic cages containing one or two threaded crown-ethers (Fig. 12c). Here, the driving force for the narcissistic and integrative self-sorting was attributed to the enthalpic benefit associated with maximising the possible host–guest interactions.
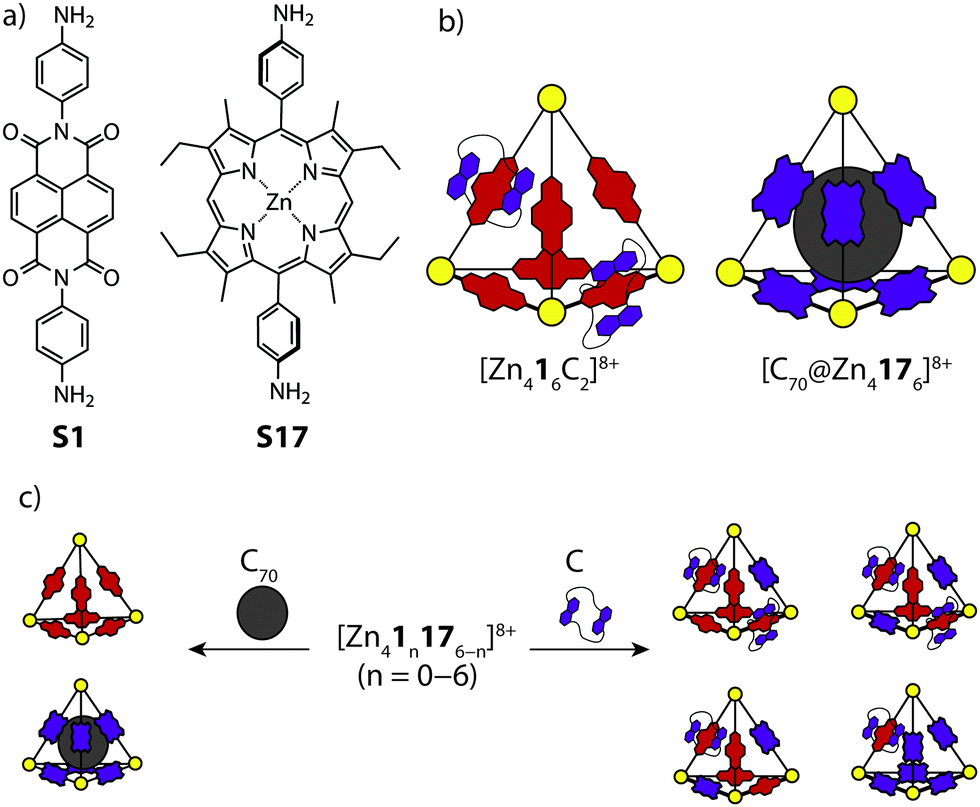 | ||
| Fig. 12 (a) Diamine sub-components S1 and S17, based on either a NDI or porphyrin backbone; (b) the corresponding tetrahedral cages showing the distinct host–guest chemistry of each cage; (c) a dynamic library which rearranges according to the type of stimulus introduced. | ||
3. Coordination-dependent integrative self-sorting
3.1. Steric constraints
The utilization of steric control around the metal centre has been widely exploited in forming both 2D and 3D heteroleptic structures.28,37,66 This concept is complementary to the aforementioned approaches of topological constraints and maximum-site occupancy, where simple enthalpic principles drive mixed-ligand structures in spite of the entropic penalty.Recently, Schmittel reported a multi-component cage structure using a combination of sub-component self-assembly and what he coined to be the “HETPHEN” approach.67 Preliminary experiments revealed that a discrete heteroleptic complex can be formed from a mixture of pyridine-2-aldehyde, a di-mesityl phenanthroline ligand and CuI cations. Furthermore, it was shown that subcomponent self-assembly is compatible with this heteroleptic motif. This observation formed a basis to access higher-order aggregates based on a prismatic cage architecture. Combining the tris-bidentate ligand 18 with subcomponents 19, S20 and CuI gave rise to a [Cu6182203]6+ prismatic cage, which is similar to that observed in Lehn's early work.68,69 To push the boundaries of cage design, the authors synthesized a tris-capping amino derivate S21, which was utilised in the same reaction (with 18 and 19) to form a doubly-partitioned cage structure [Cu618221]6+.70 Evidence for the heteroleptic assembly was obtained through multiple 2D NMR and ESI-MS experiments, all of which were consistent with the calculated structure (Fig. 13).
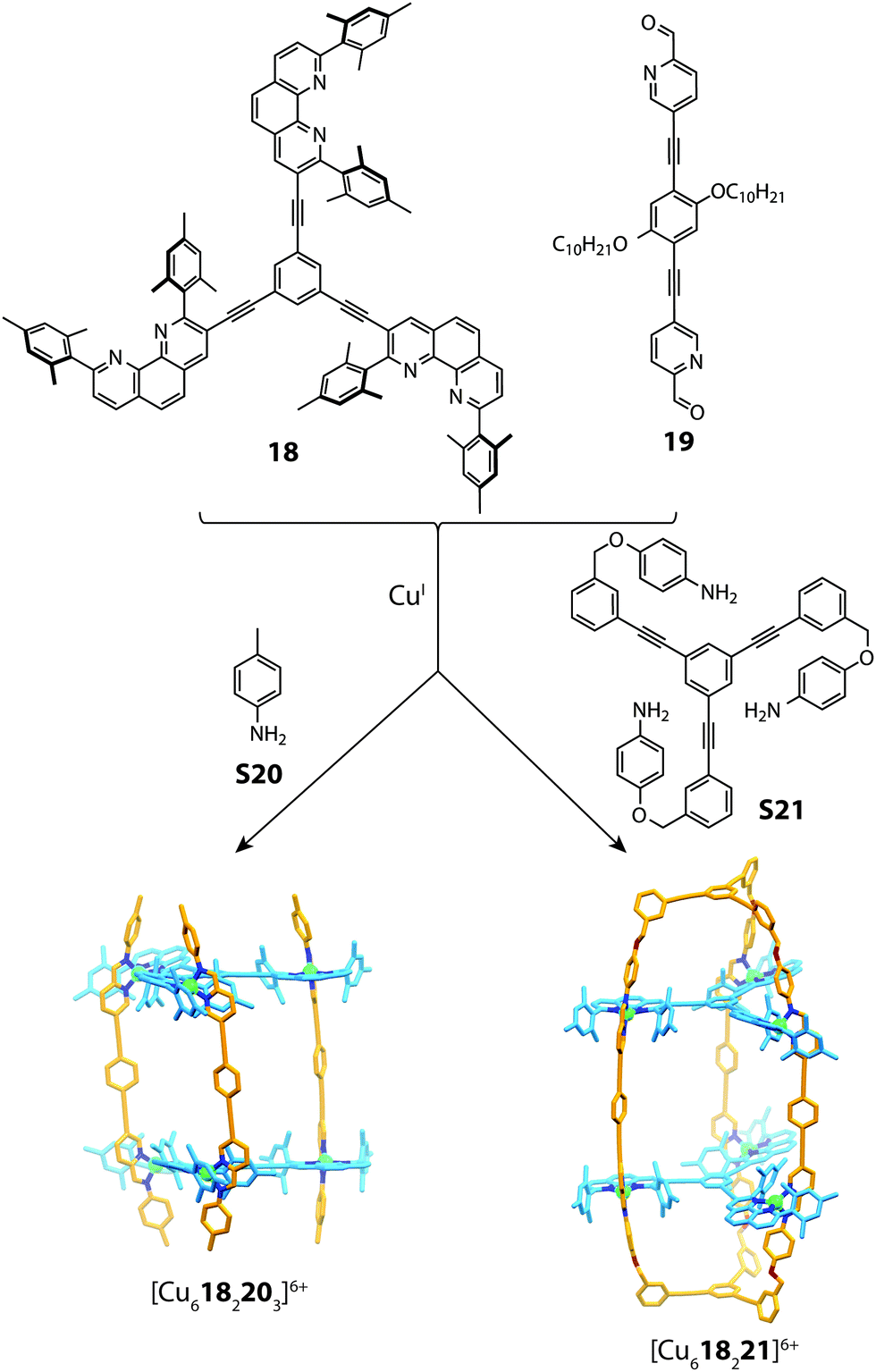 | ||
| Fig. 13 Self-assembly of a [Cu6182203]6+ prism (mediated by S20) and a doubly-partitioned cage [Cu618221]6+ mediated by S21. | ||
3.2. Hydrogen-bonding
Apart from implementing steric bulk around the metal centre, secondary interactions can also steer heteroleptic cage formation. One such example was reported by Crowley and co-workers, who utilized ortho-amino-substituted pyridyl ligands to access heteroleptic cis-[Pd2LA2LB2]4+ coordination cages through complementary hydrogen bonding (Fig. 14).71 In this study, the authors observed different outcomes upon performing ligand substitution reactions with either 3-amino (22) or 2-amino (24) substituted ligands on an unfunctionalized [Pd2234]4+ cage precursor. Addition of two equivalents of the meta-substituted ligand 22 to [Pd2234]4+ resulted in complete ligand displacement, owing to the better donor capabilities of 22.72 In contrast, the same reaction with ortho ligand 24 led to only partial displacement of the original cage, giving rise to a new heteroleptic product [Pd2232242]4+. Interestingly, the heteroleptic product could not be obtained from heating a 1:1 mixture of the homoleptic cages ([Pd2234]4+ and [Pd2244]4+) or from a 1:1:1 mixture of 23, 24 and PdII, suggesting that a specific pre-organised conformation is necessary to access the heteroleptic cage assembly. The [Pd2232242]4+ structure was supported by ESI-MS and multiple 2D NMR experiments, including 1H–1H ROESY, which revealed clear inter-ligand contacts between 23 and 24 in the heteroleptic structure. DFT calculations revealed that the cis-isomer is energetically more favourable than the trans-species, in accordance with the hypothesis that a cis-arrangement is stabilized via H-bonding; an interaction which would be diminished in the trans-conformer of the heteroleptic cage. Furthermore, the calculations also suggested that the thermodynamically favoured product is the homoleptic [Pd2244]4+ species rather than the heteroleptic assembly. Indeed, competition experiments revealed that gradually, the cis-[Pd2232242]4+ cage can undergo further ligand displacement reactions, supporting that it is a metastable kinetic product.
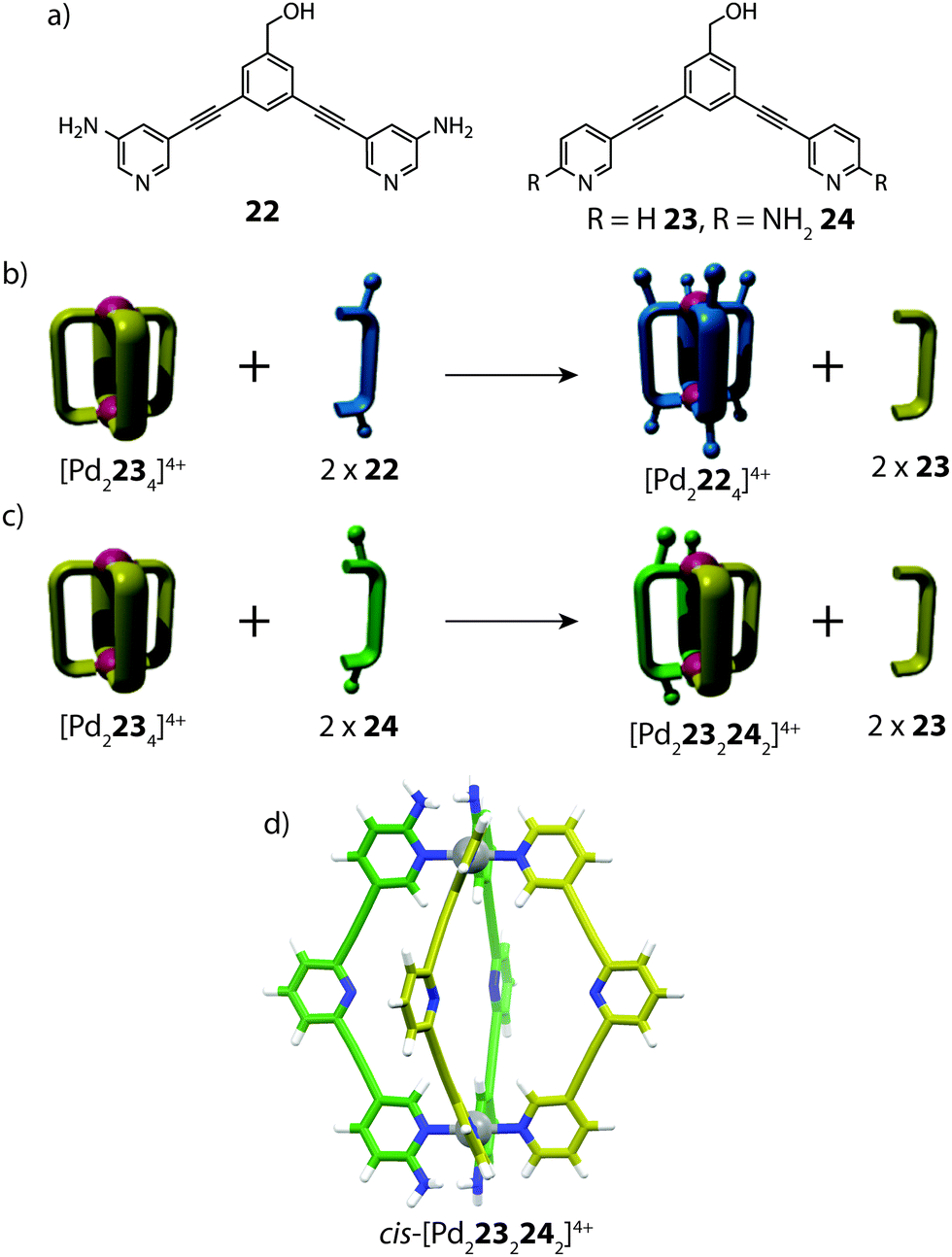 | ||
| Fig. 14 (a) Banana-shaped pyridyl ligands containing amino substituents in meta- or ortho-positions; (b) ligand substitution reaction resulting in complete ligand displacement; (c) partial ligand displacement to give a heteroleptic cis-cage; (d) a DFT-model of cis-[Pd2232242]4+. Reproduced with permission from ref. 71. | ||
4. Complementary functions
The ultimate goal of realising heteroleptic supramolecular structures is the utilization of multiple complementary functionalities in synergy. In the emerging field of multi-component self-assembly, the examples reported so far illustrate some fundamental approaches that allow a degree of control over the selective formation of heteroleptic coordination cages. Whilst exploiting multi-functionalization in heteroleptic cages is yet to be explored, the diverse principles described in this article represent a foundation for extending studies in this direction. For example, the modular combination of ligands with different catalytic functionalities in endohedral positions may increase the potency of new catalytic systems. Thus, robust strategies for the rational construction of multi-functional host compounds, and supramolecular systems in general, promise to lead to sophisticated applications.It is important to note, however, that a lack of precise control over self-assembled structures does not generally preclude the utilisation of heteroleptic cage mixtures as multi-functional systems: although appearing less attractive to the supramolecular chemist, statistical mixtures can facilitate the exploration of complementary functionalities in phenomena inaccessible to homoleptic structures. An example of this was reported by Clever and co-workers who utilised the well-known phenothiazine/anthraquinone electron-donor/acceptor functionality in the assembly of an interpenetrated cage system (Fig. 15). This study follows their previous work which demonstrated that Pd-mediated assembly of a mixture of two different bis-monodentate ligands of similar length results in [Pd4LAnLB8−n]8+ (n = 0–8) mixtures with a statistical distribution of species while preformed homoleptic double cages do not exchange ligands in solution due to kinetic hindrance.73 The realization that the interpenetrated [Pd4L8]8+ topology brings together the ligand backbones in close proximity encouraged the authors to combine the electron-donor/acceptor ligands (25 and 26) with PdII to form a [Pd425n268−n]8+ system. The mixed-ligand cages were subsequently examined by time-resolved pump–probe UV-Vis spectroscopy and their photo-physical behaviour was compared to a 1:1 mixture of the homoleptic donor- and acceptor-cages. Interestingly, only the mixed-ligand cages showed signs of an intra-assembly charge transfer, as indicated by transient spectroscopic signatures of the oxidized donor and the reduced acceptor moieties (Fig. 15).74 While this system demonstrates the potential of supramolecular assembly in the morphological control of photoactive hetero junctions, the current system still suffers from a statistical composition of the donor- and acceptor functionalities. As such, this dilemma serves as a strong motivation to foster progress in strategies for rational assembly of non-statistically assembled multi-functional systems.
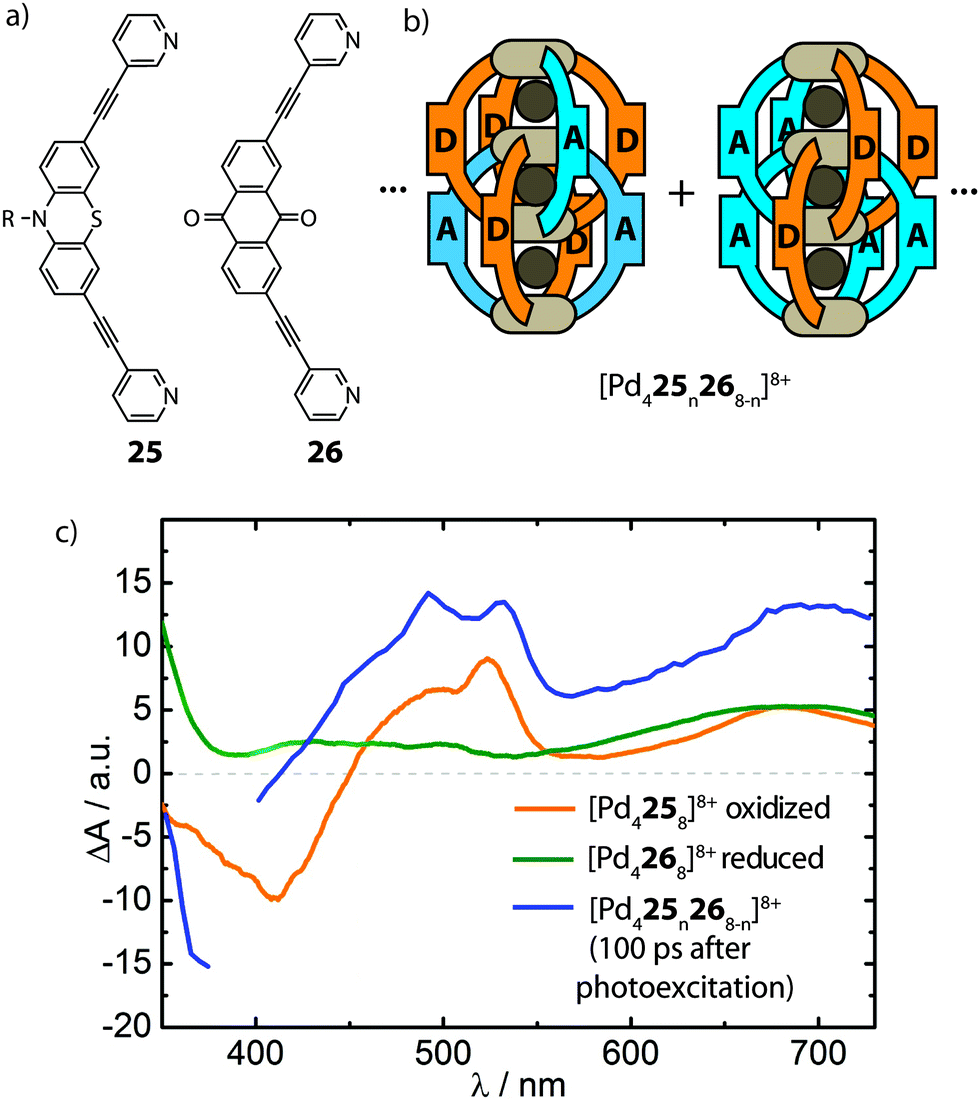 | ||
| Fig. 15 (a) Electron-donor (25) and acceptor (26) ligands; (b) representation of the statistical composition of the mixed-ligand double-cages; (c) time-resolved pump–probe UV-Vis spectrum of photo-excited mixed-ligand cages compared to the spectra of the oxidized donor and reduced acceptor cages, respectively. | ||
5. Conclusions and outlook
The design and synthesis of coordination cages is a vibrant field in which there has been much recent innovation. Whilst strategies for gaining control over the size, shape and topology are well-established,2,8,11 rational approaches to assemble heteroleptic cage structures are still in early development. Herein, we have highlighted recent progress in the design of heteroleptic cage compounds from ‘naked’ metal centres – systems which are fully-dynamic and thus require a deeper understanding of the underlying self-assembly principles, compared with traditional two-component homoleptic structures.We expect further progress in this field to focus on exploiting a variety of fine-tuneable influences such as metal–ligand bond strengths, structural strain/flexibility, electronic effects and solvent dependence. Kinetic and thermodynamic trans effects commonly observed in mononuclear coordination compounds with square-planar geometry have not yet received a lot of attention in supramolecular assembly, despite that they may be utilized to control the formation of one particular heteroleptic configuration over another (e.g. cis-[Pd2LA2LB2] vs. trans-[Pd2LA2LB2]). There is still, therefore, much scope in developing methods to control integrative self-sorting of hollow cage assemblies.
As creating multi-functional host-structures is one of the goals of this area, the approaches making use of geometric factors and/or donor-site engineering – which have shown to facilitate an accessible cavity – are the most promising. With this in mind, the next step will be to implement functionalities of a complementary nature into a single self-assembled structure. Worthy to examine function pairs would include photo-excitable donors/acceptors, catalytic centres/chiral groups or other control elements such as frustrated Lewis acids/bases or specific binding sites/reporting chromophores. We envision that a modular assembly approach under full rational control of stoichiometry and stereochemistry of all building blocks will lead to advanced host-structures, capable of stimuli-responsive catalysis, directed energy transfer, cooperative binding and more.
Acknowledgements
W. M. B. thanks the Alexander von Humboldt Foundation for a postdoctoral fellowship. We thank the Fonds der Chemischen Industrie and the European Research Council (ERC Consolidator grant 683083, RAMSES) for their support.Notes and references
- G. F. Swiegers and T. J. Malefetse, Chem. Rev., 2000, 100, 3483–3538 CrossRef CAS PubMed.
- R. Chakrabarty, P. S. Mukherjee and P. J. Stang, Chem. Rev., 2011, 111, 6810–6918 CrossRef CAS PubMed.
- T. R. Cook and P. J. Stang, Chem. Rev., 2015, 115, 7001–7045 CrossRef CAS PubMed.
- L. F. Lindoy, K.-M. Park and S. S. Lee, Chem. Soc. Rev., 2013, 42, 1713–1727 RSC.
- M. Fujita, M. Tominaga, A. Hori and B. Therrien, Acc. Chem. Res., 2005, 38, 369–378 CrossRef CAS PubMed.
- F. Würthner, C.-C. You and C. R. Saha-Möller, Chem. Soc. Rev., 2004, 33, 133–146 RSC.
- S. Zarra, D. M. Wood, D. A. Roberts and J. R. Nitschke, Chem. Soc. Rev., 2015, 44, 419–432 RSC.
- M. M. J. Smulders, I. A. Riddell, C. Browne and J. R. Nitschke, Chem. Soc. Rev., 2013, 42, 1728–1754 RSC.
- S. J. Dalgarno, N. P. Power and J. L. Atwood, Coord. Chem. Rev., 2008, 252, 825–841 CrossRef CAS.
- M. D. Ward and P. R. Raithby, Chem. Soc. Rev., 2013, 42, 1619–1636 RSC.
- M. Han, D. M. Engelhard and G. H. Clever, Chem. Soc. Rev., 2014, 43, 1848–1860 RSC.
- M. Frank, M. D. Johnstone and G. H. Clever, Chem. – Eur. J., 2016, 22, 14104–14125 CrossRef CAS PubMed.
- C. J. Brown, F. D. Toste, R. G. Bergman and K. N. Raymond, Chem. Rev., 2015, 115, 3012–3035 CrossRef CAS PubMed.
- M. Otte, ACS Catal., 2016, 6, 6491–6510 CrossRef CAS.
- Y. Ueda, H. Ito, D. Fujita and M. Fujita, J. Am. Chem. Soc., 2017, 139, 6090–6093 CrossRef CAS PubMed.
- O. Zava, J. Mattsson, B. Therrien and P. J. Dyson, Chem. – Eur. J., 2010, 16, 1428–1431 CrossRef CAS PubMed.
- S. K. Samanta, D. Moncelet, V. Briken and L. Isaacs, J. Am. Chem. Soc., 2016, 138, 14488–14496 CrossRef CAS PubMed.
- F. Schmitt, J. Freudenreich, N. P. E. Barry, L. Juillerat-Jeanneret, G. Süss-Fink and B. Therrien, J. Am. Chem. Soc., 2012, 134, 754–757 CrossRef CAS PubMed.
- W. Meng, B. Breiner, K. Rissanen, J. D. Thoburn, J. K. Clegg and J. R. Nitschke, Angew. Chem., Int. Ed., 2011, 50, 3479–3483 CrossRef CAS PubMed.
- C. García-Simón, M. Garcia-Borràs, L. Gómez, T. Parella, S. Osuna, J. Juanhuix, I. Imaz, D. Maspoch, M. Costas and X. Ribas, Nat. Commun., 2014, 5, 5557 CrossRef PubMed.
- W.-Y. Zhang, Y.-J. Lin, Y.-F. Han and G.-X. Jin, J. Am. Chem. Soc., 2016, 138, 10700–10707 CrossRef CAS PubMed.
- X. Yan, T. R. Cook, P. Wang, F. Huang and P. J. Stang, Nat. Chem., 2015, 7, 342–348 CrossRef CAS PubMed.
- P. P. Neelakandan, A. Jimenez and J. R. Nitschke, Chem. Sci., 2014, 5, 908–915 RSC.
- B. Roy, A. K. Ghosh, S. Srivastava, P. D’Silva and P. S. Mukherjee, J. Am. Chem. Soc., 2015, 137, 11916–11919 CrossRef CAS PubMed.
- P. Mal, B. Breiner, K. Rissanen and J. R. Nitschke, Science, 2009, 324, 1697–1699 CrossRef CAS PubMed.
- D. Fiedler, R. G. Bergman and K. N. Raymond, Angew. Chem., Int. Ed., 2006, 45, 745–748 CrossRef CAS PubMed.
- M. M. Safont-Sempere, G. Fernández and F. Würthner, Chem. Rev., 2011, 111, 5784–5814 CrossRef CAS PubMed.
- M. Lal Saha and M. Schmittel, Org. Biomol. Chem., 2012, 10, 4651–4684 CAS.
- Z. He, W. Jiang and C. A. Schalley, Chem. Soc. Rev., 2015, 44, 779–789 RSC.
- S. Mukherjee and P. S. Mukherjee, Chem. Commun., 2014, 50, 2239–2248 RSC.
- Y.-R. Zheng, Z. Zhao, M. Wang, K. Ghosh, J. B. Pollock, T. R. Cook and P. J. Stang, J. Am. Chem. Soc., 2010, 132, 16873–16882 CrossRef CAS PubMed.
- Y.-F. Han, W.-G. Jia, W.-B. Yu and G.-X. Jin, Chem. Soc. Rev., 2009, 38, 3419–3434 RSC.
- L. Zhang, Y.-J. Lin, Z.-H. Li and G.-X. Jin, J. Am. Chem. Soc., 2015, 137, 13670–13678 CrossRef CAS PubMed.
- D. Samanta, S. Shanmugaraju, S. A. Joshi, Y. P. Patil, M. Nethaji and P. S. Mukherjee, Chem. Commun., 2012, 48, 2298–2300 RSC.
- P. Howlader, P. Das, E. Zangrando and P. S. Mukherjee, J. Am. Chem. Soc., 2016, 138, 1668–1676 CrossRef CAS PubMed.
- D. Samanta and P. S. Mukherjee, Chem. Commun., 2014, 50, 1595–1598 RSC.
- M. Yoshizawa, M. Nagao, K. Kumazawa and M. Fujita, J. Organomet. Chem., 2005, 690, 5383–5388 CrossRef CAS.
- V. Maurizot, M. Yoshizawa, M. Kawano and M. Fujita, Dalton Trans., 2006, 2750–2756 RSC.
- M. Yamanaka, Y. Yamada, Y. Sei, K. Yamaguchi and K. Kobayashi, J. Am. Chem. Soc., 2006, 128, 1531–1539 CrossRef CAS PubMed.
- K. Kobayashi, Y. Yamada, M. Yamanaka, Y. Sei and K. Yamaguchi, J. Am. Chem. Soc., 2004, 126, 13896–13897 CrossRef CAS PubMed.
- We note that fac-capped octahedral metal centres offer three coordination sites and can be used for supramolecular cage assembly. See examples in ref. 1–3 and: S. Roche, C. Haslam, S. L. Heath and J. A. Thomas, Chem. Commun., 1998, 1681–1682 RSC.
- H. Li, Z.-J. Yao, D. Liu and G.-X. Jin, Coord. Chem. Rev., 2015, 293–294, 139–157 CrossRef CAS.
- Y.-Y. Zhang, W.-X. Gao, L. Lin and G.-X. Jin, Coord. Chem. Rev., 2017, 344, 323–344 CrossRef CAS.
- J. K. Klosterman, Y. Yamauchi and M. Fujita, Chem. Soc. Rev., 2009, 38, 1714–1725 RSC.
- D. Beaudoin, F. Rominger and M. Mastalerz, Angew. Chem., Int. Ed., 2017, 56, 1244–1248 CrossRef CAS PubMed.
- S. Klotzbach and F. Beuerle, Angew. Chem., Int. Ed., 2015, 54, 10356–10360 CrossRef CAS PubMed.
- B. Icli, E. Sheepwash, T. Riis-Johannessen, K. Schenk, Y. Filinchuk, R. Scopelliti and K. Severin, Chem. Sci., 2011, 2, 1719–1721 RSC.
- L. A. Wessjohann, O. Kreye and D. G. Rivera, Angew. Chem., Int. Ed., 2017, 56, 3501–3505 CrossRef CAS PubMed.
- T. K. Ronson, S. Zarra, S. P. Black and J. R. Nitschke, Chem. Commun., 2013, 49, 2476–2490 RSC.
- T. K. Ronson, D. A. Roberts, S. P. Black and J. R. Nitschke, J. Am. Chem. Soc., 2015, 137, 14502–14512 CrossRef CAS PubMed.
- A. M. Johnson and R. J. Hooley, Inorg. Chem., 2011, 50, 4671–4673 CrossRef CAS PubMed.
- M. Albrecht, M. Schneider and H. Röttele, Angew. Chem., Int. Ed., 1999, 38, 557–559 CrossRef CAS.
- O. Shyshov, R.-C. Brachvogel, T. Bachmann, R. Srikantharajah, D. Segets, F. Hampel, R. Puchta and M. von Delius, Angew. Chem., Int. Ed., 2017, 56, 776–781 CrossRef CAS PubMed.
- M. Yamashina, T. Yuki, Y. Sei, M. Akita and M. Yoshizawa, Chem. – Eur. J., 2015, 21, 4200–4204 CrossRef CAS PubMed.
- N. Kishi, Z. Li, K. Yoza, M. Akita and M. Yoshizawa, J. Am. Chem. Soc., 2011, 133, 11438–11441 CrossRef CAS PubMed.
- S. R. Seidel and P. J. Stang, Acc. Chem. Res., 2002, 35, 972–983 CrossRef CAS PubMed.
- B. H. Northrop, Y.-R. Zheng, K.-W. Chi and P. J. Stang, Acc. Chem. Res., 2009, 42, 1554–1563 CrossRef CAS PubMed.
- J.-R. Li and H.-C. Zhou, Nat. Chem., 2010, 2, 893–898 CrossRef CAS PubMed.
- Q.-F. Sun, J. Iwasa, D. Ogawa, Y. Ishido, S. Sato, T. Ozeki, Y. Sei, K. Yamaguchi and M. Fujita, Science, 2010, 328, 1144 LP–1147 CrossRef PubMed.
- Q.-F. Sun, S. Sato and M. Fujita, Angew. Chem., Int. Ed., 2014, 53, 13510–13513 CrossRef CAS PubMed.
- W. M. Bloch, Y. Abe, J. J. Holstein, C. M. Wandtke, B. Dittrich and G. H. Clever, J. Am. Chem. Soc., 2016, 138, 13750–13755 CrossRef CAS PubMed.
- G. H. Clever, W. Kawamura and M. Shionoya, Inorg. Chem., 2011, 50, 4689–4691 CrossRef CAS PubMed.
- R. Zhu, J. Lübben, B. Dittrich and G. H. Clever, Angew. Chem., Int. Ed., 2015, 54, 2796–2800 CrossRef CAS PubMed.
- W. M. Bloch, J. J. Holstein, W. Hiller and G. H. Clever, Angew. Chem., Int. Ed., 2017 DOI:10.1002/anie.201702573R1.
- S. P. Black, D. M. Wood, F. B. Schwarz, T. K. Ronson, J. J. Holstein, A. R. Stefankiewicz, C. A. Schalley, J. K. M. Sanders and J. R. Nitschke, Chem. Sci., 2016, 7, 2614–2620 RSC.
- S. De, K. Mahata and M. Schmittel, Chem. Soc. Rev., 2010, 39, 1555–1575 RSC.
- M. L. Saha, S. Neogi and M. Schmittel, Dalton Trans., 2014, 43, 3815–3834 RSC.
- P. Baxter, J.-M. Lehn, A. DeCian and J. Fischer, Angew. Chem., Int. Ed., 1993, 32, 69–72 CrossRef.
- P. N. W. Baxter, J.-M. Lehn, G. Baum and D. Fenske, Chem. – Eur. J., 1999, 5, 102–112 CrossRef CAS.
- M. Schmittel, M. L. Saha and J. Fan, Org. Lett., 2011, 13, 3916–3919 CrossRef CAS PubMed.
- D. Preston, J. E. Barnsley, K. C. Gordon and J. D. Crowley, J. Am. Chem. Soc., 2016, 138, 10578–10585 CrossRef CAS PubMed.
- D. Preston, S. M. McNeill, J. E. M. Lewis, G. I. Giles and J. D. Crowley, Dalton Trans., 2016, 45, 8050–8060 RSC.
- M. Frank, L. Krause, R. Herbst-Irmer, D. Stalke and G. H. Clever, Dalton Trans., 2014, 43, 4587–4592 RSC.
- M. Frank, J. Ahrens, I. Bejenke, M. Krick, D. Schwarzer and G. H. Clever, J. Am. Chem. Soc., 2016, 138, 8279–8287 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |