
A quantitative proteomic approach for unveiling novel mechanisms associated with MeHg-induced toxicity: effects on the methylation cycle
Pablo
Cabezas-Sanchez
,
Estefania
Garcia-Calvo
,
Carmen
Camara
and
Jose L.
Luque-Garcia
*
Department of Analytical Chemistry, Faculty of Chemistry, Complutense University of Madrid, 28040, Madrid, Spain. E-mail: lluque@quim.ucm.es; Tel: +34913944318
First published on 13th November 2015
Abstract
Methylmercury (MeHg) is still a major threat for human health and the environment due to its extremely high toxicity that mainly affects the nervous system. Despite the great efforts made during the last few decades, the specific molecular mechanisms involved in MeHg-induced toxicity are still not completely unveiled. In this work we explored such mechanisms using neuroblastoma cells (Neuro-2a) and SILAC as a quantitative proteomic approach. We found that exposure of Neuro-2a cells to 2 mg L−1 MeHg for 8 h decreased the cell viability to 70% and caused significant changes in the morphology of the cells, specially regarding neurite development. Our proteomic results showed different proteins altered upon MeHg exposure that helped to identify pathways related to the toxicity exerted by MeHg. Specifically, we have found that MeHg affects the methylation cycle by inhibiting the expression of key enzymes including MTHFD1 and MTR. Moreover, we demonstrate that inhibition of MTHFD1 is not observed when exposing the cells to inorganic Hg and other heavy metals such as Pb or Cu. Thus, this work sets the stage for dissecting a specific molecular mechanism for MeHg-induced toxicity.
Introduction
Mercury is one of the most hazardous environmental pollutants representing a serious risk to human health. It can be easily methylated by widespread sulfate-reducing bacteria into methylmercury (MeHg), which easily penetrates into organisms through the lipid bilayer.1 As a result of this biomethylation, MeHg distributes among tissues, the most sensitive target organ being the central nervous system.2 It is also known that ingested mercury can interact with some proteins and enzymes causing organ dysfunction.3 Some neurotoxic effects such as mental retardation, dysmorphogenesis of the cerebellum, degeneration of granule cells, neurons or dorsal root ganglia have been observed in episodes of mercury poisoning.4 Extensive research has been conducted to determine the molecular mechanisms associated with MeHg uptake and neurotoxicity. It has been suggested that MeHg enters into neural cells as a cysteine complex via the L-type neutral amino acid carrier transport (LAT) system due to the similitude between this complex and L-methionine, a substrate for the amino acid transport system.5 It is also known that MeHg inhibits several mitochondrial enzymes such as glutathione peroxidase, glutathione reductase6 and the thioredoxin system,7 and induces apoptosis and necrosis in neural cells.8,9 However, despite all the efforts made over the last half century, identification of the particular biomolecular mechanisms underlying the observed neurotoxic effects of MeHg has not yet been proposed.10With the advent of biological mass spectrometry, novel proteomic strategies have been applied to investigate toxicity mechanisms.11 Specifically, quantitative proteomics allows for the identification of differentially expressed proteins between two or more different physiological conditions, thus contributing to the discovery of candidate toxicity-specific targets and molecular pathways. Stable isotopic labeling by amino acids in cell culture (SILAC) is one of the most widely used alternatives for relative protein quantitation due to its high accuracy and because it offers the possibility for the identification and quantitation of proteins within the same experiment.12,13 SILAC involves the addition of 12C- and 13C-labeled lysine and arginine to growth media of separately cultured cells, giving rise to cells containing “light” or “heavy” proteins, respectively.
In this work, we have used the SILAC approach as a discovery platform for the identification of 125 proteins affected by MeHg exposure. The combination of this quantitative proteomic strategy with additional bioanalytical assays has allowed for the identification of mechanisms related to MeHg toxicity and to set the stage for dissecting a specific molecular mechanism in which the methylation cycle blockage upon MeHg exposure is involved.
Materials and methods
Cell culture and cytotoxicity assay
Mouse neuroblastoma cells (Neuro-2a) were maintained in Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 100 units per ml of penicillin/streptomycin in 5% CO2 at 37 °C. 2 × 104 cells were plated onto 96-well plates and incubated with methylmercury (MeHg) at different exposure times. Cell viability was determined using the MTT assay. 20 μL of MTT (5 mg L−1) were added to each well and incubated for 5 h. After this time, the MTT solution was removed and 100 μL of dimethyl sulfoxide (DMSO) were added to dissolve the insoluble purple formazan product. The cell viability was quantified by measuring the absorbance at 595 nm in a microplate absorbance reader (Sunrise, Tecan). The same procedure was used for measuring the viability when exposing the cells to the combination MeHg–vitamin B12, and for cells transfected with siRNA against MTHFD1.Determination of MeHg uptake by Neuro-2a cells
After exposing 1 × 107 cells to 2 mg L−1 MeHg for 8 h, the exposure medium was removed and preserved, and the cells were thoroughly washed with PBS and collected. The samples were digested in a microwave oven with a mixture of HNO3 (65%) and H2O2 (35%). Digested samples were introduced into a flow injection system where mercury was reduced to atomic mercury vapor with 3% stannous chloride. Determination of total mercury in both the exposure media and the cells was carried out by using cold vapor atomic fluorescence spectroscopy (CV-AFS) (Merlin 10.023). All samples were analyzed in quadruplicate. Certificated reference materials from the Commission of the European Communities (CRM) were also analyzed to ensure the quality of the results. Recoveries from CRM-029 and CRM-710 were within 5% of the certificated values. Calculated limits of detection (LOD) and quantification (LOQ) were 6 ng L−1 and 23 ng L−1, respectively.Evaluation of morphological changes
Cells (2 × 106 cells) were seeded onto coverslips 48 h before MeHg exposure in order to reach 80% confluence. At this point, cell viability was checked and no significant decrease was observed. The cells were incubated with MeHg 2 mg L−1 for 8 h and then washed with ice-cold PBS, fixed with paraformaldehyde (4% v/v) for 15 min at room temperature and permeabilized with 5 mL of Triton X-100 (0.1% v/v, 40 mM glycine). The cells were then incubated with Alexa Fluor 488-labeled phalloidin (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :60) for 20 min at room temperature. Staining of nuclei was performed in a 1 mg mL−1 solution of 4′,6-diamino-2-phenylindole (DAPI) and finally, the coverslips were mounted with Mowiol® 4–88. Fluorescence microscopy was performed with a Motic AE31 epifluorescence microscope and representative images were acquired with a 60× objective using the Motic Images Advanced 3.2 Software.
:60) for 20 min at room temperature. Staining of nuclei was performed in a 1 mg mL−1 solution of 4′,6-diamino-2-phenylindole (DAPI) and finally, the coverslips were mounted with Mowiol® 4–88. Fluorescence microscopy was performed with a Motic AE31 epifluorescence microscope and representative images were acquired with a 60× objective using the Motic Images Advanced 3.2 Software.
SILAC protein profiling
Neuro-2a cells were maintained in DMEM medium supplemented with 10% dialyzed FBS, 100 units per ml of penicillin/streptomycin and either naturally-occurring isotope abundances (“light”) or stable isotope-labeled (“heavy”) 13C6 arginine and 13C6 lysine amino acids. Culture media were refreshed every 2 days and the cells were grown for at least 6 doublings to allow full incorporation of the labeled amino acids. Two large-scale SILAC replicates (1 × 107 cells per condition) were performed. Complete incorporation of 13C-Arg and 13C-Lys into control and MeHg-exposed Neuro-2a cells after 6 cell divisions in isotopically heavy medium (direct and reverse SILAC, respectively) was verified by MS analysis of a protein digest (data not shown).After differential labeling, control and cells exposed to 2 mg L−1 MeHg for 8 h were mixed in a 1:1 ratio. Cell lysates were prepared in RIPA buffer containing protease inhibitor cocktail (Roche). Protein extracts were separated by SDS-PAGE on 10% SDS-polyacrylamide gels, visualized by Coomassie blue staining and the gel lanes were cut horizontally into 20 sections. Excised gel bands were de-stained in 50:50 25 mM ammonium bicarbonate/acetonitrile and dried. Gel pieces were rehydrated with 12.5 ng μL−1 trypsin solution in 25 mM ammonium bicarbonate and incubated overnight at 37 °C. Peptides were extracted using acetonitrile and 5% formic acid, dried by vacuum centrifugation and reconstituted in 10 μL 2% acetonitrile in 0.1% formic acid.
The peptide mixtures from in-gel digestions were analyzed using nanoflow LC-MS/MS (Eksigent). Peptides were loaded onto a 0.3 × 10 mm C18 precolumn (SGE) and separated on a reverse-phase column (75 μm × 15 cm fused silica capillary C18 HPLC PepMap column, 3 μm, 100 A, Thermo) with linear gradient of 5–95% acetonitrile in 0.1% aqueous solution of formic acid. The samples were delivered over 120 min at a flow rate of 200 nL min−1 through the analytical column to a stainless steel nano-bore emitter (Proxeon). The peptides were scanned and fragmented with an LTQ XL linear ion trap mass spectrometer (Thermo Scientific) operated in data-dependent ZoomScan and MS/MS switching mode using the three most intense precursors detected in a survey scan from 400 to 1600 u (three μscans). ZoomScan mass window was set to 12 Da enabling monitoring of the entire 12C/13C isotopic envelope of most doubly and triply charged peptides. Singly charged ions were excluded for MS/MS analysis. Normalized collision energy was set to 35% and dynamic exclusion was applied during 3 min periods to avoid repetitive fragmentation ions.
The generated .raw files were converted to .mgf files for MASCOT data search. A database containing the NCBInr Mouse sequences containing 34966 entries (as of 10-09-14) was searched using MASCOT Software (version 2.3 MatrixScience) for protein identification. Oxidation of methionine and 13C6-Arg and 13C6-Lys were specified as variable modifications, trypsin as the specific enzyme and one missed cleavage allowed. Minimum precursor and fragment-ion mass accuracies of 1.2 and 0.3 Da were used. A requirement of at least one bold (unique) red peptide (i.e. the highest scoring peptide matches to the protein with the highest total score) was required for protein identification and at least two bold red (unique) peptides were required for quantification. Cut-off values for MASCOT scores of peptides and proteins were set to 38 (p < 0.05) and 45 (p < 0.01), respectively. The false positive rate was calculated by searching the same spectra against the NCBInr Mouse decoy database. Relative quantitation ratios of identified proteins were calculated using QuiXoT (version 1.3.26). SILAC ratios were defined by the area of the heavy peptides (13C) divided by the area of light peptides (12C). Protein ratios obtained by QuiXoT were manually verified for all peptides. As observed in previous studies, a proportion of 13C6-Arg was converted to 13C5-Pro leading to a reduction in the intensity of the isotope-labeled peptide peak; this was corrected for all peptides containing one or more proline residues by adding the intensity found for the peptide containing 13C6-Arg13C5-Pro or 13C6-Lys13C5-Pro to the intensity of the peak containing only 13C6-Arg or 13C6-Lys. Molecular and cellular functions of the proteins found de-regulated by SILAC were assigned based on the biological knowledge available in Gene Ontology (GO) annotations.
Western blotting
Total protein content was extracted from Neuro-2a cells using RIPA lysis buffer and quantified with the Bradford assay using BSA as the standard. Total protein extracts were separated on 7.5% SDS-polyacrylamide gels and electrotransferred to nitrocellulose membranes (BioRad). Membranes were blocked with 3% skim dry milk in PBS-T (0.05% Tween-20) for 1 h at room temperature and incubated overnight at 4 °C with primary antibodies against: methylenetetrahydrofolate dehydrogenase 1 (MTHFD1) (1:500, Santa Cruz), methionine synthase (MTR) (1:1000, Sigma-Aldrich) and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (1:10000, Sigma-Aldrich). The blots were washed in PBS-T and incubated with horseradish peroxidase-conjugated secondary antibodies for 1 h at room temperature. Antibody binding was visualized using an enhanced chemiluminescent immunoblotting detection system (GE Healthcare).
MTHFD1 gene knock-down
Knock-down of MTHFD1 was performed by transient transfection with Lipofectamine and reduced serum media (Opti-MEM) using control (not-targeting) small interfering double-stranded RNA (siRNA) and siRNA targeted against MTHFD1. MTHFD1 silencing transfectants exposed to 10 and 30 pmol of targeted siRNA were collected at 24 h for the MTT assay as described above. MTFHD1 silencing was confirmed by immunoblotting.Results and discussion
Cytotoxicity, mercury uptake and morphological changes induced by MeHg exposure
We evaluated the cytotoxicity induced by MeHg in Neuro-2a cells using the MTT assay that measures the reducing potential of cells; while viable cells are able to reduce the MTT to formazan (a purple compound), non-viable cells are unable to do so. We selected concentrations of MeHg ranging from 0.5 to 5 mg L−1 and exposure times between 6 and 24 h. The viability of cells exposed to 0.5 mg L−1 MeHg was not affected at the exposure times tested (see Fig. 1a). However, the highest concentration assayed (5 mg L−1) caused a decrease in the cell viability close to 100% even after 6 h of exposure. On the other hand, the viability of Neuro-2a cells exposed to 2 mg L−1 MeHg decreased in a time-dependent manner. It is important to mention that since MeHg was dissolved in MeOH, we tested the potential effect of MeOH on the cell viability, and no significant changes were observed as compared to control cells (data not shown). This data is in accordance with a previous report in which HepG2 cells were exposed to MeHg. Cell viability of HepG2 cells diminished to practically the same extent at the same MeHg concentrations and exposure time tested13 as Neuro-2a cells. In order to evaluate the effect of MeHg in Neuro-2a cells but without drastically compromising the cell viability, we selected 2 mg L−1 and 8 h as the exposure time for further experiments.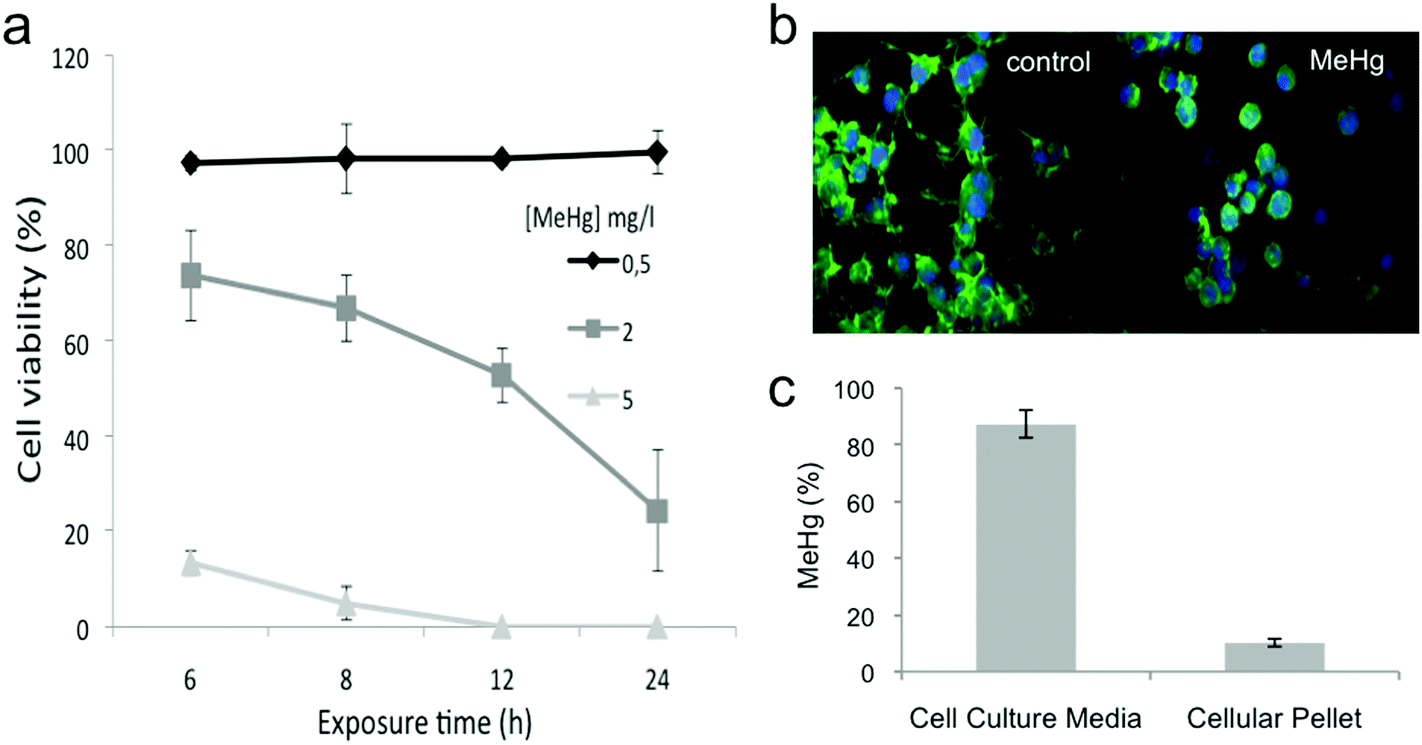 | ||
| Fig. 1 Cytotoxicity, mercury uptake, and morphological changes induced by MeHg exposure. (a) Viability of Neuro-2a cells exposed to different concentrations (0.5, 2 and 5 mg L−1) of MeHg at various exposure times (n = 10) While the viability of cells exposed to 0.5 mg L−1 MeHg was not affected at any of the exposure times tested, 5 mg L−1 MeHg caused a decrease on the cell viability close to 100% even after 6 h of exposure. We selected 2 mg L−1 and 8 h exposure for further experiments to evaluate the effect of MeHg on Neuro-2a cells without drastically compromising the cell viability. (b) Morphological changes induced after MeHg exposure visualized by fluorescence microscopy after staining with phalloidin-FITC and DAPI (n = 3). Exposure to MeHg markedly disrupted the structural integrity of neurites. (c) Determination of the total amount of MeHg internalized by the cells by atomic fluorescence spectroscopy (AFS) (n = 4). Only 10% of the total MeHg added were actually found inside the cells. | ||
We also evaluated morphological changes induced after MeHg exposure by fluorescence microscopy. Neuro-2a cells were allowed to extend neurites for 48 h before exposure to 2 mg L−1 MeHg for 8 h. Exposure to MeHg markedly disrupted the structural integrity of neurites (see Fig. 1b).
We finally determined the total amount of MeHg internalized by the cells by using atomic fluorescence spectroscopy (AFS). Our results (see Fig. 1c) showed that 10% of the total MeHg added was actually found inside the cells. These results reflect that the amount of internalized MeHg can significantly impair cell viability, functioning and morphology.
Differential protein expression of Neuro-2a cells exposed to MeHg
In order to identify novel molecular pathways related to MeHg toxicity, we have used a state-of-the-art quantitative proteomic approach (SILAC) able to quantify differentially expressed proteins in MeHg-exposed cells as compared to control Neuro-2a cells. In our SILAC experiment, we carried out two large-SILAC experiments. We identified a total of 1524 proteins (see Fig. 2a), from which 978 proteins passed the criteria established for protein quantitation. Most of the quantified proteins presented a SILAC ratio close to 1, as expected for a 1:1 mixture (see Fig. 2b). The overall false discovery rate was 1.3% being estimated by the number of hits against the reverse sequence/total hits (p < 0.01). The mean relative standard deviation (RSD) of the ratios obtained from replicates was lower than 20%, indicating good agreement between experiments. Using 1.5 as the threshold ratio, we found 125 proteins altered, 48 of which were over-expressed (Table 1) and 77 down-regulated (Table 2) upon MeHg exposure.
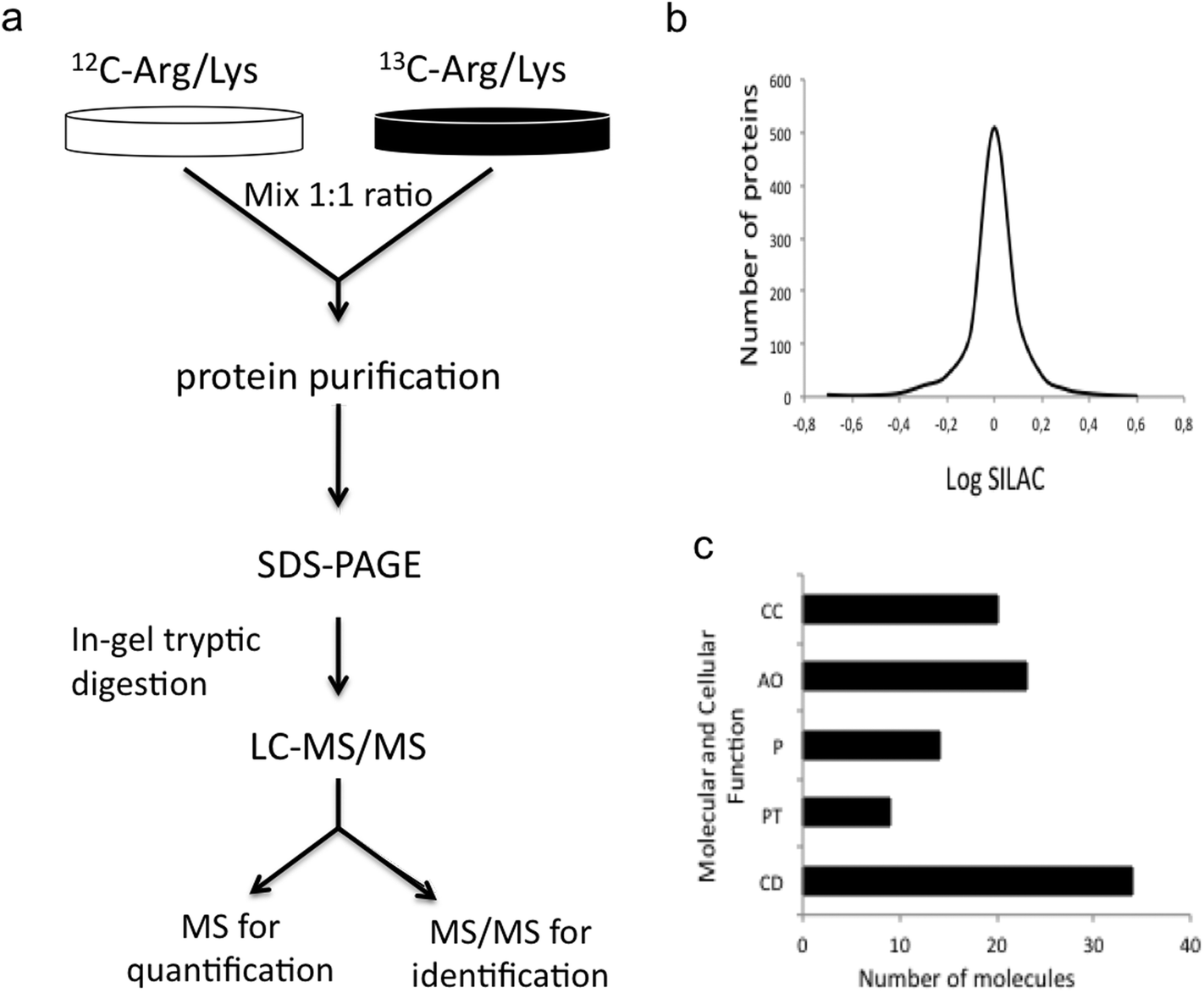 | ||
| Fig. 2 SILAC results. (a) General scheme of the SILAC procedure. (b) Distribution of the SILAC ratios for the identified proteins. Most quantified proteins presented a SILAC ratio close to 1, as expected for a 1:1 mixture. (c) Functional annotation of the 125 altered proteins upon MeHg exposure obtained from the gene ontology GO consortium website. Major molecular and cellular functions altered included cell death (CD), RNA post-transcriptional modification (PT), protein synthesis (P), cellular assembly and organization (AO), and cell cycle (CC). | ||
| Common name | Accession number | Protein name | Silac ratio | RSD | Mascot score |
|---|---|---|---|---|---|
| MCM5 | 112293273 | Minichromosome maintenance deficient 5, cell division cycle 46 | 3.71 | 4.14 | 131 |
| PIR | 51317401 | Pirin | 2.98 | 4.71 | 113 |
| ALDH2 | 6753036 | Aldehyde dehydrogenase 2, mitochondrial | 2.93 | 12.14 | 194 |
| CAD | 51093867 | Carbamoyl-phosphate synthetase 2, aspartate transcarbamylase, and dihydroorotase | 2.92 | 11.24 | 92 |
| CDK1 | 31542366 | Cell division cycle 2 homolog A | 2.80 | 1.87 | 502 |
| MO25 | 31982582 | MO25 protein | 2.68 | 8.85 | 82 |
| NDUFS3 | 58037117 | NADH dehydrogenase (ubiquinone) Fe–S protein 3 | 2.65 | 1.03 | 238 |
| RPF2 | 110347519 | Brix domain containing 1 isoform 1 | 2.53 | 10.31 | 119 |
| BAX | 6680770 | Bcl2-associated X protein | 2.52 | 12.48 | 750 |
| PRPF8 | 115583687 | pre-mRNA processing factor 8 | 2.30 | 9.28 | 320 |
| TRXR1 | 13569841 | Thioredoxin reductase 1 isoform 2 | 2.23 | 13.31 | 381 |
| CEND1 | 10946620 | Cell cycle exit and neuronal differentiation 1 | 2.10 | 17.11 | 128 |
| EIF4B | 21704128 | Eukaryotic translation initiation factor 4B | 2.05 | 16.61 | 57 |
| DUT | 21281687 | Deoxyuridine triphosphatase | 2.00 | 15.19 | 408 |
| TRX | 31543902 | Thioredoxin | 1.99 | 0.98 | 136 |
| USMG5 | 77404294 | Upregulated during skeletal muscle growth 5 | 1.91 | 4.96 | 169 |
| CUL3 | 7710014 | Cullin 3 | 1.88 | 12.53 | 98 |
| NXF1 | 31980798 | Nuclear RNA export factor 1 | 1.85 | 13.70 | 122 |
| AP1B1 | 88853578 | Adaptor protein complex AP-1, beta 1 subunit | 1.85 | 15.07 | 444 |
| MCM2 | 6678826 | Minichromosome maintenance deficient 2 mitotin | 1.85 | 16.25 | 118 |
| PSMB2 | 31981327 | Proteasome (prosome, macropain) subunit, beta type 2 | 1.83 | 14.80 | 555 |
| PDXP | 47059486 | Pyridoxal phosphate phosphatase | 1.80 | 12.33 | 272 |
| NAP1L4 | 6679012 | Nucleosome assembly protein 1-like 4 | 1.79 | 12.67 | 256 |
| RPS28 | 21426821 | Ribosomal protein S28 | 1.76 | 10.93 | 131 |
| EIF2S3X | 6753738 | Eukaryotic translation initiation factor 2, subunit 3, structural gene X-linked | 1.75 | 17.77 | 101 |
| SORD | 22128627 | Sorbitol dehydrogenase | 1.73 | 12.27 | 109 |
| EIF2S2 | 14149756 | Eukaryotic translation initiation factor 2, subunit 2 (beta) | 1.71 | 8.30 | 149 |
| PAICS | 13385434 | Phosphoribosylaminoimidazole carboxylase, phosphoribosylaminoimidazole succinocarboxamide synthetase | 1.69 | 19.50 | 258 |
| CKAP5 | 66955862 | Cytoskeleton associated protein 5 | 1.67 | 18.10 | 238 |
| PRDX5 | 6755114 | Peroxiredoxin 5 precursor | 1.67 | 0.45 | 169 |
| LMNA | 50355692 | Lamin A isoform A | 1.67 | 17.15 | 642 |
| ENO1 | 12963491 | Enolase 1, alpha non-neuron | 1.65 | 15.15 | 141 |
| RSL24D1 | 38348464 | Ribosomal protein L24-like | 1.62 | 6.94 | 280 |
| PLCXD3 | 28893495 | Phosphatidylinositol-specific phospholipase C, X domain containing 3 | 1.62 | 6.82 | 193 |
| IPO4 | 19745156 | RANBP4 | 1.61 | 9.44 | 154 |
| EIF3I | 9055370 | Eukaryotic translation initiation factor 3, subunit 2 (beta) | 1.59 | 17.18 | 621 |
| U2AF1 | 17530980 | U2 small nuclear ribonucleoprotein auxiliary factor (U2AF) 1 | 1.59 | 17.21 | 96 |
| ALG2 | 31560366 | Alpha-1,3-mannosyltransferase ALG2 | 1.58 | 9.45 | 92 |
| PSMD11 | 134053905 | Proteasome 26S non-ATPase subunit 11 | 1.58 | 10.38 | 425 |
| FAM49B | 21450053 | Hypothetical protein LOC223601 | 1.58 | 12.93 | 238 |
| TIMM23 | 12025536 | Translocase of inner mitochondrial membrane 23 homolog | 1.55 | 18.91 | 184 |
| ATAD1 | 31560168 | ATPase family, AAA domain containing 1 | 1.53 | 8.15 | 378 |
| CNOT1 | 94383743 | PREDICTED: CCR4-NOT transcription complex, subunit 1 isoform 2 | 1.53 | 0.90 | 137 |
| VAMP3 | 6678553 | Vesicle-associated membrane protein 3 | 1.51 | 2.20 | 193 |
| TSTA3 | 13654268 | Tissue specific transplantation antigen P35B | 1.51 | 11.51 | 291 |
| DNAJC8 | 27369493 | DnaJ (Hsp40) homolog, subfamily C, member 8 | 1.51 | 2.81 | 88 |
| PRPF19 | 19527358 | Nuclear matrix protein SNEV | 1.51 | 9.57 | 115 |
| ITGB1 | 45504394 | Integrin beta 1 (fibronectin receptor beta) | 1.50 | 11.37 | 152 |
| Common name | Accession number | Protein name | Silac ratio | RSD | Mascot Score |
|---|---|---|---|---|---|
| KIF5C | 45433560 | Kinesin family member 5C | −18.73 | 8.92 | 139 |
| RAB1B | 21313162 | RAB1B, member RAS oncogene family | −15.31 | 12.26 | 1199 |
| NSF | 31543349 | N-Ethylmaleimide sensitive fusion protein | −8.34 | 12.52 | 265 |
| PCM1 | 13540494 | Pericentriolar material 1 | −8.02 | 10.22 | 96 |
| SMARCA4 | 76253779 | SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily a, member 4 | −7.95 | 10.04 | 156 |
| PICALM | 32567788 | Phosphatidylinositol-binding clathrin assembly protein | −7.70 | 12.36 | 287 |
| TPPP3 | 13385968 | Hypothetical protein LOC67971 | −7.20 | 16.97 | 90 |
| TCEB1 | 21312712 | Transcription elongation factor B (SIII), polypeptide 1 | −5.94 | 5.98 | 142 |
| ACSL4 | 46518528 | Acyl-CoA synthetase long-chain family member 4 isoform 1 | −5.76 | 14.62 | 105 |
| — | 149262716 | PREDICTED: similar to hCG1813078 | −5.72 | 14.82 | 117 |
| ATP5L | 31980744 | ATP synthase, H+ transporting, mitochondrial F0 complex, subunit G | −4.93 | 9.37 | 232 |
| NT5C3B | 31980891 | 5′-Nucleotidase, cytosolic III-like | −4.73 | 4.95 | 92 |
| TUBB2B | 21746161 | Tubulin, beta | −4.48 | 8.62 | 3658 |
| PGM2 | 31980726 | Phosphoglucomutase 2 | −4.48 | 6.41 | 114 |
| CBR3 | 27413160 | Carbonyl reductase 3 | −4.34 | 6.04 | 38 |
| CTTN | 75677414 | Cortactin | −3.84 | 5.83 | 51 |
| COP9 | 7242142 | COP9 (constitutive photomorphogenic) homolog, subunit 7a | −3.45 | 5.81 | 242 |
| DDX39 | 38372907 | DEAD (Asp-Glu-Ala-Asp) box polypeptide 39 | −3.30 | 17.44 | 297 |
| NDUFB10 | 58037109 | NADH dehydrogenase (ubiquinone) 1 beta subcomplex, 10 | −2.83 | 2.15 | 433 |
| ATP5J2 | 10181184 | ATP synthase, H+ transporting, mitochondrial F0 complex, subunit f, isoform 2 | −2.73 | 11.28 | 70 |
| PSMA4 | 6755196 | Proteasome (prosome, macropain) subunit, alpha type 4 | −2.56 | 16.41 | 230 |
| TMPO | 121949765 | Thymopoietin isoform epsilon | −2.51 | 2.00 | 313 |
| KIF11 | 45476577 | Kinesin family member 11 | −2.41 | 7.51 | 87 |
| RQCD1 | 10946722 | rcd1 (required for cell differentiation) homolog 1 | −2.33 | 10.46 | 103 |
| THOP1 | 31981237 | Thimet oligopeptidase 1 | −2.29 | 3.82 | 376 |
| IPO | 112734861 | Importin | −2.24 | 13.83 | 172 |
| — | 149252028 | PREDICTED: hypothetical protein | −2.17 | 0.65 | 231 |
| SRSF6 | 31543689 | arginine/serine-rich splicing factor 6 | −2.14 | 10.06 | 109 |
| ATAD3 | 30725845 | AAA-ATPase TOB3 | −2.09 | 17.44 | 141 |
| PDLIM3 | 7948997 | PDZ and LIM domain 3 | −2.07 | 10.83 | 201 |
| ARPC3 | 9790141 | Actin related protein 2/3 complex, subunit 3 | −2.05 | 19.19 | 65 |
| COX5A | 112181182 | Cytochrome c oxidase, subunit Va | −2.02 | 13.30 | 141 |
| CHCHD3 | 149254953 | PREDICTED: hypothetical protein | −1.99 | 12.73 | 228 |
| HNRNPC | 8393544 | Heterogeneous nuclear ribonucleoprotein C | −1.98 | 6.39 | 336 |
| SLC25A1 | 23943838 | Solute carrier family 25, member 1 | −1.96 | 13.33 | 82 |
| COPE | 10946972 | Epsilon subunit of coatomer protein complex | −1.94 | 10.74 | 353 |
| TRNT1 | 33859692 | tRNA nucleotidyl transferase, CCA-adding, 1 | −1.92 | 5.80 | 187 |
| MYEF2 | 27819594 | Myelin basic protein expression factor 2, repressor | −1.91 | 3.53 | 78 |
| KRAS | 84370270 | c-K-ras2 protein | −1.91 | 0.65 | 137 |
| PFKL | 31560653 | Phosphofructokinase, liver, B-type | −1.89 | 12.56 | 88 |
| RPF2 | 110347521 | Brix domain containing 1 isoform 2 | −1.89 | 10.02 | 179 |
| HNRNPR | 33859724 | Heterogeneous nuclear ribonucleoprotein R | −1.86 | 18.16 | 226 |
| BDH1 | 31982169 | 3-Hydroxybutyrate dehydrogenase, type 1 | −1.83 | 0.56 | 132 |
| TUBA1C | 6678469 | Tubulin, alpha 1C | −1.83 | 14.45 | 507 |
| TNPO1 | 115385966 | Transportin 1 isoform 2 | −1.81 | 13.62 | 389 |
| BAG3 | 115270960 | Bcl2-associated athanogene 3 | −1.81 | 15.06 | 384 |
| H1FX | 38348566 | H1 histone family, member X | −1.81 | 16.15 | 207 |
| RBM25 | 124430762 | RNA binding motif protein 25 | −1.80 | 9.76 | 72 |
| BCAT1 | 66792792 | Branched chain aminotransferase 1, cytosolic isoform 1 | −1.78 | 3.61 | 257 |
| — | 149249564 | PREDICTED: hypothetical protein isoform 1 | −1.77 | 14.21 | 224 |
| GST | 6754086 | Glutathione S-transferase | −1.74 | 15.07 | 207 |
| PPP2R4 | 31981677 | Protein phosphatase 2A, regulatory subunit B (PR 53) | −1.73 | 0.51 | 200 |
| SRRT | 13937395 | Arsenate resistance protein 2 | −1.73 | 13.47 | 132 |
| MSN | 70778915 | Moesin | −1.72 | 4.91 | 141 |
| — | 94390383 | PREDICTED: similar to La ribonucleoprotein domain family, member 1 | −1.72 | 17.54 | 105 |
| RRM1 | 31982026 | Ribonucleotide reductase M1 | −1.71 | 16.03 | 175 |
| MEST | 6678866 | Mesoderm specific transcript | −1.70 | 3.33 | 259 |
| SNCB | 15809030 | Synuclein, beta | −1.69 | 7.68 | 208 |
| DYN | 134288917 | Dynein | −1.68 | 18.65 | 733 |
| MTHFD1 | 31559887 | Methylenetetrahydrofolate dehydrogenase (NADP+ dependent) 1 | −1.67 | 18.63 | 317 |
| AP2A1 | 6671561 | Adaptor protein complex AP-2, alpha 1 subunit isoform a | −1.66 | 6.45 | 132 |
| NASP | 125490378 | Nuclear autoantigenic sperm protein isoform 2 | −1.65 | 14.21 | 60 |
| RBMX | 6755296 | RNA binding motif protein, X-linked | −1.64 | 9.19 | 221 |
| BCL2A1 | 24496776 | BCL2-associated transcription factor 1 isoform 2 | −1.64 | 18.50 | 82 |
| — | 149265144 | PREDICTED: filamin B, beta | −1.62 | 14.83 | 422 |
| STMN1 | 9789995 | Stathmin 1 | −1.59 | 17.20 | 129 |
| SMC1A | 9790237 | SMC1 structural maintenance of chromosomes 1-like 1 | −1.58 | 14.20 | 129 |
| DDX18 | 31981163 | DEAD (Asp-Glu-Ala-Asp) box polypeptide 18 | −1.58 | 19.13 | 80 |
| PSMD5 | 134053913 | Proteasome 26S non-ATPase subunit 5 | −1.56 | 16.01 | 98 |
| SLC3A2 | 31560670 | Solute carrier family 3 (activators of dibasic and neutral amino acid transport), member 2 | −1.55 | 13.75 | 178 |
| TRA2B | 6677975 | Splicing factor, arginine/serine-rich 10 | −1.55 | 0.74 | 235 |
| LOC433762 | 149253163 | PREDICTED: hypothetical protein | −1.54 | 13.95 | 444 |
| DDX21 | 72384374 | DEAD (Asp-Glu-Ala-Asp) box polypeptide 21 | −1.53 | 1.82 | 701 |
| PRPH | 7305413 | Peripherin | −1.52 | 17.67 | 1706 |
| SNX2 | 13385878 | Sorting nexin 2 | −1.51 | 18.33 | 73 |
| TARS | 27229277 | Threonyl-tRNA synthetase | −1.51 | 10.43 | 319 |
| EIF4A3 | 20149756 | Eukaryotic translation initiation factor 4A, isoform 3 | −1.50 | 1.16 | 389 |
The functional annotation of the 125 altered proteins upon MeHg exposure was obtained from the gene ontology GO consortium website. Major molecular and cellular functions altered included cell death, RNA post-transcriptional modification, protein synthesis, cellular assembly and organization, and cell cycle (see Fig. 2c). We also looked for the molecular pathways in which altered proteins might play key roles. Such pathways included apoptosis (PIR, CDK1, CAD, MO25, BAX, CEND1, CUL3, BCL2A1, COP9), stress resistance (MCM5, MCM2, ALDH2, TRX, TRXR1), ROS generation (NDUFS3, GST), axonogenesis (IPO9, TUBB2B, MSN, DYN) and neurodegeneration (RAB1B, COPE). Alteration or activation of some of these mechanisms has already been associated with MeHg toxicity.
MeHg induces cell death through the intrinsic and extrinsic apoptotic pathways
Several previous reports have demonstrated how MeHg exposure activates the apoptotic pathway in different cell cultures and animals.8,13–15 Apoptosis is a naturally occurring process by which a cell is directed to programmed cell death. It involves the activation of caspases, a family of cysteine proteases, and a complex cascade of events that link the initiating stimuli to the final demise of the cell.16 Activation of caspases can be initiated at the plasma membrane upon ligation of a death receptor (extrinsic pathway) or at the mitochondria induced by intracellular stress (intrinsic pathway). Most previous studies on MeHg toxicity have demonstrated the activation of the apoptotic intrinsic pathway and thus, alteration of the expression of well-known apoptotic markers such as the caspases and the Bcl2 protein family after MeHg exposure.17 In agreement with these studies, we have also found alteration of some of these proteins, thus suggesting MeHg-mediated activation of the apoptotic intrinsic pathway. We show overexpression of the pro-apoptotic protein BAX (RSILAC = 2.52) and inhibition of anti-apoptotic proteins BCL2A1 (RSILAC = −1.64) and BAG3 (RSILAC = −1.81), in Neuro-2a cells exposed to MeHg. While BAX induces opening of the mitochondrial voltage-dependent anion channel (VDAC) resulting in the release of cytochrome c and other pro-apoptotic factors, BCL2A1 has the opposite role by reducing the release of pro-apoptotic cytochrome c and blocking caspase activation. BAG3 is a modulator of cellular anti-apoptotic activity that functions through its interaction with Bcl-2.18–20 We also found significant overexpression of CAD (RSILAC = 2.92), which is activated by caspase 3 and causes DNA degradation in the nucleus of apoptotic cells. Besides these well-known apoptotic markers, our SILAC approach has allowed us to find additional routes that are also being activated by MeHg and thus, should be considered for a better understanding of the mechanisms related to MeHg-induced cell death.The ubiquitin-proteasome system (UPS) constitutes a major degradation pathway for intracellular proteins and plays an important role in regulating apoptosis. In this pathway, proteins, which are going to be degraded by the proteasome, are first tagged with a polypeptide called ubiquitin (Ub) in order to be later recognized and degraded by the 26S proteasome. Ubiquitination reaction is carried out by a set of enzymes called ubiquitin ligases or E3s, which transfer the ubiquitin moiety to the substrate.21 Within this molecular machinery, there is an essential family of proteins named cullins, which provide a scaffold for ubiquitin ligases. These proteins combined with RING proteins form the Cullin-RING ubiquitin ligases (CRLs) that contain the catalytic core needed for the ubiquitination reaction. In our experiment, we have found overexpression of CUL3 (RSILAC = 1.88) in Neuro-2a cells exposed to MeHg. CUL3, which is one of the four members of the cullin protein family,22 mediates the ubiquitination of caspase-8.23 Overexpression of CUL3 might increase aggregation of CUL3-modified caspase-8, thus inducing full activation and processing of caspase-8, thereby leading to robust stimulation of effector caspases and apoptosis.24 It is also important to consider the interplay between Ub family modifiers that creates a regulatory network with the Ub family proteins. One of the best-studied crosstalks between Ub family modifiers is the stimulation of ubiquitination by Nedd8 (neural precursor cell expressed developmentally down regulated 8) modification. Neddylation of the cullin scaffolds of the CRLs, allosterically activates the transfer of Ub from the CRLs to the target substrates.25 The neddylation–deneddylation pathway of the CRLs is regulated by the COP9 signalosome (CSN). CSN has an intrinsic metalloproteinase that removes the Nedd8 from cullins (deneddylation), thus converting CRLs into an inactive state.25 Interestingly, COP9 is down-regulated in our experiment (RSILAC = −3.45), this deficiency of COP9 induced by MeHg exposure might preclude deneddylation of the CRLs, also promoting ubiquitination of caspase-8 by CUL3 and therefore, the apoptotic pathway. These results are in agreement with a recent report demonstrating that deficiency of COP9 induces ubiquitin-proteasome system impairment and apoptosis.26 Although previous reports support the idea that MeHg-induced apoptosis occurs through activation of the intrinsic apoptotic pathway;13,27 taken together, our results suggest that MeHg could induce apoptosis through both extrinsic and intrinsic pathways in Neuro-2a cells.
Considering the link between the cell cycle and apoptosis, it is not surprising that some of the proteins we found affected by MeHg exposure are related to cell cycle regulation. CDK1, also known as CDC2, is a serine/threonine protein kinase that plays a well-characterized role in cell cycle control and cellular differentiation. CDK1 is critical for G2/M transition and mitosis. Several studies have demonstrated the relevant role of CDK1 in some forms of apoptosis.28 It has been shown that CDK1-mediated phosphorylation of BCL2 family proteins enhances their pro-apoptotic function.29 Thus, the overexpression of CDK1 (RSILAC = 2.80) in cells exposed to MeHg, might explain the significant overexpression of pro-apoptotic proteins observed in our SILAC experiment. We have found a similar degree of overexpresion for CEND1 (RSILAC = 2.10), which is a neuronal-lineage specific regulator that coordinates cell cycle withdrawal and differentiation of neuronal progenitors. It is involved in histone deacetylase inhibition-mediated growth arrest of neuroblastoma cells,30 and its overexpression in fibroblasts has been shown to trigger cell cycle exit, driving cells towards a pro-apoptotic pathway.31
We have also found two additional proteins highly upregulated, MO25 (RSILAC = 2.68) and PIR (RSILAC = 2.98), whose overexpression have been previously associated with a higher degree of apoptosis. MO25 is a scaffold protein that can directly interact with certain STE20 kinases including MST4 (mammalian STE20-like kinase) increasing its kinase activity.32 MST4 regulates multiple cellular aspects such as cell polarity and proliferation. In fact, MST4 might be one of the targets for caspase 3.33 It has been demonstrated that overexpression of MO25 activates MST4 resulting in dramatically increased apoptosis.34 PIR is an iron-binding nuclear protein and transcription cofactor that stabilizes the formation of quaternary complexes between Bcl-3, NF-kB and a DNA target protein directing the NF-kB DNA binding towards a pro-apoptotic response.35 Overexpression of PIR has been associated with increased apoptosis in several cellular types.35,36
MeHg induces overexpression of proteins involved in ROS generation and stress resistance
MeHg is considered to increase reactive oxygen species (ROS) generation, thus inducing an imbalance in the normal redox state of cells, which results in damaging of cell components and structures.37 Since this process, known as oxidative stress, is directly related to apoptosis, we expected to find some proteins involved in ROS generation, oxidative stress and stress resistance, up- or down-regulated upon MeHg exposure. Actually, we found overexpression of NDUFS3 ((RSILAC = 2.65) and downregulation of GST (RSILAC = −1.74). NDUFS3 is a subunit of the NADH:ubiquinone oxidoreductase ETC complex I involved in electron transfer and coupling.38 Overexpression of NDUFS3 has been previously related to ROS generation and apoptosis.39,40 GST is a well-known antioxidant enzyme and its inhibition markedly exaggerates oxidative-stress induced apoptosis.41 In addition, it has also been demonstrated that GST inhibition is associated with increased activation of MAP kinases, a family of proteins that play crucial roles in stress response, cell survival and apoptosis.42 We also found overexpression of several antioxidant proteins such as TRXR1 (RSILAC = 2.23) and TRX (RSILAC = 1.99), which upregulated or overexpressed, protect against oxidative stress. Upregulation of these proteins shows a defense mechanism against MeHg-induced toxicity based on the disulfide reductase activity of TRX, and thereby on the supply of NADPH and the activity of TRXR1.43 Upregulation of ALDH2 (RSILAC = 2.93) shows an additional defense mechanism since ALDHs metabolize endogenous and exogenous aldehydes and thereby mitigate oxidative stress in prokaryotic and eukaryotic organisms.44 In the case of minochromosome maintenance proteins (MCMs), which are required for DNA replication in eukaryotic cells, their expression is restricted to proliferating and dedifferentiated tissues and is a typical feature of many malignant and premalignant diseases. Overexpression of MCM is one of the molecular determinants of highly mitogenic phenotypes.45 Thus, we could have expected downregulation of these proteins since Neuro-2a cells exposed to MeHg undergo apoptosis. However, we found upregulation of two MCM proteins (MCM5, RSILAC = 3.71; MCM2, RSILAC = 1.85) which can be explained as an additional cellular defense mechanism against MeHg toxicity, since it has been demonstrated that overexpression of MCM is needed for cells to withstand stress conditions.46MeHg exposure affects neurite integrity and development
Our evaluation of morphological changes in Neuro-2a cells after exposure to MeHg demonstrates a significant disruption of the structural integrity of neurites, which are precursors of either axons or dendrites (see Fig. 1b). In relationship with this observation, we found several proteins related to axonogenesis, neurite outgrowth and microtubule formation significantly inhibited. While IPO is constitutively complexed with the retrograde motor DYN, the DYN–IPO complex being essential for axonogenesis,47 the complex DYN–CTTN plays a central structural role in focal adhesion assembly in a neuronal growth cone.48 In our SILAC experiment, we found not only down-regulation of DYN (RSILAC = −1.68), which is in agreement with a previous study that demonstrated that a reduction in DYN prevented neurite formation in cultured hippocampal neurons;49 but also IPO (RSILAC = −2.24) and CTTN (RSILAC = −3.84), which forms key complexes with DYN as commented upon before, also appeared down-regulated, showing significant damage induced by MeHg on axonogenesis. We observed a similar down-regulation in the case of MSN (RSILAC = −1.72), which plays a key role in generating and maintaining the normal structure and functional organization of neuronal growth cones.50 In addition, we found other proteins directly related to neurite extension such as tubulins (TUBB2B, RSILAC = −4.48; TUBA1C, RSILAC = −1.83) down-regulated, whose assembly is essential for microtubule formation and thus, for neurite extension; and kinesins (KIF5C, RSILAC = −18.73; KIF11, RSILAC = −2.41), which have been recently recognized as microtubule motors providing the mechanical forces necessary for initial neurite extension.51 Altogether, these findings are consistent with earlier evidence demonstrating that microtubule metabolism is compromised in the presence of Hg ions,52 and confirming how MeHg directly affects the development of neurites, therefore demonstrating the detrimental effect of MeHg on axonogenesis.MeHg induces blockage of the methylation cycle mediated by downregulation of MTHFD1 and MTR
One of the down-regulated proteins that we found particularly interesting in our SILAC experiment is the enzyme methylenetetrahydrofolate dehydrogenase 1 (MTHFD1) (RSILAC = −1.67), which plays a key role in the methylation cycle. This cycle is the principal mechanism whereby vitamin B12 and folate metabolism influence brain function and DNA methylation, synthesis and repair. Folate metabolism plays a vital role in cell division and homeostasis due to the multiple enzymes53 involved in nucleic acid synthesis, methionine regeneration and reduction of carbon units required for normal metabolic regulation.54 The methylation pathway involves the conversion of homocysteine to methionine where tetrahydrofolate is converted to 5,10-methylenetetrahydrofolate (5,10-MTHF) by the enzyme methylenetetrahydrofolate dehydrogenase 1 (MTHFD1), followed by conversion of 5,10-MTHF to 5-methyltetrahydrofolate (5-MTHF) by the methylenetetrahydrofolate reductase (MTHFR). Finally, 5-MTHF reacts with homocysteine to form methionine and to regenerate tetrahydrofolate (see Fig. 3). Methionine synthase (MTR) regulates this reaction, vitamin B12 being a cofactor of MTR.55 Based on these premises and a previous study56 that correlates exposure to Hg2+ with inactivation of MTR, we decided to further study the effect of MeHg on the methylation cycle (see Fig. 3) and, particularly, on the expression of MTHFD1 and MTR. We validated the SILAC results by immunoblotting (see Fig. 4), and indeed we observed a significantly decreased expression of MTHFD1 with increasing concentrations of MeHg. A similar behavior was observed for MTR, whose expression also decreased significantly in Neuro-2a cells exposed to 2 mg L−1 of MeHg for 8 h as compared to control cells. Inhibition of these proteins has been associated with malfunction in the synthesis, methylation and reparation of DNA; thus, we hypothesize that the alteration in the expression of these enzymes could be related to the mechanism of neurotoxicity activated by MeHg exposure.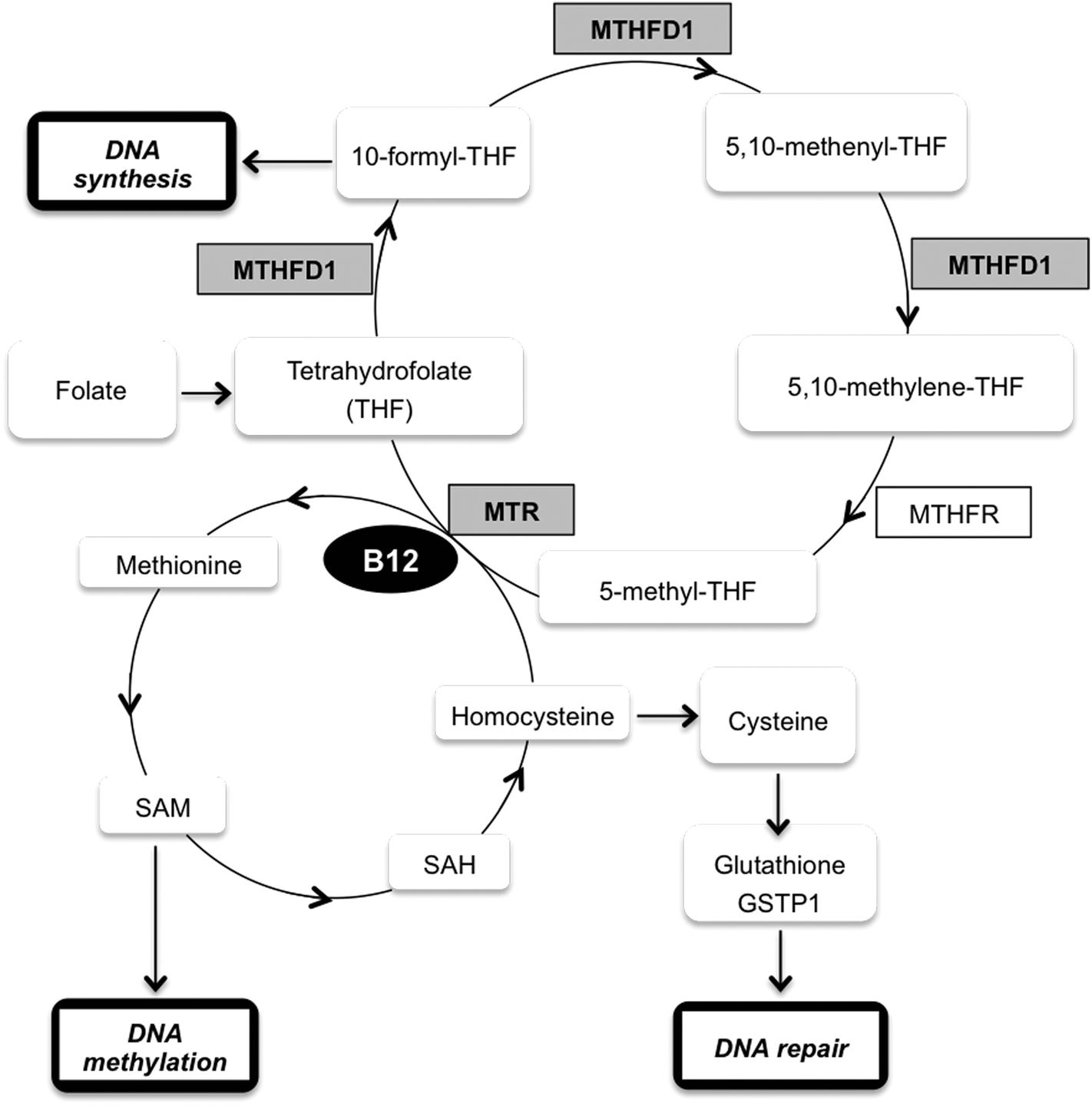 | ||
| Fig. 3 Schematic representation of the methylation cycle. Vitamin B12 (B12); glutathione S-transferase P (GSTP1); methionine synthase (MTR); methylenetetrahydrofolate dehydrogenase (MTHFD1); methylenetetrahydrofolate reductase (MTHFR); S-adenosylmethionine (SAM); S-adenosylhomocysteine (SAH); tetrahydrofolate (THF). | ||
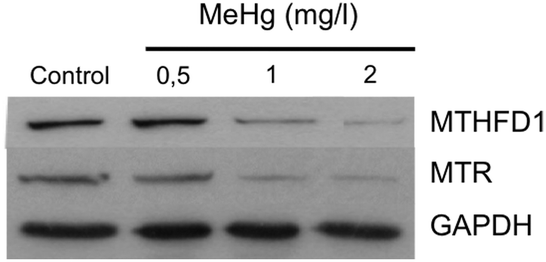 | ||
| Fig. 4 Evaluation of the levels of MTHFD1 and MTR in Neuro-2a cells exposed to different concentrations of MeHg by western blot analysis (n = 3). The expression of MTHFD1 and MTR decreased significantly with increasing concentrations of MeHg. GAPDH was used as the loading control. | ||
To support our previous results, we investigated whether siRNA-mediated gene knock-down of MTHFD1 induces a decrease in the cell viability of Neuro-2a cells similar to that induced by MeHg. After validating the suitability of the transfection (see Fig. 5a) by western blot, we measured the cell viability in transfected cells and in cells exposed to 2 mg L−1 of MeHg for 8 h using the MTT assay. The results show that MTHFD1 knock-down affected the viability of Neuro-2a cells, although the decrease in cell viability was more pronounced in cells exposed to MeHg (see Fig. 5b). This could be explained by the fact that additional mechanisms (besides the methylation cycle) are also being affected by MeHg exposure.
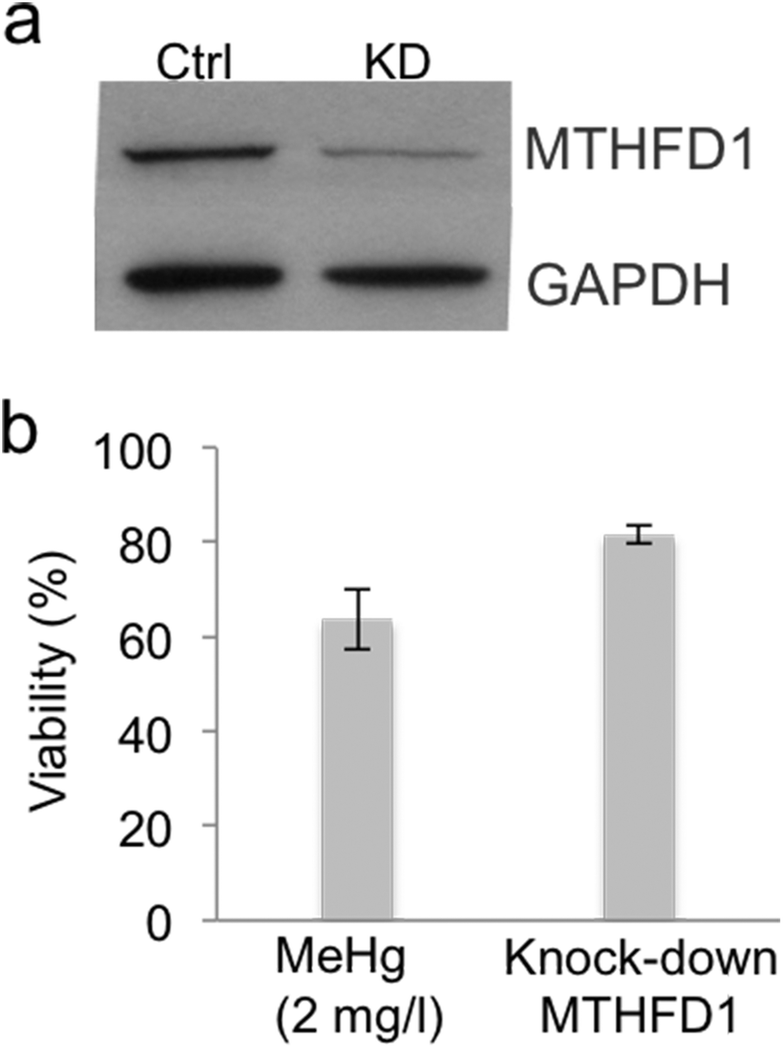 | ||
| Fig. 5 siRNA-mediated gene knock-down of MTHFD1. (a) Immunoblotting of control vs. MTHFD1 knock-down (KD) that shows the suitability of the transfection (n = 3). (b) Comparison of the viability between Neuro-2a cells transfected with 30 pmol of siRNA-MTHFD1 and cells exposed to 2 mg L−1 of MeHg for 8 h (n = 10). MTHFD1 knock-down affected the viability of Neuro-2a cells, although the decrease in cell viability was more pronounced in cells exposed to MeHg. | ||
Another important aspect to be considered is the fact that the methylation cycle requires vitamin B12, which acts as the coenzyme required for correct functioning of the methyl donation from 5-methyl-THF to THF.54 Vitamin B12 deficiency blocked the methylation cycle and induces a series of pathological conditions that are quite similar to the effects induced by Hg poisoning.57 Vitamin B12 is highly reactive and vulnerable to inactivation by oxidation of the Co atom in its structure; this oxidation inactivates the enzyme MTR and blocks the methylation cycle.58 The blockage of the methylation cycle causes an increase in levels of homocysteine that generates an increased concentration of calcium, accumulation of reactive oxygen species (ROS), activation of MAP kinases and the release of arachidonic acid from platelets that inhibits glutathione peroxidase, an antioxidant that protects cells against oxidative stress.59 Homocysteine is easily accumulated inside the cells where it undergoes autoxidation transforming it into homocysteic acid accompanied by H2O2 accumulation.60 In fact, long-term incubation of neurons with homocysteic acid induces necrotic cell death.61 It is well known that the effects on human health induced by Hg poisoning are very similar to those experienced by individuals with a significant vitamin B12 deficiency. Several years ago, a possible hypothesis of the Hg/vitamin B12 connection suggested that Hg could oxidize the Co atom in vitamin B12, thus causing denaturation of the vitamin, blockage of the methylation cycle and an increase in the levels of homocysteine.62 Although we have previously shown inhibition of MTHFD1 and MTR as the main cause for the methylation cycle to be blocked upon MeHg exposure, we decided to test whether the oxidation of vitamin B12 by MeHg could also be another factor to be considered. This would explain why both conditions caused similar symptoms, although this hypothesis has never been experimentally demonstrated. In order to demonstrate if another way by which MeHg affects the methylation cycle is by denaturizing vitamin B12, we added increasing concentrations of vitamin B12 to Neuro-2a cells exposed to MeHg. Considering the initial hypothesis, high concentration of vitamin B12 would counteract the partial oxidation of the vitamin induced by MeHg and would restore the cell viability to a certain level. Our results (see Fig. 6) show that supplementation with vitamin B12 did not ameliorate the decrease in cell viability observed in MeHg-exposed cells, not even at the higher concentration of vitamin B12 tested.
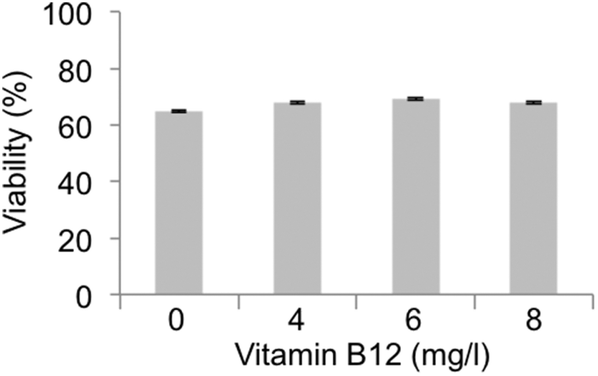 | ||
| Fig. 6 Effect of high levels of vitamin B12 supplementation to MeHg-exposed Neuro-2a cells (n = 10). Supplementation with vitamin B12 did not ameliorate the decrease in cell viability observed in MeHg-exposed cells. | ||
Comparative effect of MeHg and other heavy metals on the expression of MTHFD1 and MTR
One of the challenges in toxicoproteomics is the identification of particular molecular mechanisms or pathways associated with the toxicity exerted by a particular compound rather than general mechanisms related to stress-response and/or cell defense.63 To investigate whether blockage of the methylation cycle mediated by downregulation of MTHFD1 and MTR could also be induced by inorganic mercury or other heavy metals, we carried out a comparative Western blot analysis using Neuro-2a cells exposed to MeHg, Hg2+, Pb2+ or Cu2+. We selected concentrations of these ions that caused a decrease in the viability of the cells similar to that induced by 2 mg L−1 MeHg for 8 h. Our results show that while the four species highly affect the expression of MTR (see Fig. 7), the expression pattern for MTHFD1 was significantly different depending on the metallic species tested. Surprisingly, Hg2+ did not affect the expression of MTHFD1, while Pb2+ and Cu2+ slightly affected the expression of this enzyme but to a much lesser extent as compared to MeHg (see Fig. 7). The different behavior observed for MTHFD1 and MTR after exposure to these metals could explain why in a previous study, stimulation of MTR with dopamine and insulin resulted in the re-activation of the methylation cycle of Neuro-2a cells exposed to Cu2+ and Pb2+, but was inefficient for cells exposed to MeHg.57 This fact reinforces the specific negative interaction found between MeHg and MTHFD1.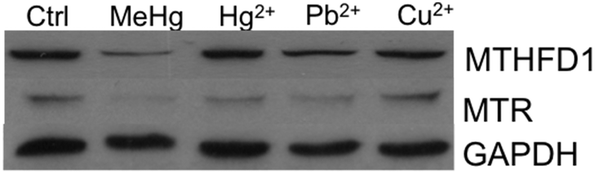 | ||
| Fig. 7 Comparative effect of MeHg and other heavy metals on the expression of MTHFD1 and MTR (n = 3). The four species highly affect the expression of MTR while the expression of MTHFD1 was significantly different depending on the metallic species tested. | ||
Conclusion
A quantitative proteomic approach (SILAC) has been used for the discovery of novel molecular pathways associated with MeHg induced neurotoxicity. We have identified a set of proteins altered upon MeHg exposure of Neuro-2a cells. Such altered proteins turned out to be involved in pathways related to apoptosis, stress resistance, ROS generation, axonogenesis and neurodegeneration. One of the main discoveries derived from our study is the MeHg-induced downregulation of key enzymes of the methylation cycle (MTHFD1 and MTR). We hypothesize that the effect of MeHg blocking the methylation cycle could be related to the mechanisms of neurotoxicity and the symptoms observed after MeHg poisoning. It could also explain the similarity observed in the symptomatology between MeHg poisoning and vitamin B12 deficiency. Moreover, we have demonstrated that inhibition of MTHFD1 is specific for MeHg as compared to inorganic Hg and other heavy metals such as Pb or Cu. Thus, this work sets the stage for dissecting a specific molecular mechanism for MeHg-induced toxicity.Acknowledgements
Authors thank the Spanish Ministry of Economy and Competitiveness (grant CTQ2014-55711-R) and the Comunidad Autónoma of Madrid (Spain) and European funding from the FEDER program (Project S2013/ABI-3028, AVENSACAL-CM). P.C. was supported by a FPU predoctoral fellowship from the Spanish Ministry of Education.References
- V. Glaser, E. M. Nazari, Y. M. R. Müller, L. Feksa, C. M. D. Wannmacher, J. B. T. Rocha, A. F. de Bem, M. Farina and A. Latini, Int. J. Dev. Neurosci., 2010, 28, 631–637 CrossRef PubMed.
- M. Farina, J. B. T. Rocha and M. Aschner, Life Sci., 2011, 89, 555–563 CrossRef CAS PubMed.
- K. Eto, Toxicol. Pathol., 1997, 25, 614–623 CrossRef CAS PubMed.
- C. Watanabe and H. Satoh, Environ. Health Perspect., 1996, 104, 367–379 CrossRef CAS PubMed.
- L. E. Kerper, N. Ballatori and T. W. Clarkson, Am. J. Physiol.: Regul., Integr. Comp. Physiol., 1992, 262, 761–765 Search PubMed.
- R. Dringen, Prog. Neurobiol., 2000, 62, 649–671 CrossRef CAS PubMed.
- V. Branco, J. Canario, A. Holmgren and C. Carvalho, Toxicol. Appl. Pharmacol., 2011, 251, 95–103 CrossRef CAS PubMed.
- M. Kunimoto, Biochem. Biophys. Res. Commun., 1994, 204, 310–317 CrossRef CAS PubMed.
- S.-H. Chang, H. J. Lee, B. Kang, K.-N. Yu, A. Minai-Tehrani, S. Lee, S. U. Kim and M.-H. Cho, J. Toxicol. Sci., 2013, 38, 823–831 CrossRef CAS PubMed.
- E. M. Faustman, R. A. Ponce, Y. C. Ou, M. A. C. Mendoza, T. Lewandowski and T. Kavanagh, Environ. Health Perspect., 2002, 110, 859–864 CrossRef CAS PubMed.
- J. L. Luque-Garcia, R. Sanchez-Diaz, I. Lopez-Heras, P. Martin and C. Camara, Trends Anal. Chem., 2013, 43, 254–268 CrossRef CAS.
- S. E. Ong and M. A. Mann, Nat. Protoc., 2006, 1, 2650–2660 CrossRef CAS PubMed.
- S. Cuello, L. Goya, Y. Madrid, S. Campuzano, M. Pedrero, L. Bravo, C. Camara and S. Ramos, Food Chem. Toxicol., 2010, 48, 1405–1411 CrossRef CAS PubMed.
- E. Fonfria, E. Dare, M. Benelli, C. Sunol and S. Ceccatelli, Eur. J. Neurosci., 2013, 16, 2013–2016 CrossRef.
- S. Cuello, P. Ximenez-Embun, I. Ruppen, H. B. Schonthaler, K. Ashman, Y. Madrid, J. L. Luque-Garcia and C. Camara, Analyst, 2012, 137, 5302–5311 RSC.
- S. Elmore, Toxicol. Pathol., 2007, 35, 495–516 CrossRef CAS PubMed.
- S. Cuello, S. Ramos, Y. Madrid, J. L. Luque-Garcia and C. Cámara, Anal. Bioanal. Chem., 2012, 404, 315–324 CrossRef CAS PubMed.
- Q. Liao, F. Ozawa, H. Friess, A. Zimmermann, S. Takayama, J. C. Reed, J. Kleeff and M. W. Büchler, FEBS Lett., 2001, 503, 151–157 CrossRef CAS PubMed.
- M. MacFarlane and A. C. Williams, EMBO Rep, 2004, 5, 674–678 Search PubMed.
- M. Vogler, Cell Death Differ., 2012, 19, 67–74 CrossRef CAS PubMed.
- C. Wójcik, J. Cell. Mol. Med., 2002, 6, 25–48 CrossRef.
- L. Grau, J. L. Luque-Garcia, P. Gonzalez-Peramato, D. Theodorescu, J. Palou, J. M. Fernandez-Gomez and M. Sanchez-Carbayo, PLoS One, 2013, 8, e53328 CAS.
- A. B. Parrish, C. D. Freel and S. Kombluth, Cold Spring Harbor Perspect. Biol., 2013, 5, a008672 CrossRef PubMed.
- Z. Jin, Y. Li, R. Pitti, D. Lawrence, V. C. Pham, J. R. Lill and A. Ashkenazi, Cell, 2009, 127, 721–735 CrossRef PubMed.
- T. Schmaler and W. Dubiel, Subcell. Biochem., 2010, 54, 57–68 CAS.
- D. Lei, F. Li, S. Huabo, J. Liu, N. Wei and X. Wang, PLoS One, 2013, 8, e67793 CAS.
- K. Sokolowski, A. Falluel-Morel, X. Zhou and E. DiCicco-Bloom, NeuroToxicology, 2011, 32, 535–544 CrossRef CAS PubMed.
- Y. Furukawa, S. Iwase, Y. Terui, J. Kikuchi, T. Sakai, M. Nakamura, S. Kitagawa and M. Kitagawa, J. Biol. Chem., 1996, 271, 28469–28477 CrossRef CAS PubMed.
- L. Zhou, X. Cai, X. Han, N. Xu and D. C. Chang, Cell Biol. Int., 2014, 38, 737–746 CrossRef CAS PubMed.
- P. K. Politis, S. Akrivou, C. Hurel, O. Papadodima and R. Matsas, FEBS Lett., 2008, 582, 741–748 CrossRef CAS PubMed.
- N. Georgopoulou, C. Hurel, P. K. Politis, M. Gaitanou, R. Matsas and D. Thomaidou, J. Biol. Chem., 2006, 281, 33606–33620 CrossRef CAS PubMed.
- B. M. Filippi, P. de los Heros, Y. Mehellou, I. Navratilova, R. Gourlay, M. Deak, L. Plater, R. Toth, E. Zeqiraj and D. R. Alessi, EMBO J., 2011, 30, 1730–1741 CrossRef CAS PubMed.
- I. Dan, S. E. Ong, N. M. Watanabe, B. Blagoev, M. M. Nielsen, E. Kajikawa, T. Z. Kristiansen, M. Mann and A. Pandey, J. Biol. Chem., 2002, 277, 5929–5939 CrossRef CAS PubMed.
- Z. Shi, S. Jiao, Z. Zhang, M. Ma, Z. Zhang, C. Chen, K. Wang, H. Wang, W. Wang, L. Zhang, Y. Zhao and Z. Zhou, Structure, 2013, 21, 449–461 CrossRef CAS PubMed.
- B. D. Gelbman, A. Heguy, T. P. O'Connor, J. Zabner and R. G. Crystal, Respir. Res., 2007, 8, 10 CrossRef PubMed.
- D. Orzaez, A. J. de Jong and E. J. Woltering, Plant Mol. Biol., 2001, 46, 459–468 CrossRef CAS PubMed.
- E. Patel and M. Reynold, Toxicol. Lett., 2013, 222, 265–272 CrossRef CAS PubMed.
- D. Martinvalet, D. M. Dykxhoorn, R. Ferrini and J. Lieberman, Cell, 2008, 133, 681–692 CrossRef CAS PubMed.
- G. Huang, Y. Chen, H. Lu and X. Cao, Cell Death Differ., 2007, 14, 327–337 CrossRef CAS PubMed.
- S. Suhane, H. Kanzaki, V. Arumugaswami, R. Murali and V. K. Ramanujan, Biol. Open, 2013, 14, 295–305 CrossRef PubMed.
- E. Röth, N. Marczin, B. Balatonyi, S. Ghosh, V. Kovács, N. Alotti, B. Borsiczky and B. Gasz, Exp. Clin. Cardiol., 2011, 16, 92–96 Search PubMed.
- T. Wada and J. Penninger, Oncogene, 2004, 23, 2838–2849 CrossRef CAS PubMed.
- E. S. J. Arner and A. Holmgren, Eur. J. Biochem., 2000, 267, 6102–6109 CrossRef CAS PubMed.
- S. Singh, C. Brocker, V. Koppaka, C. Ying, B. Jackson, A. Matsumoto, D. C. Thompson and V. Vasiliou, Free Radicals, 2013, 56, 89–101 CrossRef CAS PubMed.
- T. Guida, G. Salvatore, P. Faviana, R. Giannini, G. Garcia-Rostan, L. Provitera, F. Basolo, A. Fusco, F. Carlomagno and M. Santoro, J. Clin. Endocrinol. Metab., 2005, 90, 4703–4709 CrossRef CAS PubMed.
- M. Das, S. Singh, S. Pradhan and G. Narayan, Mol. Biol. Int., 2014, 574850 Search PubMed.
- R. Ohara, K. Hata, N. Yasuhara, R. Mehmood, Y. Yoneda, M. Nakagawa and T. Yamashita, Biochem. Biophys. Res. Commun., 2011, 405, 697–702 CrossRef CAS PubMed.
- S. Kurklinsky, J. Chen and M. A. McNiven, J. Neurochem., 2011, 177, 48–60 CrossRef PubMed.
- E. Torre, M. A. McNiven and R. Urrutia, J. Biol. Chem., 1994, 269, 32411–32417 CAS.
- G. Paglini, P. Kunda, S. Quiroga, K. Kosik and A. Caceres, J. Cell Biol., 1998, 143, 443–455 CrossRef CAS PubMed.
- E. Lu, P. Fox, M. Lakonishok, M. W. Davidson and V. I. Gelfand, Curr. Biol., 2013, 23, 1018–1023 CrossRef PubMed.
- J. C. Pendergrass, B. E. Haley, M. J. Vimy, S. A. Winfield and F. L. Lorscheider, NeuroToxicology, 1997, 18, 315–324 CAS.
- T. J. Lightfoot, C. F. Skibola, E. V. Willett, D. R. Skibola, J. M. Allan, F. Coppede, P. J. Adamson, G. J. Morgan, E. Roman and M. T. Smith, Cancer Epidemiol. Biomarkers Prev., 2005, 14, 2999–3003 CAS.
- L. B. Bailey and J. F. Gregory, J. Nutr., 1999, 129, 779–782 CAS.
- J. Y. Fowdar, M. V. Lason, A. L. Szvetko, R. A. Lea and L. R. Griffiths, Int. J. Hypertens., 2012, 190923 Search PubMed.
- M. Waly, H. Olteanu, R. Banerjee, S.-W. Choi, J. B. Mason, B. S. Parker, S. Sukumar, S. Shim, A. Sharma, J. M. Benzecry, V. Power-Charnitsky and R. C. Deth, Mol. Psychiatry, 2004, 9, 358–370 CrossRef CAS PubMed.
- M. J. Shipton and J. Thachil, Clin. Med., 2015, 15, 145–150 CrossRef PubMed.
- D. G. Weir and J. M. Scott, Br. Med. Bull., 1999, 55, 669–682 CrossRef CAS PubMed.
- C. M. Gallagher and J. R. Meliker, Sci. Total Environ., 2011, 409, 1399–1405 CrossRef CAS PubMed.
- A. Boldyrev, E. Bryushkova, A. Mashkina and E. Vladychenskaya, Aging Sci., 2013, 6, 29–36 CrossRef CAS.
- E. Ziemińska, A. Stafiej and J. W. Łazarewicz, Neurochem. Int., 2003, 43, 481–492 CrossRef.
- B. Ahlrot-Westerlund, Heavy Metal Bull., 1995, 3, 11–12 Search PubMed.
- J. L. Luque-Garcia, P. Cabezas-Sanchez and C. Camara, Trends Anal. Chem., 2011, 30, 703–716 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2016 |