
Effect of base sequence context on the conformational heterogeneity of aristolactam-I adducted DNA: structural and energetic insights into sequence-dependent repair and mutagenicity†
Preetleen
Kathuria
a,
Purshotam
Sharma
ab and
Stacey D.
Wetmore
*a
aDepartment of Chemistry and Biochemistry, University of Lethbridge, 4401 University Drive West, Lethbridge, Alberta, Canada T1K 3M4. E-mail: stacey.wetmore@uleth.ca; Fax: +1 403-329-2057; Tel: +1 403-329-2323
bCentre for Computational Sciences, Central University of Punjab, Bathinda, Punjab, India 151001
First published on 23rd October 2015
Abstract
Aristolochic acids (AAs) are nephrotoxic and potentially carcinogenic plant mutagens that form bulky DNA adducts at the exocyclic amino groups of the purines. The present work utilizes classical molecular dynamics simulations and free energy calculations to investigate the role of lesion site sequence context in dictating the conformational outcomes of DNA containing ALI-N6-dA, the most persistent and mutagenic adduct arising from the AAs. Our calculations reveal that the anti base-displaced intercalated conformer is the lowest energy conformer of damaged DNA in all sequence contexts considered (CXC, CXG, GXC and GXG). However, the experimentally-observed greater mutagenicity of the adduct in the CXG sequence context does not correlate with the relative thermodynamic stability of the adduct in different sequences. Instead, AL-N6-dA adducted DNA is least distorted in the CXG sequence context, which points toward a possible differential repair propensity of the lesion in different sequences. Nevertheless, the structural deviations between adducted DNA with different lesion site sequences are small, and therefore other factors (such as interactions between the adducted DNA and lesion-bypass polymerases during replication) are likely more important for dictating the observed sequence-dependent mutagenicity of ALI-N6-dA.
1. Introduction
Aristolochic acids (AAs) I and II (Fig. 1a) are plant toxins commonly found in various species of Aristolochia and Aasrum genera.1 The consumption of traditional herbal medicines containing extracts of Aristolochia fangchi has been linked to a chronic renal disease known as Chinese Herbal Nephropathy2 (more commonly referred to as Aristolochic Acid Nephropathy (AAN)),3 which can further develop into urothelial carcinoma.4–6 In addition, a closely related renal condition called Balkan Endemic Nephropathy (BEN)7–9 has been associated with the development of upper urinary tract carcinoma,10 and traced to the consumption of bread prepared from wheat locally grown in the Balkan region and contaminated with seeds of Aristolochia clematitis.11,12 Based on sufficient evidence of the involvement of AAs in the etiology of AAN, BEN and related carcinogenesis,13 the International Agency for Research on Cancer (IARC) has categorized herbal medicines containing plant species of the Aristolochia genus as Group 1 (human) carcinogens.14 | ||
| Fig. 1 Structure of the (a) aristolochic acid and (b) AL-N6-dA adduct (R = OCH3 for ALI and H for ALII). The θ (∠(N1C6N6C10)) and ϕ (∠(C6N6C10C11)) dihedral angles determine the orientation of the AL moiety with respect to A, while the χ dihedral angle (∠(O4′C1′N9C4)) dictates the glycosidic bond orientation to be syn (χ = 0 ± 90°) or anti (χ = 180 ± 90°). | ||
The genotoxic mechanism of action of AA-induced carcinogenesis involves the formation of DNA adducts (addition products), which has been supported by their identification in the renal tissue of AA-exposed humans.12,15 AAs are metabolically activated in cells to form N-hydroxylaristolactams (N-hydroxyl ALs),16,17 which hydrolyze to yield nitrenium ions that react with the amino groups of the purines.18 Among the resulting AL-N6-dA and AL-N2-dG adducts, the adenine lesions (Fig. 1b) are more persistent and hence more mutagenic in the affected tissues.19 Furthermore, despite differing by a single methoxy group, the ALI-N6-dA adduct is more abundant,20–22 persistent23 and nephrotoxic24 compared to the ALII-N6-dA adduct. The mutagenicity of the AL-N6-dA adducts has been associated with AA-induced “signature” A → T mutations that are dominant in the TP53 tumor suppressor gene,25 the FGFR3 and HRAS oncogenes,26 and T/CXG (X = adduct) trinucleotide sequence motifs within the genome.27,28
In order to understand the structural basis for the observed differential biological outcomes associated with the mutagenic AL-N6-dA adducts, it is vital to understand the conformational preferences of (damaged) DNA containing these lesions. In this context, our previous computational work studied both ALI-N6-dA and ALII-N6-dA in the 5′-CGTA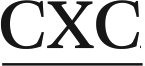 ATGC 11-mer sequence,29 which contains the CXC motif that experiments predict to yield the most stable ALII-N6-dA adducted duplexes.30 Our unrestrained molecular dynamics (MD) simulations revealed that both ALI and ALII-N6-dA adducted DNA prefer a base-displaced intercalated structure, which contains an anti adduct glycosidic orientation (Fig. 1b), displaces the opposing thymine into the major groove, and completely disrupts the lesion site Watson–Crick hydrogen-bonding.29 These results agree with the most stable ALII-N6-dA conformation deduced from NMR based studies in the same sequence,30 thereby verifying the robustness of our computational approach. Overall, the conformational preferences of the AL-N6-dA adducts markedly differ from those of the well-studied N6-linked dA adducts derived from polycyclic aromatic hydrocarbons (PAHs) within the same (CXC) sequence context.31–35 Specifically, whereas the linker atom in the PAH adducts is tetrahedral, which allows the adduct to maintain either partial or complete hydrogen bonding with the opposing base, the carcinogenic moiety is directly attached to the exocyclic amino group in the AL-N6-dA adducts and the lesions cannot form interactions with the opposing base. Thus, the differences in the flexibility within the AL-nucleobase linker bond leads to differences in the intercalated conformations of the PAH and AL-N6-dA adducts.
ATGC 11-mer sequence,29 which contains the CXC motif that experiments predict to yield the most stable ALII-N6-dA adducted duplexes.30 Our unrestrained molecular dynamics (MD) simulations revealed that both ALI and ALII-N6-dA adducted DNA prefer a base-displaced intercalated structure, which contains an anti adduct glycosidic orientation (Fig. 1b), displaces the opposing thymine into the major groove, and completely disrupts the lesion site Watson–Crick hydrogen-bonding.29 These results agree with the most stable ALII-N6-dA conformation deduced from NMR based studies in the same sequence,30 thereby verifying the robustness of our computational approach. Overall, the conformational preferences of the AL-N6-dA adducts markedly differ from those of the well-studied N6-linked dA adducts derived from polycyclic aromatic hydrocarbons (PAHs) within the same (CXC) sequence context.31–35 Specifically, whereas the linker atom in the PAH adducts is tetrahedral, which allows the adduct to maintain either partial or complete hydrogen bonding with the opposing base, the carcinogenic moiety is directly attached to the exocyclic amino group in the AL-N6-dA adducts and the lesions cannot form interactions with the opposing base. Thus, the differences in the flexibility within the AL-nucleobase linker bond leads to differences in the intercalated conformations of the PAH and AL-N6-dA adducts.
Although the currently available (computational) structural details of ALI-N6-dA adducted DNA have only been obtained in a single (CXC) sequence context,29 previous studies on a variety of carcinogenic adducts, including those formed from aromatic amines,36–39 PAHs,35,40–43 and ochratoxin A,44,45 point towards the sequence dependence of the conformational preference of damaged DNA. Specifically, changes in the sequence at the lesion site can affect key structural features and alter the stability of the preferred conformational theme of adducted DNA.35,37,38,40,44 For example, although the 10S (+)-trans-anti-B[a]P-N2-dG PAH adduct preferentially induces a minor groove conformation at G6 or G7 in the 5′-CATGCG6G7CCTAC oligonucleotide, greater distortion occurs with the adduct at G7 because of intrastrand steric clashes with the 5′-flanking guanine.40 Furthermore, within the 5′-CTC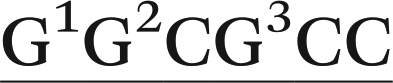 ATC oligonucleotide containing the NarI mutational hotspot sequence (underlined),46 the base-displaced intercalated conformer (the bulky moiety in the syn damaged base stacks in the helix and renders the opposing base extrahelical) of AF-C8-dG aromatic amine adducted DNA is most stabilized with the lesion at G3 due to better stacking between the bulky moiety and the flanking bases in the opposing strand than at G1 or G2.37,38 Additionally, due to the formation of discrete hydrogen-bonding interactions at the lesion site, DNA containing the (monoanionic) C-linked C8-bonded adduct of dechlorinated ochratoxin A (OTB) and dG (OTB-dG adduct) at G3 in the NarI sequence exhibits greater stabilization of the minor groove conformer compared to the major groove and base-displaced intercalated conformations.44 In addition to affecting the structure and stability of the preferred conformational theme, changing the flanking bases can even completely alter the conformational preference of damaged DNA. For example, the NarI sequence-containing adducted DNA acquires a minor groove conformation when the IQ-C8-dG aromatic amine adduct is located at G1 or G2, but a base-displaced intercalated conformation with the lesion at G3.36
ATC oligonucleotide containing the NarI mutational hotspot sequence (underlined),46 the base-displaced intercalated conformer (the bulky moiety in the syn damaged base stacks in the helix and renders the opposing base extrahelical) of AF-C8-dG aromatic amine adducted DNA is most stabilized with the lesion at G3 due to better stacking between the bulky moiety and the flanking bases in the opposing strand than at G1 or G2.37,38 Additionally, due to the formation of discrete hydrogen-bonding interactions at the lesion site, DNA containing the (monoanionic) C-linked C8-bonded adduct of dechlorinated ochratoxin A (OTB) and dG (OTB-dG adduct) at G3 in the NarI sequence exhibits greater stabilization of the minor groove conformer compared to the major groove and base-displaced intercalated conformations.44 In addition to affecting the structure and stability of the preferred conformational theme, changing the flanking bases can even completely alter the conformational preference of damaged DNA. For example, the NarI sequence-containing adducted DNA acquires a minor groove conformation when the IQ-C8-dG aromatic amine adduct is located at G1 or G2, but a base-displaced intercalated conformation with the lesion at G3.36
Since changing the sequence context can alter the preferred conformation of adducted DNA, it is not surprising that the sequence can also influence the biological consequences of adduct formation. Specifically, the sequence context has been shown to play an important role in modulating the repair propensity of DNA lesions.40–43,47–52 Bulky DNA lesions, including those formed from aristolochic acids, are most commonly repaired through the nucleotide excision repair (NER) pathway,53 which may proceed either through a transcription-coupled repair (TCR) or a global genome repair (GGR) mechanism.54–56 Whereas TCR only deals with transcriptionally-active regions,56 GGR operates on lesions spanning the entire genome.55 Furthermore, although lesion recognition in TCR is associated with inhibition of transcription (stalling of an RNA polymerase), GGR recognition involves specific damage recognition factors (e.g., eukaryotic XPC-RAD23B). Nevertheless, the subsequent repair steps are similar in both mechanisms. Although a number of bulky DNA adducts are commonly repaired by GGR,57–59 experimental studies have determined that the AL-N6-dA adducts are resistant to GGR, and are exclusively repaired by TCR.60 However, the structural basis of this GGR resistance and the related sequence dependence of the repair propensity of the AL-N6-dA adducts is not well understood.
An abundance of experimental and computational evidence has led to proposals that the primary GGR recognition step, including binding of the damage recognition factor and opening of the damaged DNA (flipping of the base opposing the lesion into the active site), involves identification of structural perturbations at the lesion site.42,51,61–68 As a result, the relative repair efficiency of DNA adducts in different sequences can be altered by differences in distortions and/or dynamics at the lesion site. For example, the increased repair susceptibility of the 10S (+)-trans-anti-B[a]P-N6-dA PAH adduct in the CXC compared to the CXA sequence context has been attributed to the formation of a sequence-specific hydrogen bond that increases the roll and bend at the CXC lesion site.41 Similarly, the 10S (+)-trans-anti-B[a]P-N2-dG PAH adduct induces a flexible kink when located at G7 in 5′-CATGCG6G7CCTAC due to steric interactions with the 5′-flanking guanine, which leads to better recognition by NER machinery at G7 compared to G6.40 Alternatively, the 10S (+)-trans-anti-B[a]P-N2-dG PAH adduct exhibits greater helical dynamics and associated repair propensity in the TXT than CXC sequence due to the reduced intrinsic stability of T:A compared to C:G flanking base pairs, as well as the reduced stability of the T–G compared to the C–G base steps involving damaged G.42,47
In addition to influencing the GGR repair propensity, the identity of the bases flanking the lesion site,69–71 as well as the next nearest neighbors,72 has been shown to affect the mutagenic profile of DNA lesions. For example, the mutational frequency of the PhIP-C8-dG aromatic amine adduct is greater in the 5′-TCCTCCTNXCCTCTC oligonucleotide when N = C or G compared to A or T.71 Additionally, the greater stability of the base-displaced intercalated conformer associated with AF-dG in the CXA over the TXA sequence context causes enhanced misincorporation in the CXA motif.69 Alternatively, the slippage prone active site of the model translesion synthesis (TLS) polymerase Dpo4 leads to different mutagenic outcomes for the 10S (+)-trans-anti-B[a]P-N2-dG PAH adduct, which exhibits dGMP insertion opposite the lesion in the CXG sequence context, and dAMP insertion in the TXG motif.70
Despite observed variations in adduct conformational preferences, repair propensities, and mutational outcomes with sequence context, it is currently unclear how these effects depend on the chemical structure of the adduct. In the case of the AL-N6-dA lesions, a previous study indicates that AAII-damaged DNA is thermodynamically more stable when the adduct is present in the CXC sequence context compared to T/CXG motifs.30 Nevertheless, recent studies on patients exposed to AAs have revealed that signature A → T mutations most commonly occur in T/CXG motifs.27,28 Unfortunately, no other information is available to date regarding the sequence effects on the stability of AL-adducted DNA or the associated biological consequences of AA-induced damage. For example, the conformation and stability of DNA strands containing AL-N6-dA in a purine–X–purine or purine–X–pyrimidine sequence is not currently available. Thus, it is not surprising that the molecular basis of the observed sequence dependence associated with AL-N6-dA formation has yet to be explored.
In the present work, we utilize classical (unrestrained) MD simulations and post-processing free energy calculations to investigate the role of sequence context in dictating the conformational outcomes of ALI-N6-dA, the most persistent and mutagenic DNA adduct arising from the AAs. Specifically, ALI-N6-dA (X) is incorporated into three 11-mer oligonucleotides (i.e., 5′-CGTA ATGC, 5′-CGTA
ATGC, 5′-CGTA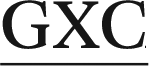 ATGC, and 5′-CGTA
ATGC, and 5′-CGTA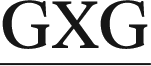 ATGC), which differ in the identity of the bases flanking the lesion (underlined). Two of the oligonucleotides considered place a purine (G) and pyrimidine (C) on either the 5′ or 3′ side of the lesion, while a purine (G) flanks both the 5′ and 3′ sides of the lesion in the third oligonucleotide. The conformational outcomes within these sequences are compared to our previous study on the 5′-CGTA
ATGC), which differ in the identity of the bases flanking the lesion (underlined). Two of the oligonucleotides considered place a purine (G) and pyrimidine (C) on either the 5′ or 3′ side of the lesion, while a purine (G) flanks both the 5′ and 3′ sides of the lesion in the third oligonucleotide. The conformational outcomes within these sequences are compared to our previous study on the 5′-CGTA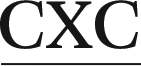 ATGC oligonucleotide, which places a pyrimidine (C) on both sides of the lesion.29 We focus on different purine and pyrimidine sequence contexts generated by varying the combination of G:C flanking base pairs, since a previous experimental study determined that the TXG and CXG motifs containing the related ALII-N6-dA adduct have similar stabilities, while the CXC motif leads to the most stable helices.30 Due to the similarity in the conformational preferences of ALII-N6-dA and ALI-N6-dA predicted in our previous study,29 it is reasonable to assume that the sequence-dependent conformational preferences of ALI-N6-dA will not be significantly different from ALII-N6-dA in the absence of experimental data on the sequence-dependent stability of ALI-N6-dA adducted DNA. Our detailed comparison of the preferred conformations of ALI-N6-dA adducted DNA in different lesion site sequence contexts provides valuable clues about the origin of the experimentally-observed sequence-dependent strand stability. Furthermore, our work explores the relationship between the sequence context and potential biological outcomes of adduct formation, including the mutagenicity and repair propensity, and thereby provides insight into the sequence-dependent persistence of this potent lesion.
ATGC oligonucleotide, which places a pyrimidine (C) on both sides of the lesion.29 We focus on different purine and pyrimidine sequence contexts generated by varying the combination of G:C flanking base pairs, since a previous experimental study determined that the TXG and CXG motifs containing the related ALII-N6-dA adduct have similar stabilities, while the CXC motif leads to the most stable helices.30 Due to the similarity in the conformational preferences of ALII-N6-dA and ALI-N6-dA predicted in our previous study,29 it is reasonable to assume that the sequence-dependent conformational preferences of ALI-N6-dA will not be significantly different from ALII-N6-dA in the absence of experimental data on the sequence-dependent stability of ALI-N6-dA adducted DNA. Our detailed comparison of the preferred conformations of ALI-N6-dA adducted DNA in different lesion site sequence contexts provides valuable clues about the origin of the experimentally-observed sequence-dependent strand stability. Furthermore, our work explores the relationship between the sequence context and potential biological outcomes of adduct formation, including the mutagenicity and repair propensity, and thereby provides insight into the sequence-dependent persistence of this potent lesion.
2. Computational details
The ALI-N6-dA adduct was considered in four 11-mer DNA sequences: (i) 5′-CGTA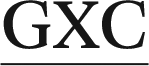 ATGC, (ii) 5′-CGTA
ATGC, (ii) 5′-CGTA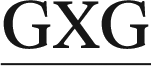 ATGC and (iii) 5′-CGTA
ATGC and (iii) 5′-CGTA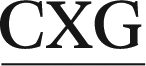 ATGC, as well as the previously studied (iv) 5′-CGTA
ATGC, as well as the previously studied (iv) 5′-CGTA ATGC (X = adduct).29 The B-form of canonical DNA was initially built for the 5′-CGTACXCATGC (X = A) sequence using the NAB73 module of AMBER 11.74 The ALI moiety was attached to the N6 position of A at the X position using GaussView.75 The previously characterized lowest energy anti and syn nucleotide orientations (with θ ∼ 0°, Fig. 1b) were incorporated into DNA,29 while avoiding steric clashes with the surrounding nucleotides. The damaged base was paired against complementary T. Initial structures for simulations were built by replacing the flanking base pairs in the representative structures obtained from the simulation trajectories of each of the six conformers belonging to the previously studied 5′-CGTA
ATGC (X = adduct).29 The B-form of canonical DNA was initially built for the 5′-CGTACXCATGC (X = A) sequence using the NAB73 module of AMBER 11.74 The ALI moiety was attached to the N6 position of A at the X position using GaussView.75 The previously characterized lowest energy anti and syn nucleotide orientations (with θ ∼ 0°, Fig. 1b) were incorporated into DNA,29 while avoiding steric clashes with the surrounding nucleotides. The damaged base was paired against complementary T. Initial structures for simulations were built by replacing the flanking base pairs in the representative structures obtained from the simulation trajectories of each of the six conformers belonging to the previously studied 5′-CGTA ATGC (X = ALI-N6-dA) adducted DNA oligonucleotide.29 All force field parameters, as well as the simulation protocol, were adopted from our previous study of ALI-N6-dA adducted DNA.29 Final unrestrained production MD simulations were subsequently run at 300 K for 20 ns using the PMEMD76 module of AMBER 12.77 The choice of room temperature for simulations ensures the viability of the harmonic approximation in the bonded terms of the force field, while remaining close to physiological conditions. The simulation time is justified by our previous study of AL-N6-dA adducted DNA in the CXC sequence, which illustrated that extending the simulation time to 320 ns does not significantly change lesion site structural parameters (Tables S1 and S2, ESI†).29 Indeed, the root-mean-square deviation (RMSD) in the position of all heavy atoms in the damaged base pair, and the 5′ and 3′-flanking base pairs (including the associated sugar-phosphate backbone), from the 20 and 320 ns simulations on the CXC sequence in the (most stable) base-displaced conformations falls between 0.8 and 1.2 Å (Table S1, ESI†). Furthermore, each production simulation for each sequence context gave rise to a single stable conformation, which did not significantly deviate from the initial structure. Specifically, the maximum standard deviation in the backbone RMSD for each final production simulation is approximately 1.5 Å (Table S2, ESI†). Additionally, unimodal distributions were obtained with respect to the χ, θ and ϕ dihedral angles of the adducted nucleotide (Fig. 1b) throughout each simulation (Fig. S1–S3, ESI†). Therefore, a single representative structure was extracted by clustering each simulation trajectory with respect to the position of the atoms forming χ, θ, and ϕ using the PTRAJ module of AMBER 12.
ATGC (X = ALI-N6-dA) adducted DNA oligonucleotide.29 All force field parameters, as well as the simulation protocol, were adopted from our previous study of ALI-N6-dA adducted DNA.29 Final unrestrained production MD simulations were subsequently run at 300 K for 20 ns using the PMEMD76 module of AMBER 12.77 The choice of room temperature for simulations ensures the viability of the harmonic approximation in the bonded terms of the force field, while remaining close to physiological conditions. The simulation time is justified by our previous study of AL-N6-dA adducted DNA in the CXC sequence, which illustrated that extending the simulation time to 320 ns does not significantly change lesion site structural parameters (Tables S1 and S2, ESI†).29 Indeed, the root-mean-square deviation (RMSD) in the position of all heavy atoms in the damaged base pair, and the 5′ and 3′-flanking base pairs (including the associated sugar-phosphate backbone), from the 20 and 320 ns simulations on the CXC sequence in the (most stable) base-displaced conformations falls between 0.8 and 1.2 Å (Table S1, ESI†). Furthermore, each production simulation for each sequence context gave rise to a single stable conformation, which did not significantly deviate from the initial structure. Specifically, the maximum standard deviation in the backbone RMSD for each final production simulation is approximately 1.5 Å (Table S2, ESI†). Additionally, unimodal distributions were obtained with respect to the χ, θ and ϕ dihedral angles of the adducted nucleotide (Fig. 1b) throughout each simulation (Fig. S1–S3, ESI†). Therefore, a single representative structure was extracted by clustering each simulation trajectory with respect to the position of the atoms forming χ, θ, and ϕ using the PTRAJ module of AMBER 12.
To analyze the structural features at the lesion site for each distinct adducted DNA conformation, base step parameters were calculated using a pseudostep, which consists of the base pairs 5′ and 3′ with respect to the lesion. Free energy calculations were performed on each simulation trajectory using the molecular mechanics-Poisson Boltzmann surface area (MM-PBSA) method.78 The entropy term was evaluated using the normal mode approximation at a temperature of 298.15 K, which is close to the temperature used for simulations. Snapshots for the free energy calculations were taken from each simulation at 50 ps intervals (400 frames in total). The relative total free energy (Grel) is reported with respect to the adducted DNA conformation with the lowest (most negative) Gtotal. Furthermore, although all adducted DNA oligonucleotides studied in the present work have different nucleobase sequences, the chemical composition of these strands is the same (i.e., the lesion pair is always flanked by a G:C base pair). Hence, the reported relative stabilities of the damaged DNA strands are compared using the relative total free energies (ΔGtotal) for the energetically accessible (base-displaced intercalated) conformer with respect to the conformation with the lowest Gtotal (regardless of the sequence context). We emphasize that these free energy differences do not reflect the strand destabilization due to the ALI moiety, which cannot be extracted from our data.
3. Results and discussion
3.1. The base-displaced intercalated conformational theme leads to the smallest helical distortion regardless of the sequence context
Three distinct conformational themes, namely the base-displaced intercalated, 5′-intercalated and 3′-intercalated conformations, were characterized for both the anti and syn adducted nucleotide orientations in each lesion site sequence context (denoted as GXC, GXG, CXG and CXC). Salient features associated with the three conformational themes are outlined below. In this discussion, all references to the 5′ and 3′ directions are made with respect to the damaged base.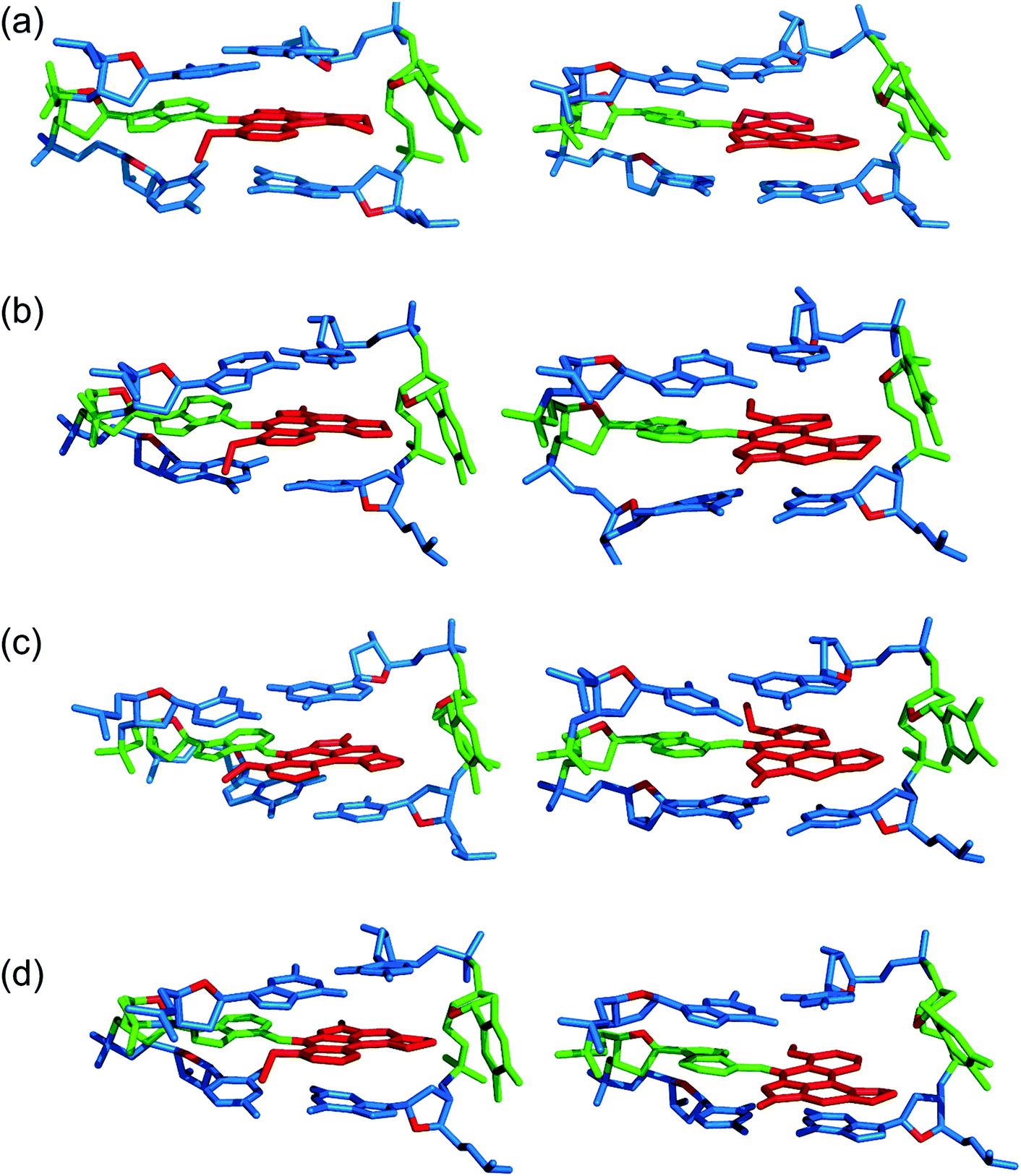 | ||
| Fig. 2 Representative structures from MD simulations with the anti (left) and syn (right) orientation of the ALI-N6-dA adduct paired opposite thymine in the (a) CXC, (b) GXG, (c) CXG and (d) GXC sequence contexts for the base-displaced intercalated DNA conformation. | ||
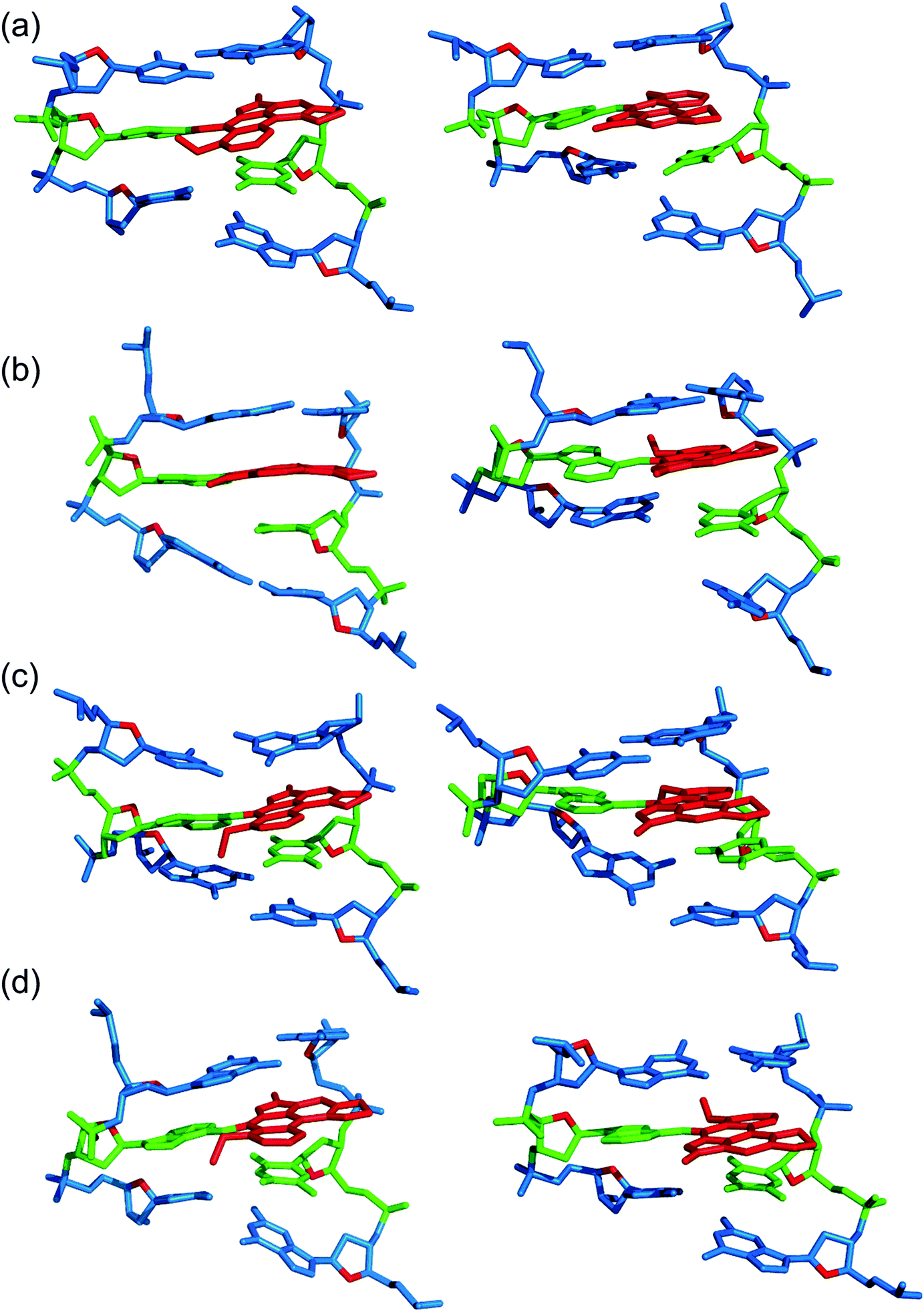 | ||
| Fig. 3 Representative structures from MD simulations with the anti (left) and syn (right) orientation of the ALI-N6-dA adduct paired opposite thymine in the (a) CXC, (b) GXG, (c) CXG and (d) GXC sequence contexts for the 5′-intercalated DNA conformation. | ||
 | ||
| Fig. 4 Representative structures from MD simulations with the anti (left) and syn (right) orientation of the ALI-N6-dA adduct paired opposite thymine in the (a) CXC, (b) GXG, (c) CXG and (d) GXC sequence context for the 3′-intercalated DNA conformation. | ||
Overall, the base-displaced intercalated conformer adopts a similar structure regardless of the sequence considered, with the only exception being the syn T opposing the syn adduct in the CXG sequence. In contrast, the structural features of the 5′-intercalated and 3′-intercalated conformers can vary with sequence context, mainly in the hydrogen-bonding patterns of the flanking bases and the magnitude of helical distortion. More importantly, regardless of the sequence considered, the ALI-N6-dA lesion consistently causes fewer helical perturbations when adducted DNA adopts the base-displaced intercalated orientation. This is largely due to the extrahelical position of the opposing T in this conformational theme, which allows the flanking pairs to remain intact, compared to the intrahelical opposing T in the other (5′-intercalated and 3′-intercalated) conformational themes, which disrupts one or two of the flanking base pairs depending on the sequence context.
3.2. The anti base-displaced intercalated orientation is the most stable ALI-N6-dA adducted DNA conformational theme regardless of the flanking bases
Despite the range of structurally distinct conformers isolated from the MD simulations, free energy calculations reveal that the 5′-intercalated and 3′-intercalated conformational themes are ∼25–70 kJ mol−1 less stable than the base-displaced intercalated orientation of ALI-N6-dA adducted DNA for all sequence contexts (Table 1). This finding correlates with the greatest helical distortion for these adducted DNA conformations (Table S4, ESI†). Furthermore, these structural distortions in the 3′ or 5′-intercalated conformers decrease the van der Waals (stacking) interactions involving the damaged base by ∼10–45 kJ mol−1 compared to the base-displaced intercalated conformers (Table 1). As a result, the 5′-intercalated and 3′-intercalated conformers are likely energetically inaccessible to ALI-N6-dA adducted DNA for each sequence considered.| Sequential motif | ||||||||
|---|---|---|---|---|---|---|---|---|
| CXC | GXG | CXG | GXC | |||||
| Conformation |
G
rel![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a a |
E
vdWb |
G
rela |
E
vdWb |
G
rela |
E
vdWb |
G
rela |
E
vdWb |
| a Relative total free energy calculated with respect to the most stable conformation of adducted DNA in each sequence context. b Total van der Waals interaction energy between the adduct and both flanking base pairs. | ||||||||
| anti Base-displaced | 0.0 | −171.8 ± 6.8 | 0.0 | −173.0 ± 6.3 | 0.0 | −173.5 ± 6.7 | 0.0 | −167.2 ± 6.3 |
| anti 5′-Intercalated | 36.1 | −148.1 ± 10.9 | 54.0 | −153.4 ± 10.9 | 27.9 | −134.4 ± 7.1 | 44.3 | −155.4 ± 8.8 |
| anti 3′-Intercalated | 51.6 | −148.9 ± 12.4 | 38.7 | −135.2 ± 7.1 | 56.5 | −127.7 ± 6.7 | 52.1 | −139.0 ± 9.2 |
| syn Base-displaced | 2.1 | −166.3 ± 7.1 | 16.2 | −169.2 ± 7.5 | 14.8 | −167.6 ± 7.1 | 22.1 | −163.8 ± 7.1 |
| syn 5′-Intercalated | 42.0 | −144.8 ± 10.5 | 69.4 | −149.5 ± 18.5 | 46.5 | −127.3 ± 11.3 | 56.4 | −146.1 ± 9.2 |
| syn 3′-Intercalated | 49.1 | −135.6 ± 7.6 | 47.5 | −139.9 ± 10.9 | 52.2 | −126.0 ± 11.3 | 47.8 | −136.5 ± 7.1 |
On the other hand, the base-displaced intercalated conformers are stabilized by intact hydrogen bonds in the 5′ and 3′-flanking base pairs (Table S3, ESI†), significant van der Waals interactions between the lesion and the flanking bases (Table 1), lesion pair interactions (primarily C–H⋯π, Table S5, ESI†), and minimal distortions to the DNA step parameters at the lesion site (Table S4, ESI†). Since these factors stabilize the base-displaced conformation for both the anti and syn glycosidic orientations of the adduct, both conformers lie very close in energy (deviating by a maximum of ∼22 kJ mol−1 depending on the sequence, Table 1). This suggests that both base-displaced intercalated conformers may be thermodynamically accessible regardless of the bases flanking the lesion. Nevertheless, the stabilizing effects are greater for the anti than the syn base-displaced intercalated conformation in each sequence. For example, the interactions between the ALI moiety and the opposing thymine are ∼8–10 kJ mol−1 stronger for the anti than the syn adduct conformation (Table S5, ESI†), which primarily reflects changes in the C–H⋯π contact. Additionally, the van der Waals interactions between the adduct and the flanking base pairs are (up to ∼6 kJ mol−1) greater in the anti base-displaced intercalated conformers (Table 1). Most importantly, the experimentally-observed non-accessibility of the syn base-displaced intercalated conformation for ALII-N6-dA adducted DNA suggests that the syn conformer is kinetically prohibited due to a large anti/syn energy barrier for rotation about the adduct glycosidic angle, a possibility that remains viable even in the case of ALI-N6-dA adducted DNA. Regardless, the anti base-displaced intercalated adducted DNA conformer is the most stable among the six conformations characterized in the present work irrespective of the sequential motif.
3.3. Calculated relative free energies of the most stable anti base-displaced intercalated adducted DNA conformer correlate with experimentally-predicted strand stabilities
A previous experimental study carried out by Lukin et al. examined the stability of ALII-N6-dA adducted DNA in three sequence contexts,30 two of which (CXG and CXC) were also considered in the present work. It was found that adducted DNA has a melting temperature (Tm) of 39.9 °C with the lesion in the CXG sequence context, but a Tm of 45.2 °C when the adduct is in the CXC sequence, and the Gibbs free energy of duplex formation is ∼7 kJ mol−1 more stable for CXC. Furthermore, Lukin et al. hypothesized that ALI-N6-dA and ALII-N6-dA adducted DNA will have similar conformational preferences due to the solvent exposure of ring I of the AL moiety in the most stable anti base-displaced intercalated conformer, which contains the methoxy group that differentiates the lesions.30 This proposal was substantiated by our previous computational work comparing the structure of the ALI and ALII-adducted DNA.29 Therefore, in the absence of analogous experimental data on the ALI-N6-dA adduct, it is anticipated that DNA containing either ALI-N6-dA or ALII-N6-dA will have a similar dependence of the strand stabilities on the sequence context. Although it is not possible to quantitatively compare the MM-PBSA free energies determined in the present study to the previously reported free energies of duplex formation, our calculations similarly predict that ALI-N6-dA adducted DNA is ∼9 kJ mol−1 more stable in the CXC than CXG sequence context (Table 2). This direct correlation between the trends in the sequence dependence of the strand stabilities illustrates the robustness of our computational approach.| Components of free energy | CXC | GXG | CXG | GXC |
|---|---|---|---|---|
| a Internal energy emerging from deviation of bonds (Ebond). b Internal energy emerging from deviation of angles (Eangle). c Internal energy emerging from deviation of dihedral angles (Edihedral). d van der Waals interaction energy (EvdW). e Electrostatic interaction energy (Eelec). f Solvation free energy (Gsol). g Entropy term (–TS). h Total free energy (Gtotal). i Relative total free energies with respect to the conformation with the lowest (most negative) Gtotal regardless of the sequence context (ΔGtotal). | ||||
|
E
bonda |
808.2 | 807.9 | 812.4 | 807.5 |
|
E
angleb |
1727.1 | 1723.6 | 1720.0 | 1722.3 |
|
E
dihedralc |
2085.5 | 2087.0 | 2100.8 | 2086.0 |
|
E
vdWd |
−813.0 | −830.8 | −821.6 | −814.8 |
|
E
elece |
1257.1 | 1351.8 | 1364.6 | 1227.6 |
|
G
Solf |
−22535.5 |
−22617.8 |
−22641.2 |
−22502.2 |
| −TSg | −2464.6 | −2461.3 | −2461.4 | −2464.0 |
|
G
totalh |
−19935.1 |
−19939.8 |
−19926.2 |
−19937.7 |
| ΔGtotali |
4.7 | 0.0 | 13.6 | 2.1 |
3.4. Biological consequences of the effects of sequence context on the conformational preferences of ALI-N6-dA adducted DNA
Since the overall composition of the ALI-N6-dA adducted DNA remains the same irrespective of the sequence context considered in the present work, the relative stabilities of the adducted DNA conformations across different sequences can be directly compared (ΔGtotal, Table 2). Our calculations reveal that the stability of the anti base-displaced intercalated conformer varies with sequence context according to GXG > GXC > CXC > CXG. Hence, the anti base displaced intercalated conformer of ALI-N6-dA adduct is not the most stable in the CXG sequence, but rather falls at least ∼14 kJ mol−1 higher in energy than the most stable GXG sequence. Therefore, the experimentally-observed higher frequency of A → T mutations associated with the CXG sequence is not thermodynamically driven by the stability of the mutagenic ALI-N6-dA adducted DNA conformation(s). Instead, other factors must dictate the sequence-dependent mutagenicity of AL-N6-dA. For example, interactions between the adduct and the TLS polymerase could be important. Indeed, such interactions were determined to be responsible for the sequence-dependent mutagenicity of PhIP-dG.70 Alternatively, a higher repair resistance of the most abundant ALI-N6-dA adduct in the CXG sequence may play a role in the sequence-dependent mutagenicity, which is considered in the next section.
Although greater lesion site dynamics and an associated higher GGR propensity in the TXT than CXC sequence context has been reported for 10S (+)-trans-anti-B[a]P-N2-dG PAH adducted DNA,42,47 the lesion site dynamics remain unaltered in all base-displaced intercalated conformers of ALI-N6-dA adducted DNA compared to the corresponding unmodified DNA regardless of the sequence (Table S4, ESI†). In addition, although a decrease in the lesion van der Waals (stacking) energy when AF-dG is at G2 compared to G1 or G3 in the NarI sequence explains the observed higher adduct repair propensity at G2,48 the lesion van der Waals interactions in the most persistent anti base-displaced intercalated conformer of ALI-N6-dA adducted DNA is only 0.5 kJ mol−1 more stabilizing for the most mutagenic CXG sequence compared to the non-mutagenic GXG sequence (Table 1). Hence, neither dynamics nor the lesion site van der Waals energy likely afford a sequence-dependent GGR propensity for ALI-N6-dA. Nevertheless, sequence-dependent distortions have also been shown to affect the GGR propensity of DNA adducts, including AAF-dG,48 10S (+)-trans-anti-B[a]P-dA41 and 10S (+)-trans-anti-B[a]P-dG.47 In the case of ALI-N6-dA adducted DNA, the magnitude of the distortion at the lesion site also varies with the sequence (Fig. 5). Specifically, the most persistent anti base-displaced intercalated conformer in the most mutagenic CXG sequence27,28 exhibits the least lesion site distortions, with the minor groove width changing by 0.4 Å and the helix untwisting by ∼6° relative to undamaged DNA. In contrast, the most persistent adducted DNA conformers in the other sequence contexts show minor groove widening of up to 0.8 Å and helix untwisting of up to 9° (Fig. 5 and Table S4, ESI†). These differences point toward the possibility of a differential repair propensity that is consistent with the experimentally-observed greater mutagenicity in the CXG sequence. Nevertheless, the structural differences between the adducted DNA conformers are small, falling within the dynamics of the natural helix (Table S4, ESI†), and may not be sufficient to lead to different repair susceptibilities. Therefore, other factors (such as interactions between the adducted DNA and lesion-bypass polymerases during replication) likely have a greater influence on the observed mutagenic profile of the ALI-N6-dA adduct and additional biochemical experiments are required to further explore the toxicity of this important lesion.
 | ||
| Fig. 5 The pseudostep parameters and minor groove dimensions for the anti base-displaced intercalated confomers of ALI-N6-dA adducted DNA in the (a) CXC, (b) GXG, (c) CXG and (d) GXC sequence contexts, as well as the values for the corresponding unmodified strand. | ||
4. Conclusions
The present computational study provides structural details of the possible conformers of ALI-N6-dA adducted DNA in different sequence contexts (namely, GXC, GXG, CXG and CXC). Our calculations reveal that the conformational outcomes of ALI-N6-dA adducted DNA are independent of the identity of the flanking bases. Specifically, the anti base-displaced intercalated conformer is the lowest energy conformer in all sequences considered. Our analysis reveals that the experimentally-observed greater mutagenicity of the CXG sequence is not thermodynamically driven by the stability of the adducted DNA as reported for other adducts. However, comparison of the structural perturbations upon lesion formation in different sequence contexts indicates that less stable lesion site van der Waals (stacking) interactions and greater distortions likely lead to a dependence of the adduct repair propensity on the lesion site sequence context. Most importantly, ALI-N6-dA adducted DNA in the CXG sequence context is the least distorted helix, which might contribute to a greater repair resistance that is consistent with the experimentally-observed higher mutagenicity in this sequential motif. Nevertheless, since the differences in distortions are small across different sequence contexts, other factors (such as interactions with the lesion-bypass polymerases) likely more greatly contribute to the experimentally-observed sequence-dependent mutagenicity of this adduct.Conflict of interest
The authors declare no competing financial interest.Abbreviations
| dA | 2′-Deoxyadenosine |
| dG | 2′-Deoxyguanosine |
| MD | Molecular dynamics |
| AA | Aristolochic acid |
| AL | Aristolactam |
| ALII-N6-dA | Aristolactam II-dA adduct |
| ALI-N6-dA | Aristolactam I-dA adduct |
| RMSD | Root mean square deviation |
| QM | Quantum mechanical |
| DFT | Density functional theory |
| AAF-dG | Acetylaminofluorene-dG adduct |
| AF-dG | Aminofluorene-dG adduct |
| PAH | Polycyclic aromatic hydrocarbons |
| OTB-dG | C-linked ochratoxin-dG adduct |
| PhOH-dG | Phenoxl-dG adduct |
| GAFF | General AMBER force field |
| IARC | International agency for research on cancer |
| NER | Nucleotide excision repair |
| AMBER | Assisted model building and energy refinement |
| B[a]P | Benzo[a]pyrene |
| DB[a,l]P | Dibenzo[a,l]pyrene |
| IQ | 2-Amino-3-methylimidazo[4,5-f]quinolone |
| PhIP | 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine |
Acknowledgements
Calculations were conducted on the New Up-scale Cluster for Lethbridge to Enable Innovative Chemistry (NUCLEIC). Additional computational resources were provided by WestGrid and Compute/Calcul Canada. Financial support for this research was provided by Canada Foundation for Innovation (CFI), Natural Sciences and Engineering Research Council (NSERC) of Canada, the Canada Research Chair (CRC) program, and the University of Lethbridge Research Fund. P. K. thanks the University of Lethbridge for financial support. P. S. thanks the Department of Science and Technology (DST), Government of India for financial support under the DST-INSPIRE Faculty scheme (no. DST/INSPIRE/04/2014/001855).References
- V. M. Arlt, M. Stiborova and H. H. Schmeiser, Mutagenesis, 2002, 17, 265–277 CrossRef CAS PubMed.
- J. L. Vanherweghem, C. Tielemans, D. Abramowicz, M. Depierreux, R. Vanhaelen-Fastre, M. Vanhaelen, M. Dratwa, C. Richard, D. Vandervelde, D. Verbeelen and M. Jadoul, Lancet, 1993, 341, 387–391 CrossRef CAS.
- F. D. Debelle, J. L. Vanherweghem and J. L. Nortier, Aristolochic acid nephropathy: A worldwide problem, Kidney Int., 2008, 74, 158–169 CrossRef CAS PubMed.
- J. P. Cosyns, M. Jadoul, J. P. Squifflet, F. X. Wese and C. van Ypersele de Strihou, Am. J. Kidney Dis., 1999, 33, 1011–1017 CrossRef CAS PubMed.
- J. L. Nortier and J. L. Vanherweghem, Toxicology, 2002, 181–182, 577–580 CrossRef CAS PubMed.
- J. L. Nortier, M. C. M. Martinez, H. H. Schmeiser, V. M. Arlt, C. A. Bieler, M. Petein, M. F. Depierreux, L. De Pauw, D. Abramowicz, P. Vereerstraeten and J. L. Vanherweghem, N. Engl. J. Med., 2000, 342, 1686–1692 CrossRef CAS PubMed.
- V. Batuman, Kidney Int., 2006, 669, 644–646 CrossRef PubMed.
- L. Djukanović and Z. Radovanović, Balkan endemic nephropathy, in Clinical Nephrotoxins, ed. M. de Broe, G. Porter, W. Bennett and G. Verpooten, Springer, Netherlands, 2003, pp. 587–601 Search PubMed.
- M. E. De Broe, Kidney Int., 2012, 81, 513–515 CrossRef CAS PubMed.
- V. Stefanovic and Z. Radovanovic, Nat. Clin. Pract. Urol., 2008, 5, 105–112 CrossRef PubMed.
- M. Ivić, Lijec. Vjesn., 1969, 91, 1273 Search PubMed.
- A. P. Grollman, S. Shibutani, M. Moriya, F. Miller, L. Wu, U. Moll, N. Suzuki, A. Fernandes, T. Rosenquist, Z. Medverec, K. Jakovina, B. Brdar, N. Slade, R. Turesky, A. K. Goodenough, R. Rieger, M. Vukelić and B. Jelaković, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 12129–12134 CrossRef CAS PubMed.
- V. M. Arlt, V. Alunni-Perret, G. Quatrehomme, P. Ohayon, L. Albano, H. Gaïd, J. F. Michiels, A. Meyrier, E. Cassuto, M. Wiessler, H. H. Schmeiser and J. P. Cosyns, Int. J. Cancer, 2004, 111, 977–980 CrossRef CAS PubMed.
- IARC monographs-100 A Plants containing aristolochic acids, 2002, pp. 347–361 Search PubMed.
- H. H. Schmeiser, C. A. Bieler, M. Wiessler, C. van Ypersele de Strihou and J. P. Cosyns, Cancer Res., 1996, 56, 2025–2028 CAS.
- V. S. Sidorenko, S. Attaluri, I. Zaitseva, C. R. Iden, K. Dickman, F. Johnson and A. P. Grollman, Carcinogenesis, 2014, 35, 1814–1822 CrossRef PubMed.
- M. Stiborová, J. Mareîs, E. Frei, V. M. Arlt, V. Martínek and H. H. Schmeiser, Environ. Mol. Mutagen., 2011, 52, 448–459 CrossRef PubMed.
- W. Pfau, H. H. Schmeiser and M. Wiessler, Carcinogenesis, 1990, 11, 313–319 CrossRef CAS PubMed.
- S. Attaluri, R. R. Bonala, I. Y. Yang, M. A. Lukin, Y. Wen, A. P. Grollman, M. Moriya, C. R. Iden and F. Johnson, Nucleic Acids Res., 2010, 38, 339–352 CrossRef CAS PubMed.
- V. Martinek, B. Kubickova, V. M. Arlt, E. Frei, H. H. Schmeiser, J. Hudecek and M. Stiborova, Neuroendocrinol. Lett., 2010, 32, 57–70 Search PubMed.
- C. A. Bieler, M. Stiborova, M. Wiessler, J. P. Cosyns, C. van Ypersele de Strihou and H. H. Schmeiser, Carcinogenesis, 1997, 18, 1063–1067 CrossRef CAS PubMed.
- W. Pfau, H. H. Schmeiser and M. Wiessler, Carcinogenesis, 1990, 11, 1627–1633 CrossRef CAS PubMed.
- H. H. Schmeiser, J. L. Nortier, R. Singh, G. Gamboa da Costa, J. Sennesael, E. Cassuto-Viguier, D. Ambrosetti, S. Rorive, A. Pozdzik and D. H. Phillips, Int. J. Cancer, 2014, 135, 502–507 CrossRef CAS.
- S. Shibutani, H. Dong, N. Suzuki, S. Ueda, F. Miller and A. P. Grollman, Drug Metab. Dispos., 2007, 35, 1217–1222 CrossRef CAS PubMed.
- M. Moriya, N. Slade, B. Brdar, Z. Medverec, K. Tomic, B. Jelaković, L. Wu, S. Truong, A. Fernandes and A. P. Grollman, Int. J. Cancer, 2011, 129, 1532–1536 CrossRef CAS PubMed.
- C. H. Chen, K. G. Dickman, M. Moriya, J. Zavadil, V. S. Sidorenko, K. L. Edwards, D. V. Gnatenko, L. Wu, R. J. Turesky, X. R. Wu, Y. S. Pu and A. P. Grollman, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 8241–8246 CrossRef CAS PubMed.
- S. L. Poon, S. T. Pang, J. R. McPherson, W. Yu, K. K. Huang, P. Guan, W. H. Weng, E. Y. Siew, Y. Liu, H. L. Heng, S. C. Chong, A. Gan, S. T. Tay, W. K. Lim, I. Cutcutache, D. Huang, L. D. Ler, M. L. Nairismägi, M. H. Lee, Y. H. Chang, K. J. Yu, W. Chan-on, B. K. Li, Y. F. Yuan, C. N. Qian, K. F. Ng, C. F. Wu, C. L. Hsu, R. M. Bunte, M. R. Stratton, P. A. Futreal, W. K. Sung, C. K. Chuang, C. K. Ong, S. G. Rozen, P. Tan and B. T. Teh, Sci. Transl. Med., 2013, 5, 197ra101 CrossRef PubMed.
- M. L. Hoang, C. H. Chen, V. S. Sidorenko, J. He, K. G. Dickman, B. H. Yun, M. Moriya, N. Niknafs, C. Douville, R. Karchin, R. J. Turesky, Y. S. Pu, B. Vogelstein, N. Papadopoulos, A. P. Grollman, K. W. Kinzler and T. A. Rosenquist, Sci. Transl. Med., 2013, 5, 197ra102 CrossRef PubMed.
- P. Kathuria, P. Sharma, M. Abendong and S. D. Wetmore, Biochemistry, 2015, 54, 2414–2428 CrossRef CAS PubMed.
- M. Lukin, T. Zaliznyak, F. Johnson and C. de los Santos, Nucleic Acids Res., 2012, 40, 2759–2770 CrossRef CAS PubMed.
- Y. Cai, S. Ding, N. E. Geacintov and S. Broyde, Chem. Res. Toxicol., 2011, 24, 522–531 CrossRef CAS PubMed.
- Z. Li, H. Y. Kim, P. J. Tamura, C. Harris, T. M. Harris and M. P. Stone, Biochemistry, 1999, 38, 16045–16057 CrossRef CAS PubMed.
- Z. Li, H. Mao, H. Y. Kim, P. J. Tamura, C. M. Harris, T. M. Harris and M. P. Stone, Biochemistry, 1999, 38, 2969–2981 CrossRef CAS PubMed.
- Z. Li, P. J. Tamura, A. S. Wilkinson, C. M. Harris, T. M. Harris and M. P. Stone, Biochemistry, 2001, 40, 6743–6755 CrossRef CAS PubMed.
- I. S. Zegar, P. Chary, R. J. Jabil, P. J. Tamura, T. N. Johansen, R. S. Lloyd, C. M. Harris, T. M. Harris and M. P. Stone, Biochemistry, 1998, 37, 16516–16528 CrossRef CAS PubMed.
- F. Wang, C. E. Elmquist, J. S. Stover, C. J. Rizzo and M. P. Stone, Biochemistry, 2007, 46, 8498–8516 CrossRef CAS PubMed.
- B. Mao, B. E. Hingerty, S. Broyde and D. J. Patel, Biochemistry, 1998, 37, 95–106 CrossRef CAS PubMed.
- B. Mao, B. E. Hingerty, S. Broyde and D. J. Patel, Biochemistry, 1998, 37, 81–94 CrossRef CAS PubMed.
- S. Patnaik and B. P. Cho, Chem. Res. Toxicol., 2010, 23, 1650–1652 CrossRef CAS PubMed.
- K. Kropachev, M. Kolbanovskii, Y. Cai, F. Rodríguez, A. Kolbanovskii, Y. Liu, L. Zhang, S. Amin, D. Patel, S. Broyde and N. E. Geacintov, J. Mol. Biol., 2009, 386, 1193–1203 CrossRef CAS PubMed.
- S. Yan, M. Wu, T. Buterin, H. Naegeli, N. E. Geacintov and S. Broyde, Biochemistry, 2003, 42, 2339–2354 CrossRef CAS PubMed.
- Y. Cai, D. J. Patel, N. E. Geacintov and S. Broyde, J. Mol. Biol., 2007, 374, 292–305 CrossRef CAS PubMed.
- F. A. Rodríguez, Y. Cai, C. Lin, Y. Tang, A. Kolbanovskiy, S. Amin, D. J. Patel, S. Broyde and N. E. Geacintov, Nucleic Acids Res., 2007, 35, 1555–1568 CrossRef PubMed.
- P. Sharma, R. A. Manderville and S. D. Wetmore, Nucleic Acids Res., 2014, 42, 11831–11845 CrossRef PubMed.
- P. Sharma, R. A. Manderville and S. D. Wetmore, Chem. Res. Toxicol., 2013, 26, 803–816 CrossRef CAS PubMed.
- R. P. P. Fuchs, N. Schwartz and M. P. Daune, Nature, 1981, 294, 657–659 CrossRef CAS PubMed.
- Y. Cai, D. J. Patel, S. Broyde and N. E. Geacintov, J. Nucleic Acids, 2010, 2010, 9 Search PubMed.
- H. Mu, K. Kropachev, L. Wang, L. Zhang, A. Kolbanovskiy, M. Kolbanovskiy, S. Broyde and N. E. Geacintov, Nucleic Acids Res., 2012, 40, 9675–9690 CrossRef CAS PubMed.
- V. Jain, B. Hilton, S. Patnaik, Y. Zou, M. P. Chiarelli and B. P. Cho, Nucleic Acids Res., 2012, 40, 3939–3951 CrossRef CAS PubMed.
- Y. Cai, N. E. Geacintov and S. Broyde, Biochemistry, 2012, 51, 1486–1499 CrossRef CAS PubMed.
- V. Jain, B. Hilton, B. Lin, S. Patnaik, F. Liang, E. Darian, Y. Zou, A. D. MacKerell and B. P. Cho, Nucleic Acids Res., 2013, 41, 869–880 CrossRef CAS PubMed.
- S. Meneni, S. M. Shell, Y. Zou and B. P. Cho, Chem. Res. Toxicol., 2007, 20, 6–10 CrossRef CAS PubMed.
- C. P. Rubbi and J. Milner, Carcinogenesis, 2001, 22, 1789–1796 CrossRef CAS PubMed.
- W. L. de Laat, N. G. J. Jaspers and J. H. J. Hoeijmakers, Genes Dev., 1999, 13, 768–785 CrossRef CAS PubMed.
- L. C. J. Gillet and O. D. Schärer, Chem. Rev., 2005, 106, 253–276 CrossRef PubMed.
- P. C. Hanawalt and G. Spivak, Transcription-Coupled DNA repair: Two Decades of Progress and Surprises, Nat. Rev. Mol. Cell Biol., 2008, 9, 958–970 CrossRef CAS PubMed.
- K. Kropachev, M. Kolbanovskiy, Z. Liu, Y. Cai, L. Zhang, A. G. Schwaid, A. Kolbanovskiy, S. Ding, S. Amin, S. Broyde and N. E. Geacintov, Chem. Res. Toxicol., 2013, 26, 783–793 CrossRef CAS PubMed.
- Y. Cai, D. J. Patel, S. Broyde and N. E. Geacintov, J. Mol. Biol., 2009, 385, 30–44 CrossRef CAS PubMed.
- B. Hang, J. Nucleic Acids, 2010, 2010, 29 Search PubMed.
- V. S. Sidorenko, J. E. Yeo, R. R. Bonala, F. Johnson, O. D. Schärer and A. P. Grollman, Nucleic Acids Res., 2012, 40, 2494–2505 CrossRef CAS PubMed.
- K. Kropachev, S. Ding, M. A. Terzidis, A. Masi, Z. Liu, Y. Cai, M. Kolbanovskiy, C. Chatgilialoglu, S. Broyde, N. E. Geacintov and V. Shafirovich, Nucleic Acids Res., 2014, 42, 5020–5032 CrossRef CAS PubMed.
- O. Maillard, U. Camenisch, F. C. Clement, K. B. Blagoev and H. Naegeli, Trends Biochem. Sci., 2007, 32, 494–499 CrossRef CAS PubMed.
- K. B. Blagoev, B. S. Alexandrov, E. H. Goodwin and A. R. Bishop, DNA Repair, 2006, 5, 863–867 CrossRef CAS PubMed.
- R. J. Isaacs and H. P. Spielmann, DNA Repair, 2004, 3, 455–464 CrossRef CAS PubMed.
- X. Chen, Y. Velmurugu, G. Zheng, B. Park, Y. Shim, Y. Kim, L. Liu, B. Van Houten, C. He, A. Ansari and J. H. Min, Nat. Commun., 2015, 6 DOI:10.1038/ncomms6849.
- D. A. Reeves, H. Mu, K. Kropachev, Y. Cai, S. Ding, A. Kolbanovskiy, M. Kolbanovskiy, Y. Chen, J. Krzeminski and S. Amin, Nucleic Acids Res., 2011, 39, 8752–8764 CrossRef CAS PubMed.
- W. Yang, DNA Repair, 2006, 5, 654–666 CrossRef CAS PubMed.
- A. Janićijević, K. Sugasawa, Y. Shimizu, F. Hanaoka, N. Wijgers, M. Djurica, J. H. J. Hoeijmakers and C. Wyman, DNA Repair, 2003, 2, 325–336 CrossRef.
- V. G. Vaidyanathan and B. P. Cho, Biochemistry, 2012, 51, 1983–1995 CrossRef CAS PubMed.
- P. Xu, L. Oum, Y. C. Lee, N. E. Geacintov and S. Broyde, Biochemistry, 2009, 48, 4677–4690 CrossRef CAS PubMed.
- S. Shibutani, A. Fernandes, N. Suzuki, L. Zhou, F. Johnson and A. P. Grollman, J. Biol. Chem., 1999, 274, 27433–27438 CrossRef CAS PubMed.
- V. Jain, V. G. Vaidyanathan, S. Patnaik, S. Gopal and B. P. Cho, Biochemistry, 2014, 53, 4059–4071 CrossRef CAS PubMed.
- D. A. Case, T. A. Darden, T. E. Cheatham, C. L. Simmerling, J. Wang, R. E. Duke, R. Luo, M. Crowley, R. C. Walker, W. Zhang, K. M. Merz, B. Wang, S. Hayik, A. Roitberg, G. Seabra, I. Kolossvary, K. F. Wong, F. Paesani, J. Vanicek, X. Wu, S. R. Brozell, T. Steinbrecher, H. Gohlke, L. Yang, C. Tan, J. Mongan, V. Hornak, G. Cui, D. H. Mathews, M. G. Seetin, C. Sagui, V. Babin and P. A. Kollman, AMBER Tools, University of California, San Francisco, 2008 Search PubMed.
- D. A. Case, T. A. Darden, T. E. Cheatham, C. L. Simmerling, J. Wang, R. E. Duke, R. Luo, M. Crowley, R. C. Walker, W. Zhang, K. M. Merz, B. Wang, S. Hayik, A. Roitberg, G. Seabra, I. Kolossvary, K. F. Wong, F. Paesani, J. Vanicek, X. Wu, S. R. Brozell, T. Steinbrecher, H. Gohlke, L. Yang, C. Tan, J. Mongan, V. Hornak, G. Cui, D. H. Mathews, M. G. Seetin, C. Sagui, V. Babin and P. A. Kollman, AMBER 11, University of California, San Francisco, CA, 2010 Search PubMed.
- D. T. Roy and J. M. Keith, GaussView Version 5, 2009 Search PubMed.
- D. A. Case, T. E. Cheatham, T. Darden, H. Gohlke, R. Luo, K. M. Merz, A. Onufriev, C. Simmerling, B. Wang and R. J. Woods, J. Comput. Chem., 2005, 26, 1668–1688 CrossRef CAS PubMed.
- D. A. Case, T. A. Darden, T. E. Cheatham, C. L. Simmerling, J. Wang, R. E. Duke, R. Luo, R. C. Walker, W. Zhang, K. M. Merz, B. Roberts, S. Hayik, A. Roitberg, G. Seabra, J. Swails, A. W. Goetz, I. Kolossváry, K. F. Wong, F. Paesani, J. Vanicek, R. M. Wolf, J. Liu, X. Wu, S. R. Brozell, T. Steinbrecher, H. Gohlke, Q. Cai, X. Ye, J. Wang, M. J. Hsieh, G. Cui, D. R. Roe, D. H. Mathews, M. G. Seetin, R. Salomon-Ferrer, C. Sagui, V. Babin, T. Luchko, S. Gusarov, A. Kovalenko and P. A. Kollman, AMBER 12, University of California, San Francisco, CA, 2012 Search PubMed.
- B. R. Miller, T. D. McGee, J. M. Swails, N. Homeyer, H. Gohlke and A. E. Roitberg, J. Chem. Theory Comput., 2012, 8, 3314–3321 CrossRef CAS.
- S. Broyde, L. Wang, L. Zhang, O. Rechkoblit, N. E. Geacintov and D. J. Patel, Chem. Res. Toxicol., 2007, 21, 45–52 CrossRef PubMed.
- M. Sproviero, A. M. R. Verwey, K. M. Rankin, A. A. Witham, D. V. Soldatov, R. A. Manderville, M. I. Fekry, S. J. Sturla, P. Sharma and S. D. Wetmore, Nucleic Acids Res., 2014, 42, 13405–13421 CrossRef PubMed.
- J. S. Mei Kwei, I. Kuraoka, K. Horibata, M. Ubukata, E. Kobatake, S. Iwai, H. Handa and K. Tanaka, Biochem. Biophys. Res. Commun., 2004, 320, 1133–1138 CrossRef PubMed.
- A. Dimitri, A. K. Goodenough, F. P. Guengerich, S. Broyde and D. A. Scicchitano, J. Mol. Biol., 2008, 375, 353–366 CrossRef CAS PubMed.
- G. E. Damsma, A. Alt, F. Brueckner, T. Carell and P. Cramer, Nat. Struct. Mol. Biol., 2007, 14, 1127–1133 CAS.
- F. Brueckner, U. Hennecke, T. Carell and P. Cramer, Science, 2007, 315, 859–862 CrossRef CAS PubMed.
- W. Vermeulen and M. Fousteri, Mammalian Transcription-Coupled Excision Repair, Cold Spring Harbor Perspect. Biol., 2013, 5, a012625 Search PubMed.
- N. E. Geacintov, S. Broyde, T. Buterin, H. Naegeli, M. Wu, S. Yan and D. J. Patel, Biopolymers, 2002, 65, 202–210 CrossRef CAS PubMed.
- O. Rechkoblit, A. Kolbanovskiy, L. Malinina, N. E. Geacintov, S. Broyde and D. J. Patel, Nat. Struct. Mol. Biol., 2010, 17, 379–388 CAS.
- O. D. Schärer, Mol. Cell, 2007, 28, 184–186 CrossRef PubMed.
- K. Sugasawa, T. Okamoto, Y. Shimizu, C. Masutani, S. Iwai and F. Hanaoka, Genes Dev., 2001, 15, 507–521 CrossRef CAS PubMed.
- R. Jankowiak, F. Ariese, A. Hewer, A. Luch, D. Zamzow, N. C. Hughes, D. Phillips, A. Seidel, K.-L. Platt and D. Oesch, Chem. Res. Toxicol., 1998, 11, 674–685 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Radar plots for the probability distributions of χ, θ and ϕ (Fig. S1–S3); RMSD of all heavy atoms in the lesion site trimer in the base-displaced intercalated adducted DNA conformations during 20 and 320 ns MD simulations in the CXC sequence context (Table S1); backbone RMSD for all adducted DNA conformations in different sequence contexts (Table S2); the hydrogen-bonding occupancies in the trimers composed of the damaged base pair and the 3′ and 5′-flanking base pairs in different sequence contexts (Table S3); pseudostep parameters for all adducted DNA conformers in different sequence contexts (Table S4); interaction energies between the damaged base and its opposing thymine for different base-displaced intercalated conformations of ALI-N6-dA adducted DNA (Table S5). See DOI: 10.1039/c5tx00302d |
| This journal is © The Royal Society of Chemistry 2016 |