
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Novel chemical probes for the investigation of nonribosomal peptide assembly†
Y. T. Candace
Ho
a,
Daniel J.
Leng
a,
Francesca
Ghiringhelli
ab,
Ina
Wilkening
a,
Dexter P.
Bushell
a,
Otto
Köstner
ac,
Elena
Riva
a,
Judith
Havemann
a,
Daniele
Passarella
 b and
Manuela
Tosin
*a
b and
Manuela
Tosin
*a
aDepartment of Chemistry, University of Warwick, Library Road, CV4 7AL, UK. E-mail: M.Tosin@warwick.ac.uk; Tel: +442476572878
bDepartment of Chemistry, Universita' degli Studi di Milano, Via Golgi, 19 20133 Milano, Italy
cInstitut für Organische Chemie, Universität Wien, Währinger Str., 38 1090 Wien, Austria
First published on 19th June 2017
Abstract
Chemical probes were devised and evaluated for the capture of biosynthetic intermediates involved in the bio-assembly of the nonribosomal peptide echinomycin. Putative intermediate peptide species were isolated and characterised, providing fresh insights into pathway substrate flexibility and paving the way for novel chemoenzymatic approaches towards unnatural peptides.
Peptidic molecules are the most abundant and versatile chemical entities in nature: from proteins to small peptides, they display countless architectures and exert key roles in almost every known biological process. In the context of secondary metabolism, peptide natural products comprise a vast array of bioactive molecules that regulate interspecies communication and organism survival. Peptide natural products can be either biosynthesised by the ribosome (and hence known as ribosomal peptides, RiPPs) or by the multifunctional enzymes nonribosomal peptide synthetases (NRPSs).1
Amongst NRPs we encounter potent anticancer agents, such as bleomycin, and antibiotics of last resort, such as vancomycin and teicoplanin.2 In NRP biosynthesis aminoacyl units, anchored as thioesters to peptidyl carrier proteins (PCPs) via the phosphopantetheine cofactor,3 are joined together through peptide bond formation by condensation (C) domains (Fig. 1A). Similarly to polyketide and fatty acid synthases (PKSs and FASs, respectively), NRPSs generate growing enzyme-bound biosynthetic intermediates which are variably processed and ultimately converted to the final products. The ability of NRPSs to process proteinogenic and non-proteinogenic amino acids, fatty acids and α-hydroxy acids, linking them in different ways, give rise to astonishing diversity in product structure and bioactivity. Key domains utilised in peptide elaboration throughout assembly include methyltransferases, epimerases4 and heterocyclases, whereas tailoring enzymes acting in post-NRPS processes comprise oxidases,5 halogenases6 and glycosyltransferases.7
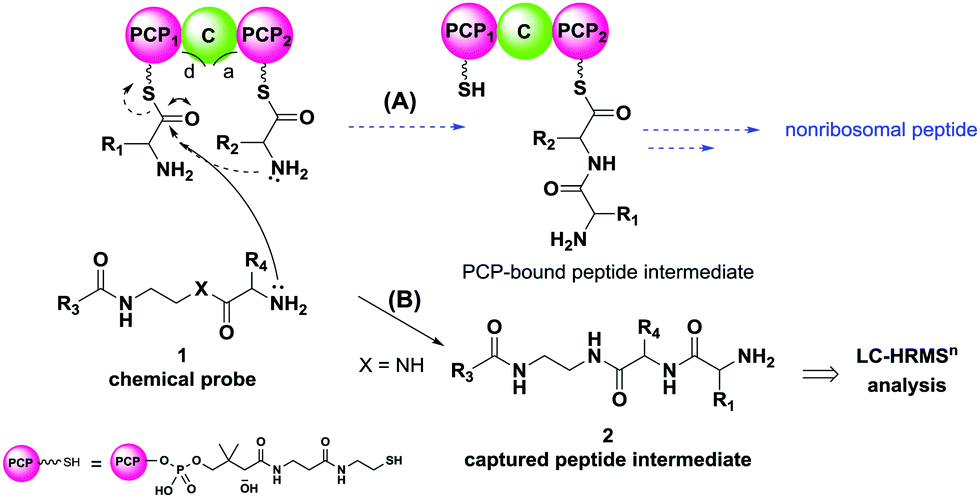 | ||
| Fig. 1 (A) General mechanism of nonribosomal peptide assembly; (B) proposed capture of peptide biosynthetic intermediates (2) via newly devised chain termination probes (1) based on nonhydrolysable analogues (X = NH) of PCP-bound amino acids. PCP = peptidyl carrier protein; C = condensation domain; d = aminoacyl donor site; a = aminoacyl acceptor site. R3 = variable alkyl moiety; R4 = variable amino acid side chain. | ||
In NRPS assembly, adenylation (A) domains act as ‘gate-keepers’ by selecting specific amino acids and activating them as adenosine monophosphate (AMP) esters for their loading onto the phosphopantetheine cofactors of PCPs.8 C domains catalyse peptide bond formation and present two distinct substrate-binding sites: a ‘donor site’ for upstream PCP-bound amino acids, and an ‘acceptor site’ for downstream PCP-bound amino acids; the free amino groups of the latter act as nucleophiles towards the thioester moiety of PCP-bound upstream substrates to generate extended PCP-bound intermediates (Fig. 1A), which are subsequently transferred to other sites for further extension and elaboration.9 Eventually peptide chain assembly is terminated, mostly by thioesterase (TE) domains promoting peptide hydrolysis or cyclisation;10 additional mechanisms of peptide chain termination and release include reduction of the final thioester bond to an aldehyde or an alcohol by R domains.11 As PKSs and FASs, NRPSs can be modular or iterative enzymes: the former comprise multiple sets of domains (modules), with each module catalysing at least one round of peptide chain extension and a direct correspondence between protein predicted function and product in most cases.12 Conversely, iterative synthases are constituted by modules that are used more than once to catalyse peptide chain growth and processing,13 hence the nature of their products is not fully predictable.
The engineering of NRPS enzymes and pathways remains a major synthetic biology focus in view of novel peptide production.14–17 Despite the much-improved ability to engineer NRPSs, many details on peptide assembly catalysed by these enzymes remain unknown, limiting our ability to fully exploit existing NRPSs and to design de novo enzymes of improved versatility and efficiency.
Over the years mechanistic insights on NRP assembly have been gathered from isotopic labelling of aminoacyl building blocks,18 genetic manipulation of in vivo biosynthetic pathways (e.g. by domain inactivation, deletion, etc.),19 NRPS activity reconstitution in vitro,11a,20 mass spectrometry of metabolites and enzyme-bound precursors,20c,21 and enzyme conformational/structural elucidation.22 Nonetheless we still lack a comprehensive and dynamic picture of how NRPS machineries work in vivo as a whole in processing substrates and successfully conveying intermediates to the end of a biosynthetic process. A major hurdle in gathering this information is given by the covalent tethering of NRP intermediates to their biosynthetic enzymes throughout peptide assembly.
In the past few years our group has developed ‘chain termination’ probes for the investigation of polyketide biosynthesis: nonhydrolysable small molecule mimics of malonate building blocks recruited in polyketide formation were devised to interfere in the key decarboxylative Claisen condensation step leading to polyketide chain assembly, thereby ‘capturing’ transient biosynthetic intermediates for characterisation.23–26 These tools have proved successful for intermediate isolation and characterisation of intermediate species from modular23b,24,26 and iterative PKS enzymes,23a,25 gathering in vitro and in vivo key information on the timing and the mechanism of catalytic events otherwise inaccessible, and unveiling novel opportunities for natural product diversification.24c
Given the similarity between PKSs and NRPSs in processing carrier protein-bound species, we reasoned that chemical probes mimicking PCP-bound amino acids could be developed to interfere in NRPS-catalysed peptide bond formation and capture intermediate peptide species for further characterisation. Hence we prepared a library of putative probes (3–17, Table 1) for the investigation of echinomycin assembly in the soil bacterium S. lasaliensis. Echinomycin (18, Fig. 2) is an antitumor antibiotic that acts as DNA bis-intercalator due to its two-fold symmetry and quinoxaline-derived moieties. It is biosynthesised by an iterative NRPS that assembles two identical peptide chains from 2-quinoxaline carboxylic acid, L-serine, alanine, cysteine and valine (Fig. 2): a terminal thioesterase domain catalyses depsipeptide formation and lactonisation, followed post-NRPS oxidative processing.27
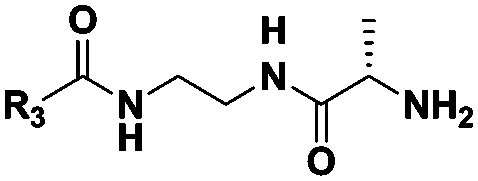
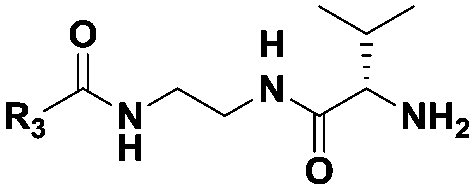
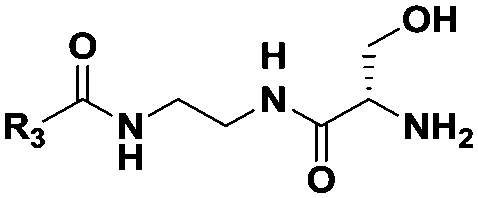
| Probe structure | R3 | Compound number | Captured species |
|---|---|---|---|
| a Major species. b Detected in minor amounts (see ESI). c Displaying cytotoxicity above 1 mM. | |||
|
CH3 | 3 | n.d. |
| (CH2)2CH3 | 4 | Dipeptideb | |
| (CH2)5CH3 | 5 | Di-, tri-, tetrapeptideb | |
| (CH2)8CH3 | 6 | n.d. | |
|
(CH2)2CH3 | 7 | Di-, tripeptideb |
| (CH2)5CH3 | 8 | Di-, tri-, tetrapeptideb | |
|
(CH2)5CH3 | 9 | Di-,a tripeptideb |
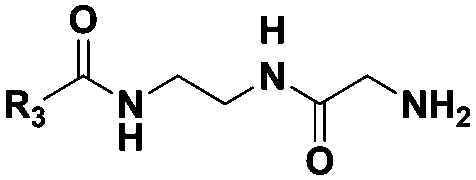
|
(CH2)2CH3 | 10 | Dipeptidea, tri-, tetra-, pentapeptidesb |
| (CH2)5CH3 | 11 | Dipeptidea, tri-, tetrapeptidesb | |
| (CH2)8CH3 | 12 | n.d. | |
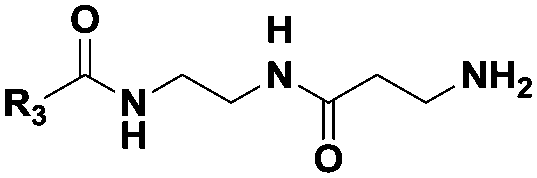
|
(CH2)2CH3 | 13 | Dipeptidea, tri-, tetrapeptideb |
| (CH2)5CH3 | 14 | Dipeptidea, tripeptideb | |
| (CH2)8CH3 | 15 | n.d. | |
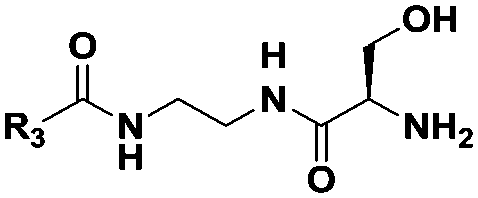
|
(CH2)5CH3 | 16 | Dipeptideb |
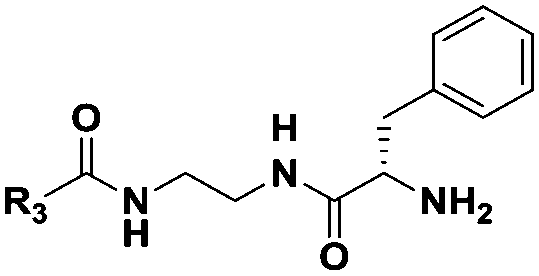
|
(CH2)5CH3 | 17 | n.d. |
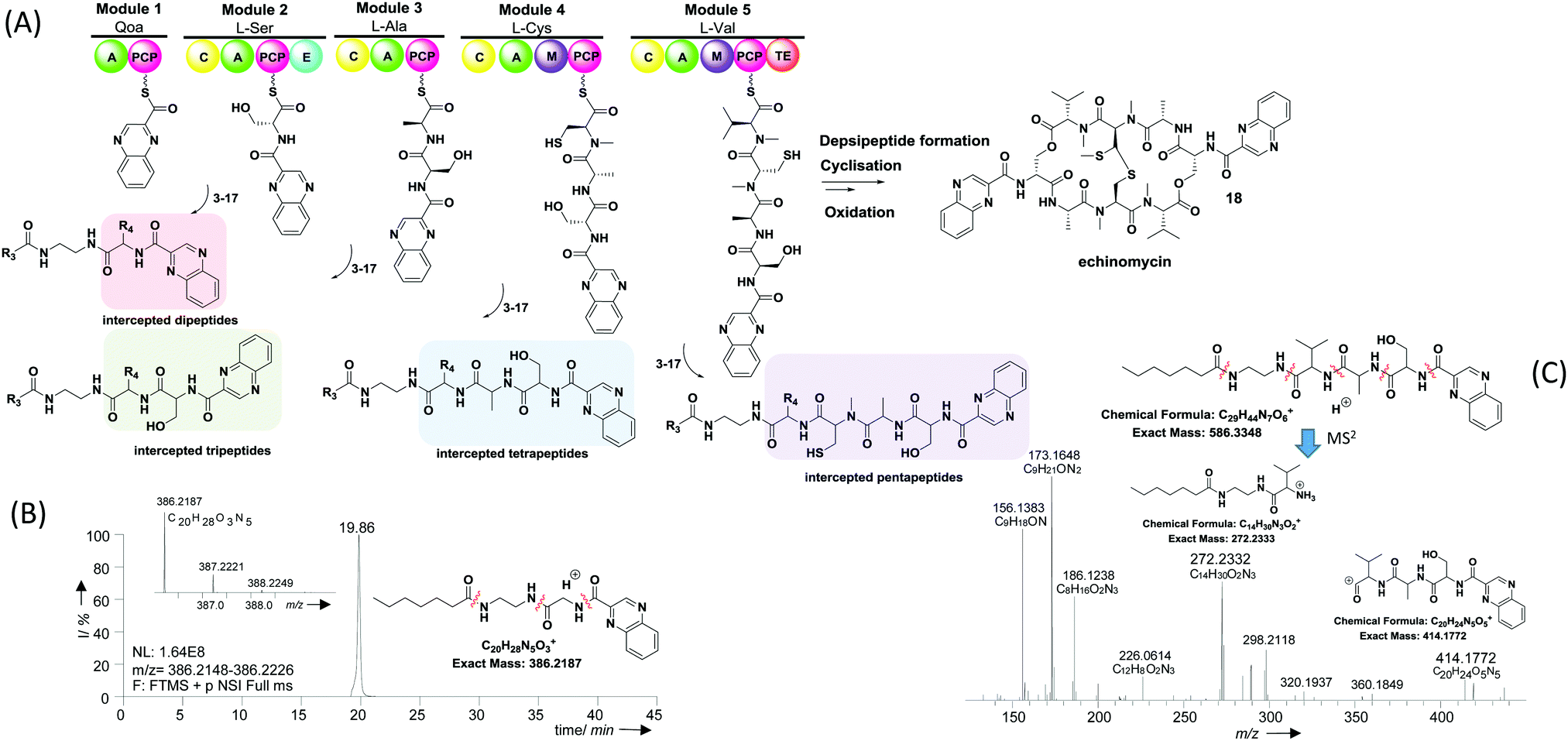 | ||
| Fig. 2 (A) Overview of echinomycin nonribosomal peptide assembly and in vivo capture of putative biosynthetic intermediates via chemical probes 3–17 (note: the general intermediate structures apply to all substrates with the exception of β-alanine-based probes 13–15, ESI†); (B) extracted ion chromatogram of a dipeptide resulting from the capture of quinoxaline 2-carboxylic acid by probe 11 (observed MS2 fragments indicated, ESI†); (C) HR-MS2 analyses of a tetrapeptide intermediate resulting from the capture of an enzyme-bound tripeptide by probe 8. The stereochemistry of captured intermediates is yet to be established. | ||
The probes devised by us were N-acyl cysteamine derivatives28 that mimic PCP-bound aminoacyl moieties and feature nonhydrolysable amide bonds in place of cleavable thioester bonds (1, X = NH, Fig. 1) in order to prevent substrate re-loading onto NRPSs. We reasoned that, once within NRPS active sites, these molecules should be recognised as acceptor substrates by C domains and hence compete with PCP-bound aminoacyl units for peptide bond formation, ultimately resulting in the off-loading and capture of prematurely truncated peptides (2, Fig. 1B).
We initially prepared N-acetyl substrates based on cognate aminoacyl moieties such as the alanine derivative 3 (Table 1) via standard peptide synthesis procedures (ESI†). These molecules were administered in variable concentrations to liquid and solid cultures of S. lasaliensis ACP12 (S970A), an engineered strain of S. lasaliensis NRRL 3382R incapable of producing the polyketide lasalocid A24a but still capable of generating the nonribosomal peptide echinomycin in relevant levels. Preliminary LC-HRMSn analysis of organic extracts of microbial fermentations showed that the probes were present and hydrolytically stable but no evidence of feasible intermediates (data not shown). We reasoned that perhaps the hydrophilicity of peptide intermediates could hamper their isolation via organic extraction.
Therefore we prepared probes of variable N-acyl chain length (R3, Fig. 1 and Table 1) in order to increase their hydrophobicity and cellular uptake. Besides, variable side chain moieties (variable R4, Fig. 1) and amino acid scaffold motifs (e.g. α versus β-amino acids, Table 1) were also included in the probe structure in order to assess the substrate flexibility of the echinomycin NRPS machinery in vivo.
An overview of echinomycin putative intermediates captured from S. lasaliensis ACP12 (S970A) via the newly devised chemical probes is given in Table 1 and Fig. 2. A whole range of captured species spanning from dipeptides to pentapeptides (whose putative structures are represented in Fig. 2) were isolated and characterised by HR-MSn: these were identified as putative echinomycin intermediates by MSn fragment peaks (obtained from amide cleavages) featuring quinoxaline 2-carboxylic acid and other amino acid constituents of echinomycin in the expected sequence/order (as shown in Fig. 2C and in the ESI†). The putative species were absent in control samples and substantially varied in amount and distribution according to the probe utilised (ESI†).
Besides the expected species captured from probes based on cognate substrates, additional species deriving from non-cognate pseudo-substrates were detected (Table 1, Fig. 2 and the ESI†). For instance, dipeptides allegedly deriving from the off-loading of the starter quinoxaline 2-carboxylic acid were detected in significant amounts from experiments utilising non-cognate glycine (Fig. 2B) and β-alanine probes (Fig. 30S, ESI†) as well as the cognate serine substrates (Fig. 17S, ESI†). Further advanced species (from tri- to pentapeptides) were most efficiently detected and characterised in extracts deriving from bacterial fermentations in the presence of N-butyroyl and N-heptanoyl glycine (Fig. 21S–23S, 26S and 27S, ESI†), alanine (Fig. 8S, ESI†), β-alanine (Fig. 31S and 32S, ESI†), and valine probes (Fig. 2C). No intermediate species were captured utilising the aromatic L-phenylalanine pseudo-substrate 17.
Aminoacyl N-acetylcysteamine (SNAc) thioesters28 have been often utilised to reconstitute the activity of C domains in vitro and have shown that C acceptor sites generally exhibit strong stereoselectivity (L- versusD-), together with some selectivity towards the side chain of amino acids. Variants of nonribosomal peptides resulting from the incorporation of different amino acids can be observed in vivo,9b and this has been utilised for precursor-directed biosynthesis purposes.14 However, to the best of our knowledge, the current study constitutes the first report of in vivo probing of nonribosomal peptide assembly utilising aminoacyl N-acetylcysteamine substrate mimics.
The overall results gathered by us seem to indicate that: (1) the echinomycin biosynthetic machinery possesses some flexibility towards the processing of ‘unnatural’ substrates in the correspondence of specific C domains, possibly due to flexible pseudo-substrate positioning at the enzyme active site during peptide bond formation9d and/or probe bioavailability in vivo: this seems particularly true for Gly and β-alanine substrates, which lack side-chain stereochemistry and steric hindrance; (2) dipeptides accumulate preferentially in comparison to more advanced intermediate species (see ESI† figures): this suggests that the first condensation step might be the slowest amongst all those taking place throughout echinomycin peptide chain assembly.
A more in-depth assessment and dissection of these in vivo findings will require separate in vitro experiments with recombinant C domains, as well as the development of advanced analytical tools29 capable of deconvoluting the acquired LC-MS data in a quantitative fashion. Nonetheless the preliminary experiments herein reported demonstrate that the in vivo profiling of NRP assembly via chain termination probes is now possible, with important implications for future biosynthetic pathway engineering. The screening of natural product bio-assembly can indeed provide not only preliminary information on substrate recognition but also insights on the kinetics of natural product assembly,26 constituting the rational for devising novel chemoenzymatic approaches towards unnatural peptide production.
In summary, we have herein gathered a first direct view of substrate processing for an iterative NRPS in vivo through the use of newly devised nonhydrolysable ‘chain termination’ probes. Further applications of these tools for the investigation of nonribosomal peptide pathways will be reported in due course.
We gratefully acknowledge BBSRC (project grant BB/J007250/1 to M. T. and MIBTP studentship to D. J. L.); the Erasmus programme (exchange bursaries to F. G. and O. K.); FP7 (Marie Curie Intraeuropean Fellowship to I. W.); the Institute of Advanced Studies at Warwick (Postdoctoral Fellowship to E. R.); Dr Cleidiane Zampronio (School of Life Sciences, Warwick) for assistance with LC-HRMSn Orbitrap Fusion analyses; Dr Lijiang Song for preliminary MS data acquired on a Bruker MaXis Impact instrument; Prof. Peter F. Leadlay (Cambridge) for the kind gift of S. lasaliensis ACP12 (S970A); and COST Action CM1407 for networking funding and opportunities.
Notes and references
- (a) M. A. Marahiel and L. O. Essen, Methods Enzymol., 2009, 458, 337–351 CAS; (b) G. H. Hur, C. R. Vickery and M. D. Burkart, Nat. Prod. Rep., 2012, 29, 1074–1098 RSC.
- E. A. Felnagle, E. E. Jackson, Y. A. Chan, A. M. Podevels, A. D. Berti, M. D. McMahon and M. G. Thomas, Mol. Pharmaceutics, 2008, 5, 191–211 CrossRef CAS PubMed.
- (a) F. Rusnak, M. Sakaitani, D. Drueckhammer, J. Reichert and C. T. Walsh, Biochemistry, 1991, 30, 2916–2927 CrossRef CAS PubMed; (b) M. A. Fischbach and C. T. Walsh, Chem. Rev., 2006, 106, 3468–3496 CrossRef CAS PubMed.
- (a) L. Luo, R. M. Kohli, M. Onishi, U. Linne, M. A. Marahiel and C. T. Walsh, Biochemistry, 2002, 41, 9184–9196 CrossRef CAS PubMed; (b) S. A. Samel, P. Czodrowski and L.-O. Essen, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2014, D70, 1442–1452 Search PubMed; (c) W.-H. Chen, K. Li, N. S. Guntaka and S. D. Bruner, ACS Chem. Biol., 2016, 11, 2293–2303 CrossRef CAS PubMed.
- M. Peschke, K. Haslinger, C. Brieke, J. Reinstein and M. J. Cryle, J. Am. Chem. Soc., 2016, 138, 6746–6753 CrossRef CAS PubMed.
- B. Bister, D. Bischoff, G. J. Nicholson, S. Stockert, J. Wink, C. Brunati, S. Donadio, S. Pelzer, W. Wohlleben and R. D. Süssmuth, ChemBioChem, 2003, 7, 658–662 CrossRef PubMed.
- T. L. Li, F. Huang, S. F. Haydock, T. Mironenko, P. F. Leadlay and J. B. Spencer, Chem. Biol., 2004, 11, 107–119 CAS.
- T. Stachelhaus, H. D. Mootz and M. A. Marahiel, Chem. Biol., 1999, 6, 493–505 CrossRef CAS PubMed.
- (a) T. A. Keating, C. G. Marshall, C. T. Walsh and A. E. Keating, Nat. Struct. Biol., 2002, 9, 522–526 CAS; (b) S. Lautru and G. L. Challis, Microbiology, 2004, 150, 1629–1636 CrossRef CAS PubMed; (c) K. Bloudoff, D. Rodionov and T. M. Schmeing, J. Mol. Biol., 2013, 425, 3137–3150 CrossRef CAS PubMed; (d) K. Bloudoff, D. A. Alonzo and T. M. Schmeing, Cell Chem. Biol., 2016, 23, 331–339 CrossRef CAS PubMed.
- (a) S. D. Bruner, T. Weber, R. M. Kohli, D. Schwarzer, M. A. Marahiel, C. T. Walsh and M. T. Stubbs, Structure, 2002, 10, 301–310 CrossRef CAS PubMed; (b) M. E. Horsman, T. P. A. Hari and C. N. Boddy, Nat. Prod. Rep., 2016, 33, 183–202 RSC.
- (a) N. Gaitatzis, B. Kunze and R. Müller, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 11136–11141 CrossRef CAS PubMed; (b) A. Chhabra, A. S. Haque, R. K. Pal, A. Goyal, R. Rai, S. Joshi, S. Panjikar, S. Pasha, R. Sankaranarayanan and R. S. Gokhalea, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 5681–5686 CrossRef CAS PubMed.
- S. C. Wenzel, P. Meiser, T. M. Binz, T. Mahmud and R. Müller, Angew. Chem., Int. Ed., 2006, 45, 2296–2301 CrossRef CAS PubMed.
- M. Juguet, S. Lautru, F.-X. Francou, Š. Nezbedová, P. Leblond, M. Gondry and J.-L. Pernodet, Chem. Biol., 2009, 16, 421–431 CrossRef CAS PubMed.
- M. Winn, J. K. Fyans, Y. Zhuo and J. Micklefield, Nat. Prod. Rep., 2016, 33, 317–347 RSC.
- (a) C. Milne, A. Powell, J. Jim, M. Al Nakeeb, C. P. Smith and J. Micklefield, J. Am. Chem. Soc., 2006, 128, 11250–11259 CrossRef CAS PubMed; (b) H. Kries, R. Wachtel, A. Pabst, B. Wanner, D. Niquille and D. Hilvert, Angew. Chem., Int. Ed., 2014, 53, 10105–10108 CrossRef CAS PubMed; (c) M. Crüsemann, C. Kohlhaasa and J. Piel, Chem. Sci., 2013, 4, 1041–1045 RSC.
- (a) A. D. Roy, S. Grüschow, N. Cairns and R. J. M. Goss, J. Am. Chem. Soc., 2010, 132, 12243–12245 CrossRef CAS PubMed; (b) R. A. Lewis, L. Nunns, J. Thirlway, K. Carroll, C. P. Smith and J. Micklefield, Chem. Commun., 2011, 47, 1860–1862 RSC.
- (a) R. H. Baltz, P. Brian, V. Miao and S. K. Wrigley, J. Ind. Microbiol. Biotechnol., 2006, 33, 66–74 CrossRef CAS PubMed; (b) K. T. Nguyen, D. Ritz, J. Q. Gu, D. Alexander, M. Chu, V. Miao, P. Brian and R. H. Baltz, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 17462–17467 CrossRef CAS PubMed; (c) H. Kries, D. L. Niquille and D. Hilvert, Chem. Biol., 2015, 22, 640–648 CrossRef CAS PubMed.
- (a) H. B. Bode, D. Reimer, S. W. Fuchs, F. Kirchner, C. Dauth, C. Kegler, W. Lorenzen, A. O. Brachmann and P. Grün, Chem. – Eur. J., 2012, 18, 2342–2348 CrossRef CAS PubMed; (b) H. B. Bode, A. O. Brachmann, K. B. Jadhav, L. Seyfarth, C. Dauth, S. W. Fuchs, M. Kaiser, N. R. Waterfield, H. Sack, S. H. Heinemann and H.-D. Arndt, Angew. Chem., Int. Ed., 2015, 54, 10352–10355 CrossRef CAS PubMed.
- (a) H. D. Mootz, N. Kessler, U. Linne, K. Eppelmann, D. Schwarzer and M. A. Marahiel, J. Am. Chem. Soc., 2002, 124, 10980–10981 CrossRef CAS PubMed; (b) D. Butz, T. Schmiederer, B. Hadatsch, W. Wohlleben, T. Weber and R. D. Süssmuth, ChemBioChem, 2008, 9, 1195–1200 CrossRef CAS PubMed.
- (a) K. Qiao, H. Zhou, W. Xu, W. Zhang, N. Garg and Y. Tang, Org. Lett., 2011, 13, 1758–1761 CrossRef CAS PubMed; (b) C. Zhang, L. Kong, Q. Liu, X. Lei, T. Zhu, J. Yin, B. Lin, Z. Deng and D. You, PLoS One, 2013, 8, e56772 CAS; (c) A. W. Goering, J. Li, R. A. McClure, R. J. Thomson, M. C. Jewett and N. L. Kelleher, ACS Synth. Biol., 2017, 6, 39–44 CrossRef CAS PubMed.
- (a) L. M. Hicks, M. T. Mazur, L. M. Miller, P. C. Dorrestein, N. A. Schnarr, C. Khosla and N. L. Kelleher, ChemBioChem, 2006, 7, 904–907 CrossRef CAS PubMed; (b) D. Meluzzi, W. H. Zheng, M. Hensler, V. Nizet and P. C. Dorrestein, Bioorg. Med. Chem. Lett., 2008, 18, 3107–3111 CrossRef CAS PubMed.
- (a) T. Kittilä, A. Mollo, L. K. Charkoudian and M. J. Cryle, Angew. Chem., Int. Ed., 2016, 55, 9834–9840 CrossRef PubMed; (b) E. J. Drake, B. R. Miller, C. Shi, J. T. Tarrasch, J. A. Sundlov, C. L. Allen, G. Skiniotis, C. C. Aldrick and A. M. Gulick, Nature, 2016, 529, 235–238 CrossRef CAS PubMed; (c) K. Haslinger, M. Peschke, C. Brieke, E. Maximowitsch and M. J. Cryle, Nature, 2015, 521, 105–109 CrossRef CAS PubMed.
- (a) M. Tosin, D. Spiteller and J. B. Spencer, ChemBioChem, 2009, 10, 1714–1723 CrossRef CAS PubMed; (b) M. Tosin, L. Betancor, E. Stephens, W. M. A. Li, J. B. Spencer and P. F. Leadlay, ChemBioChem, 2010, 11, 539–546 CrossRef CAS PubMed.
- (a) M. Tosin, Y. Demydchuk, J. S. Parascandolo, C. Blasco-Per, F. J. Leeper and P. F. Leadlay, Chem. Commun., 2011, 47, 3460–3462 RSC; (b) M. Tosin, L. Smith and P. F. Leadlay, Angew. Chem., Int. Ed., 2011, 50, 11930–11933 CrossRef CAS PubMed; (c) E. Riva, I. Wilkening, S. Gazzola, W. M. A. Li, L. Smith, P. F. Leadlay and M. Tosin, Angew. Chem., Int. Ed., 2014, 53, 11944–11949 CrossRef CAS PubMed.
- (a) J. S. Parascandolo, J. Havemann, H. K. Potter, F. Huang, E. Riva, J. Connolly, I. Wilkening, L. Song, P. F. Leadlay and M. Tosin, Angew. Chem., Int. Ed., 2016, 55, 3463–3467 CrossRef CAS PubMed; (b) H. Kage, E. Riva, J. S. Parascandolo, M. F. Kreutzer, M. Tosin and M. Nett, Org. Biomol. Chem., 2015, 13, 11414–11417 RSC; (c) J. Havemann, M. E. Yurkovich, R. Jenkins, S. Harringer, W. Tao, S. Wen, Y. Sun, P. F. Leadlay and M. Tosin, Chem. Commun., 2017, 53, 1912–1915 RSC; (d) M. E. Yurkovich, R. Jenkins, Y. Sun, M. Tosin and P. F. Leadlay, Chem. Commun., 2017, 53, 2182–2185 RSC.
- I. Wilkening, S. Gazzola, E. Riva, J. S. Parascandolo, L. Song and M. Tosin, Chem. Commun., 2016, 52, 10392–10395 RSC.
- (a) M. Sato, T. Nakazawa, Y. Tsunematsu, K. Hotta and K. Watanabe, Curr. Opin. Chem. Biol., 2013, 17, 537–545 CrossRef CAS PubMed; (b) K. Watanabe, K. Hotta, A. P. Praseuth, K. Koketsu, A. Migita, C. N. Boddy, C. C. C. Wang, H. Oguri and H. Oikawa, Nat. Chem. Biol., 2006, 2, 423–428 CrossRef CAS PubMed.
- D. E. Ehmann, J. W. Trauger, T. Stachelhaus and C. T. Walsh, Chem. Biol., 2000, 7, 765–772 CrossRef CAS PubMed.
- R. H. Wills, M. Tosin and P. B. O'Connor, Anal. Chem., 2012, 84, 8863–8870 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: General methods for the synthesis of chemical probes and LC-HRMSn analysis of the biosynthetic intermediates isolated from S. lasaliensis strains. See DOI: 10.1039/c7cc02427d |
| This journal is © The Royal Society of Chemistry 2017 |