
Identification of Id1 as a downstream effector for arsenic-promoted angiogenesis via PI3K/Akt, NF-κB and NOS signaling†
Chun-Hao
Tsai‡
ab,
Ming-Hui
Yang‡
ac,
Amos C.
Hung
a,
Shou-Cheng
Wu
d,
Wen-Chin
Chiu
e,
Ming-Feng
Hou
fg,
Yu-Chang
Tyan
ahij,
Yun-Ming
Wang
*d and
Shyng-Shiou F.
Yuan
*abcklm
aTranslational Research Center, Kaohsiung Medical University Hospital, Kaohsiung Medical University, Kaohsiung, Taiwan
bGraduate Institute of Medicine, College of Medicine, Kaohsiung Medical University, Kaohsiung, Taiwan. E-mail: yuanssf@ms33.hinet.net; Tel: +886-7-3121101 Ext2557
cDepartment of Medical Research, Kaohsiung Medical University Hospital, Kaohsiung Medical University, Kaohsiung, Taiwan
dDepartment of Biological Science and Technology and Institute of Molecular Medicine and Bioengineering, National Chiao Tung University, Hsinchu, Taiwan. E-mail: ymwang@mail.nctu.edu.tw; Tel: +886-3-5712121 Ext56972
eDivision of Thoracic Surgery, Department of Surgery, Kaohsiung Medical University Hospital, Kaohsiung Medical University, Kaohsiung, Taiwan
fCancer Center, Kaohsiung Medical University Hospital, Kaohsiung Medical University, Kaohsiung, Taiwan
gDepartment of Surgery, Kaohsiung Municipal Ta-Tung Hospital, Kaohsiung, Taiwan
hDepartment of Medical Imaging and Radiological Sciences, Kaohsiung Medical University, Kaohsiung, Taiwan
iCenter for Infectious Disease and Cancer Research, Kaohsiung Medical University, Kaohsiung, Taiwan
jInstitute of Medical Science and Technology, National Sun Yat-sen University, Kaohsiung, Taiwan
kDepartment of Obstetrics and Gynecology, Kaohsiung Medical University Hospital, Kaohsiung Medical University, Kaohsiung, Taiwan
lFaculty and College of Medicine, Kaohsiung Medical University, Kaohsiung, Taiwan
mCenter for Lipid and Glycomedicine Research, Kaohsiung Medical University, Kaohsiung, Taiwan
First published on 5th October 2015
Abstract
Exposure to arsenic is known to be a risk factor for various types of cancer. Apart from its carcinogenic activity, arsenic also shows promoting effects on angiogenesis, a crucial process for tumor growth. Yet, the mechanism underlying arsenic-induced angiogenesis is not fully understood. In this study, we aimed at investigating the involvement of inhibitor of DNA binding 1 (Id1) and the associated signal molecules in the arsenic-mediated angiogenesis. Our initial screening revealed that treatment with low concentrations of arsenic (0.5–1 μM) led to multiple cellular responses, including enhanced endothelial cell viability and angiogenic activity as well as increased protein expression of Id1. The arsenic-induced angiogenesis was suppressed in the Id1-knocked down cells compared to that in control cells. Furthermore, arsenic-induced Id1 expression and angiogenic activity were regulated by PI3K/Akt, NF-κB, and nitric oxide synthase (NOS) signaling. In summary, our current data demonstrate for the first time that Id1 mediates the arsenic-promoted angiogenesis, and Id1 may be regarded as an antiangiogenesis target for treatment of arsenic-associated cancer.
Introduction
Arsenic (As) is a metalloid widely distributed in nature via its multiple forms in association with other elements.1 Despite an ecological role, arsenic exposure has been shown to be pernicious to human health.2 For instance, intake of arsenic-contaminated drinking water, food, or air causes renal and urinary dysfunction and elevated cancer incidence.3–5 Exposure to arsenic during early gestation is also adversely correlated with long-term health issues, including increased incidence of carcinogenesis in later life.6–8Angiogenesis is implicated in many pathological conditions such as rheumatoid arthritis,9 wound healing,10 cerebral ischemia,11 and cardiovascular or peripheral artery diseases.12,13 In addition, tumorigenesis is largely dependent on neovascular formation, and therefore antiangiogenic therapy has become one of the main therapeutic strategies to control tumor growth.14,15 While most of the on-market antiangiogenic agents target the vascular endothelial growth factor (VEGF) and its downstream signaling cascade, the clinical response to these agents is modest and new targeted therapies remain to be developed.16
Inhibitor of DNA binding 1 (Id1), also known as inhibitor of differentiation 1, is a member of basic helix-loop-helix (bHLH) transcription factor proteins.17,18 Structurally, Id1 lacks the basic region that is adjacent to the HLH domain and essential for DNA binding.17 Without the capacity of DNA binding by itself, Id1 forms heterodimers with other bHLH transcription factors which regulate the expression of genes involved in cell proliferation and differentiation.18 Notably, upregulation of Id1 expression has been correlated with cancer progression, suggesting that Id1 may constitute an important target for anticancer therapeutics.19–22
It has been shown that arsenic exerts a dual effect on angiogenesis and carcinogenesis.23–25 Intriguingly, Id1 also plays a regulatory role in tumor angiogenesis.26–28 However, whether or not Id1 is involved in arsenic-associated angiogenesis has not been elucidated. In this study, we report that knockdown of Id1 expression compromises the arsenic-promoted angiogenesis. In addition, the arsenic-induced Id1 expression and angiogenesis are suppressed by inhibition of the PI3K/Akt, NF-κB and nitric oxide synthase (NOS) signaling pathway. Together, our current results provide a novel finding for the involvement of Id1 in arsenic-associated angiogenesis.
Materials and methods
Materials
Sodium arsenite, wortmannin, QNZ, DETA-NONOate, 1400W dihydrochloride, L-NAME hydrochloride, basic FGF, and heparin were purchased from Sigma-Aldrich (St Louis, MO, USA). Matrigel was purchased from BD Biosciences (San Jose, CA, USA). Dulbecco's modified Eagle's medium (DMEM) was purchased from Gibco (Carlsbad, CA, USA). Fetal bovine serum, penicillin, streptomycin, and amphotericin B were purchased from Biological Industries (Beit-Haemek, Israel). Other reagents employed in this study were indicated separately wherever suitable.Cell culture
Mouse pancreatic endothelial cells (MS1) and human umbilical vein endothelial cells (HUVEC) were obtained from the American Type Culture Collection (Manassas, VA, USA). MS1 cells were cultured in DMEM medium supplemented with 10% fetal bovine serum, 100 U mL−1 penicillin, 100 μg mL−1 streptomycin, and 0.25 μg mL−1 amphotericin B. HUVEC cells were cultured in EndoGRO-LS complete medium (Millipore, MA, USA). All cells were maintained at 37 °C in a 5% CO2 incubator.Cell viability assay
MS1 or HUVEC cells were seeded at a density of 3 × 103 cells per well into 96-well plates for 24 h prior to treatment with the indicated concentrations of arsenic. After 72 h of treatment, the number of viable cells was determined by the XTT assay (Sigma-Aldrich) according to the manufacturer's instructions.Western blot
Protein extracts of cells were prepared in lysis buffer (50 mM Tris-HCl (pH 7.6), 120 mM NaCl, 1 mM EDTA, 0.5% Nonidet P-40, 1 mM β-mercaptoethanol, 50 mM NaF, and 1 mM Na3VO4), followed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and immunoblotting analysis as described previously.29 The antibody against Id1 was purchased from Santa Cruz Biotechnology (Dallas, TX, USA). Antibodies against STAT3, Akt, phospho-Akt (Ser473), p65, phospho-p65 (Ser536), iNOS, and β-actin were from GeneTex (Irvine, CA, USA). Antibodies against phospho-STAT3 (Tyr705), phospho-STAT3 (Ser727), phosphor-eNOS (Ser1177), and eNOS were from Cell Signaling Technology (Danvers, MA, USA).In vitro angiogenic tube formation assay
The in vitro angiogenic tube formation assay was carried out according to a previous report.30 Briefly, the 48-well plate coated with growth factor-reduced Matrigel (150 μl per well) was allowed to polymerize at 37 °C for 60 min. Cells (3 × 104 cells per well) pretreated with arsenic for 24 h were then plated onto the well containing polymerized Matrigel. After 12 h of incubation, the morphology of cells was imaged using a Nikon Eclipse 80i microscope (Tokyo, Japan). The degree of tube formation in each group was estimated with the presence of total length analyzed by using ImageJ (http://rsbweb.nih.gov/ij/).Knockdown of Id1 protein expression
The short hairpin RNAs (shRNAs) targeting human Id1 (#1, 5′-CCTACTAGTCACCAGAGACTT-3′; #2, 5′-CTACGACATGAACGGCTGTTA-3′) were cloned into a pLKO.1 vector (Id1-pLKO.1) derived from the National RNAi Core Facility (Academia Sinica, Taiwan). A parental pLKO.1 vector without shRNA sequence was used as an empty vector control. Lentiviruses were prepared by transfecting three plasmids (the packing plasmid pCMV8.91, the envelope plasmid pMD.G, and either the shRNA plasmid Id1-pLKO.1 or the control plasmid pLKO.1) into 293 T cells using Lipofectamine 2000 (Invitrogen, CA, USA) as described previously.31 The MS1 cells were infected with the lentiviral particles containing either Id1-pLKO.1 or pLKO.1 collected from the corresponding cell culture medium.In vivo angiogenic Matrigel plug assay
For the in vivo Matrigel plug experiment in Fig. 1A, female immunodeficient mice (Foxn1nu/Foxn1nu) were injected subcutaneously with 500 μL Matrigel containing basic FGF (1 ng mL−1), heparin (10 U mL−1), and either arsenic (0.5 μM) or phosphate buffered saline (PBS) as the vehicle control. After 14 days, the mice were sacrificed and the Matrigel plugs were dissected out for the quantitation of hemoglobin by using Drabkin's reagent (Sigma-Aldrich) according to the manufacturer's instructions. Regarding the separate experiment of the in vivo Matrigel plug assay in Fig. 4C and D, the procedures were identical as described above except for that the Matrigel prepared likewise was additionally mixed with either 1 × 106 Id1-knocked down MS1 cells (Id1-KD) or with the same number of empty vector control MS1 cells (EV). The mice experiments carried out in this study were approved by the Institutional Animal Care and Use Committee of Kaohsiung Medical University.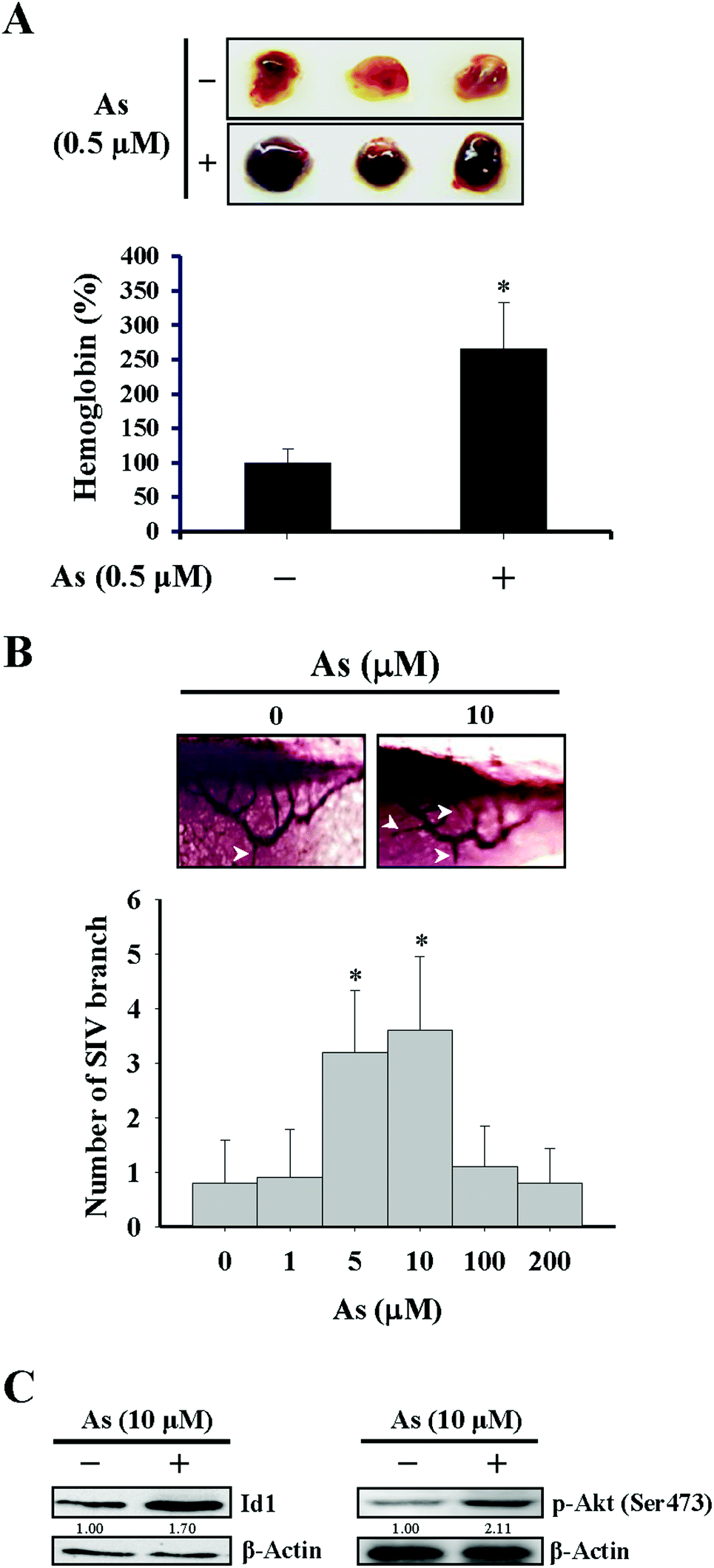 | ||
| Fig. 1 Effects of arsenic on in vivo angiogenesis. (A) Matrigels containing bovine FGF (1 ng mL−1), heparin (10 U mL−1), and either with or without arsenic (0.5 μM) were injected subcutaneously into the female immunodeficient mice. After 14 days, the Matrigel plugs were dissected out and the hemoglobin levels were determined by using Drabkin's reagent. (B) Embryos of zebrafish were incubated with different doses of arsenic for 72 h. After anesthetization, the larvae were stained and imaged, and the number of branches of sub-intestinal vessel (SIV) was quantitated. The arrows showed the typical appearance of the SIV branches under different conditions of arsenic treatment. *Significant difference of P < 0.05 compared with the untreated control by Student's t test. (C) Arsenic induced protein expression of Id1 and phospho-Akt (Ser473) in zebrafish. Embryos of zebrafish were incubated either with or without arsenic (10 μM) for 72 h. After anesthetization, the total proteins of larvae were extracted and subjected to western blot analysis. | ||
In vivo angiogenesis in the zebrafish model
The in vivo angiogenic assay using embryos of zebrafish was carried out according to a previous report.32 In brief, approximately 100 embryos were generated per pair of zebrafish via natural pairwise mating. The embryos were then incubated with the indicated concentrations of arsenic at 27 °C. After 72 h post-fertilization (hpf), the larvae were anesthetized with 0.5 g L−1 ethyl 3-aminobenzoate methanesulfonate (Sigma-Aldrich) for 30 min and fixed in 4% paraformaldehyde for 2 h, followed by staining for endogenous alkaline phosphatase activity. The branches of the sub-intestinal vessel (SIV) were imaged using a Nikon Eclipse 80i microscope. Experiments involving zebrafish in this study were approved by the Institutional Animal Care and Use Committee of Kaohsiung Medical University.Nitric oxide formation assay
Nitric oxide formation was estimated by the method of Griess reagent according to the procedures of Total Nitric Oxide and Nitrate/Nitrite Parameter Assay Kits (R&D Systems, MN, USA) and expressed as total nitrite/nitrate concentrations by comparing with a standard curve.Nitric oxide staining
MS1 cells in 12-well plates were treated with or without 0.5 μM As for 4 h followed by FA-OMe (10 μM) staining for 8 h according to a previous report33 and MitoTracker Red (Life Technologies) staining according to the manufacturer's instructions. Images were photographed using a Multiphoton and Confocal Microscope System (Leica, Germany) (FA-OMe ex/em: 460/524 nm; MitoTracker Red ex/em: 579/599 nm).Statistical analysis
Quantitative data were presented as mean ± SD. Two-sided Student's t test or one-way ANOVA with post-hoc Dunnett's test was used to determine the significant differences between different groups. P < 0.05 was considered statistically significantly different from at least three independent experiments.Results
Effects of arsenic on angiogenesis and Id1 expression
The effect of arsenic on cell viability was analyzed by the XTT assay in the mouse pancreatic endothelial cells (MS1) and human umbilical vein endothelial cells (HUVEC). As shown in ESI Fig. 1A,† low concentrations of arsenic (0.5–1 μM) promoted cell viability, while it was inhibited by a high concentration of arsenic (10 μM). The results suggested that there was a biphasic effect of arsenic on the endothelial cell viability.To evaluate the angiogenic activity of arsenic, the in vitro tube formation assay was employed. As shown in ESI Fig. 1B and C,† low concentrations of arsenic (0.5–1 μM) increased the tube formation, whereas a high concentration of arsenic (10 μM) reduced the tube formation in both MS1 and HUVEC cells, implying that arsenic also had a biphasic effect on the angiogenesis. Together, our data showed agreement with previous reports in that low concentrations promoted while high concentrations suppressed cell viability and angiogenesis by arsenic in endothelial cells.34–36
The effect of arsenic on in vivo angiogenesis was examined in a mouse model implanted with a Matrigel plug and in a zebrafish model. We observed that the level of hemoglobin from the plug containing arsenic in mice was significantly higher than that from the control plug (Fig. 1A), suggesting that new vascular formation was potentiated in the presence of arsenic. The other approach using zebrafish as a model showed that the number of branches of the sub-intestinal vessel (SIV) increased with 5–10 μM arsenic treatment and decreased with 100–200 μM arsenic treatment (Fig. 1B), in agreement with a recent paper that demonstrated a perturbed vascular development with a high dose arsenic treatment.37
Since Id1 plays a regulatory role for tumor angiogenesis,26–28 we next examined the role of Id1 and involved signaling in the arsenic-promoted angiogenesis. As shown in Fig. 2A and B, low concentrations of arsenic (0.5–1 μM) induced Id1 protein expression in MS1 cells. The protein levels of vascular endothelial growth factor (VEGF) were also increased under arsenic treatment (Fig. 2A). In addition, the phosphorylation of both Akt and NF-κB (p65 subunit) was enhanced by arsenic (Fig. 2A), while the expression of total form and phosphorylated STAT3, a transcription activator that may participate in tumor angiogenesis,38 was unaffected in the presence of arsenic (ESI Fig. 2A†). The arsenic-induced Id1 expression and Akt phosphorylation were also observed in an in vivo zebrafish model (Fig. 1C).
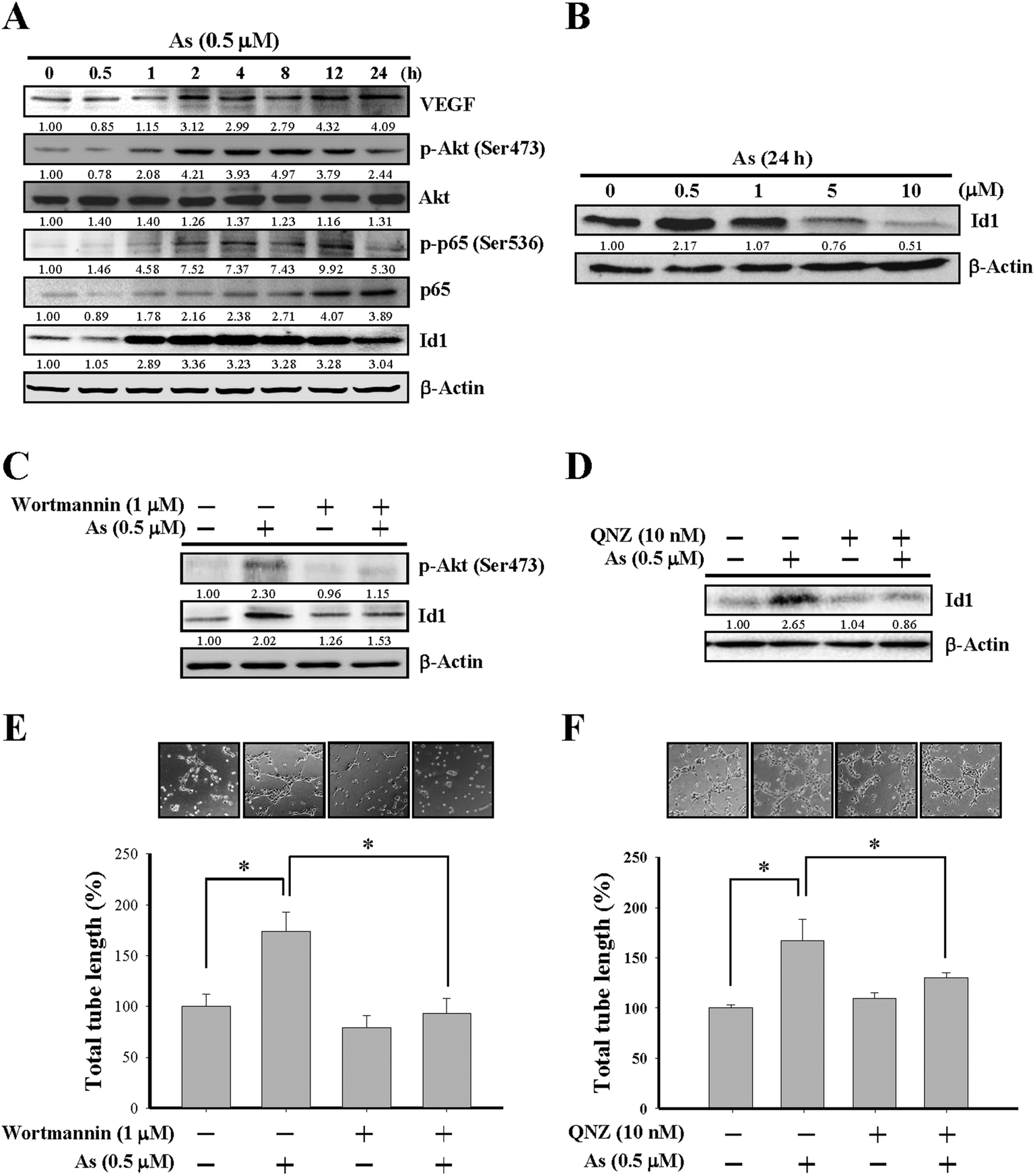 | ||
| Fig. 2 Involvement of PI3K/Akt signaling in arsenic-induced Id1 protein expression and in vitro angiogenesis. (A) MS1 cells were treated with arsenic (0.5 μM) for the indicated lengths of time, followed by western blot analysis for protein expression of VEGF, phospho-Akt (Ser473), Akt, phospho-p65 (Ser536), p65, and Id1. (B) MS1 cells were treated with different doses of arsenic for 24 h, followed by western blot analysis for Id1 protein expression. (C) MS1 cells were pretreated with or without the PI3K/Akt inhibitor wortmannin (1 μM) for 2 h, followed by arsenic treatment (0.5 μM) for 24 h. The protein expression of phospho-Akt (Ser473) and Id1 was analyzed by western blot. (D) MS1 cells were pretreated with or without NF-κB inhibitor QNZ (10 nM) for 2 h, followed by arsenic treatment (0.5 μM) for 24 h. The protein expression of Id1 was analyzed by western blot. (E) MS1 cells were pretreated with or without wortmannin (1 μM) for 2 h, followed by the in vitro tube formation assay in the presence or absence of arsenic (0.5 μM) for 24 h. (F) MS1 cells were pretreated with or without QNZ (10 nM) for 2 h, followed by the in vitro tube formation assay in the presence or absence of arsenic (0.5 μM) for 24 h. *Significant difference of P < 0.05 by one-way ANOVA with post-hoc Dunnett's test. | ||
Involvement of PI3K/Akt and NF-κB in arsenic-induced Id1 expression and tube formation
The involvement of PI3K/Akt and NF-κB in arsenic-induced Id1 expression was further examined by wortmannin, a PI3K/Akt inhibitor, and by QNZ, an inhibitor of NF-κB. As shown in Fig. 2C, the arsenic-induced Akt phosphorylation was inhibited in MS1 cells treated with wortmannin. Notably, the Id1 expression induced by arsenic was suppressed in the presence of wortmannin (Fig. 2C) or QNZ (Fig. 2D). Moreover, the arsenic-promoted in vitro tube formation was inhibited in the presence of wortmannin (Fig. 2E) or QNZ (Fig. 2F). These data suggested that the activation of PI3K/Akt and NF-κB signaling might play a regulatory role in the arsenic-induced Id1 expression and angiogenesis.Involvement of nitric oxide synthase (NOS) in arsenic-induced Id1 expression and tube formation
Although the effect of arsenic on nitric oxide production35,39 and eNOS activation40 were reported, the role of nitric oxide in Id1-mediated angiogenesis induced by arsenic has not been elucidated before. In the present study, MS1 cells treated with arsenic consistently showed an increased nitric oxide formation (Fig. 3A and ESI Fig. 2B†), and treatment with DETA-NONOate, a nitric oxide donor, was able to upregulate the Id1 expression and in vitro tube formation in these cells (Fig. 3B and C), implying that nitric oxide might play a role upstream of Id1 in this process. Furthermore, we found that treatment of arsenic increased the protein expression of phospho-eNOS (Ser1177) and iNOS (Fig. 3D). The involvement of nitric oxide in arsenic-stimulated Id1 expression and tube formation were further examined by two nitric oxide synthase inhibitors, L-NAME and 1400W. As shown in Fig. 3E and F, the arsenic-induced Id1 expression was suppressed in the presence of L-NAME and 1400W, respectively. In addition, the arsenic-promoted tube formation was inhibited by L-NAME (Fig. 3E) or 1400W (Fig. 3F). Collectively, the data suggested that the arsenic-induced Id1 expression and angiogenesis were mediated through a signaling pathway involving nitric oxide.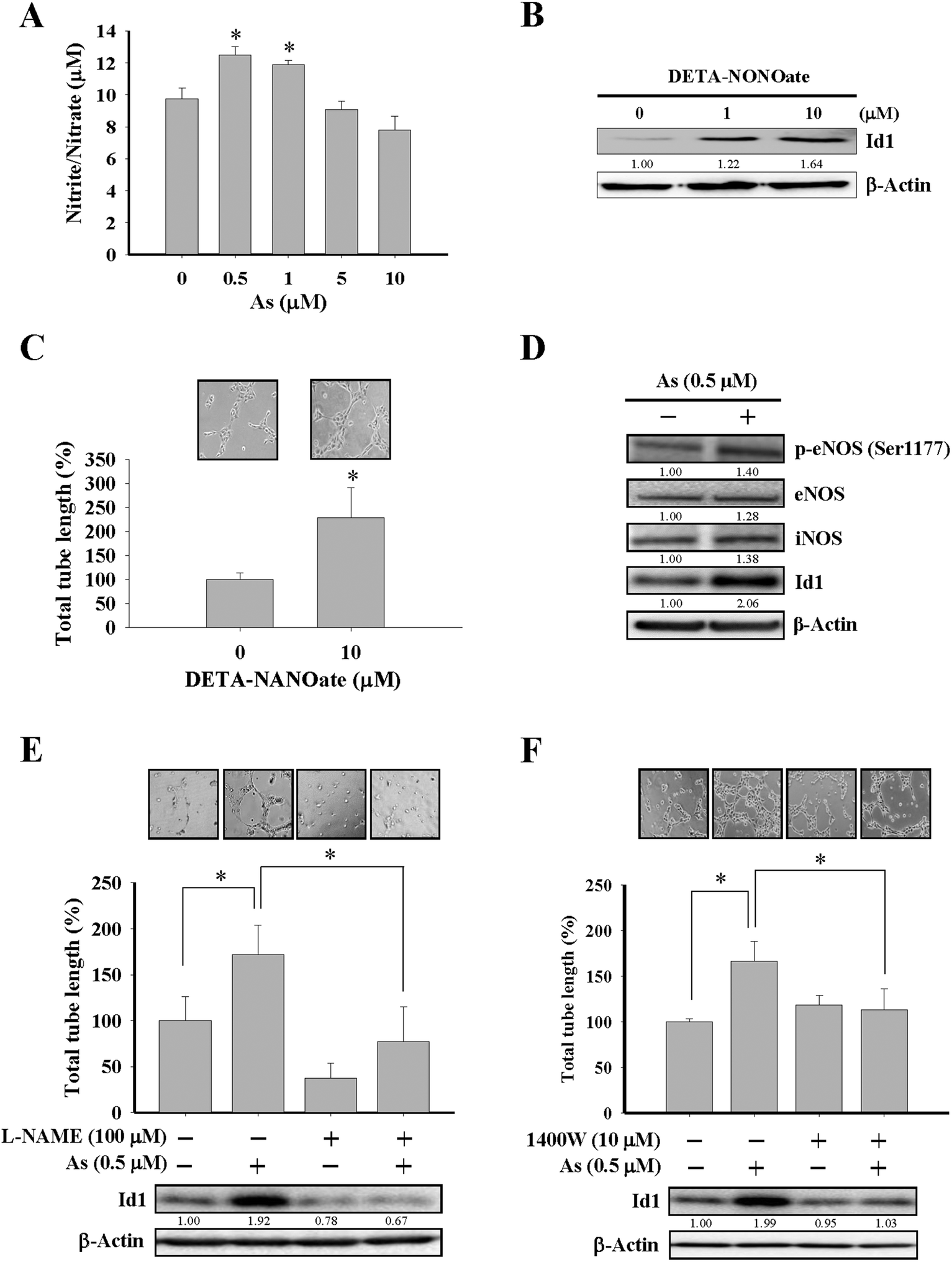 | ||
| Fig. 3 Involvement of nitric oxide in arsenic-induced Id1 protein expression and in vitro angiogenesis. (A) MS1 cells were treated with different doses of arsenic for 48 h, and the cell culture media were collected for the nitric oxide formation assay. (B) MS1 cells were treated with different doses of DETA-NONOate for 24 h, followed by western blot analysis for Id1 protein expression. (C) MS1 cells were treated with DETA-NONOate (10 μM) for 24 h, followed by the in vitro tube formation assay. *Significant difference of P < 0.05 by Student's t test. (D) MS1 cells were treated with arsenic (0.5 μM) for 24 h, followed by western blot analysis for protein expression of phospho-eNOS (Ser1177), eNOS, iNOS and Id1. (E) MS1 cells were pretreated with or without the nitric oxide synthase inhibitor L-NAME (100 μM) for 2 h, followed by the presence or absence of arsenic treatment (0.5 μM) for 24 h. The protein expression of Id1 and the tube formation ability were analyzed. (F) MS1 cells were pretreated with or without the nitric oxide synthase inhibitor 1400W (10 μM) for 2 h, followed by the presence or absence of arsenic treatment (0.5 μM) for 24 h. The protein expression of Id1 and the tube formation ability were analyzed. *Significant difference of P < 0.05 by one-way ANOVA with post-hoc Dunnett's test. | ||
Id1 as the downstream effector for arsenic-induced angiogenesis
To further investigate the role of Id1 in arsenic-induced angiogenesis, the endogenous expression of Id1 was knocked down by a lentiviral shRNA approach (Fig. 4A). As shown in Fig. 4A and B, the arsenic-induced in vitro tube formation was suppressed in the Id1-knocked down (Id1-KD) MS1 cells. In addition, the level of arsenic-promoted in vivo angiogenesis was reduced in the presence of Id1-KD cells (Fig. 4C and D). Together, the results suggested that Id1 acted as an important factor mediating the arsenic-promoted angiogenesis.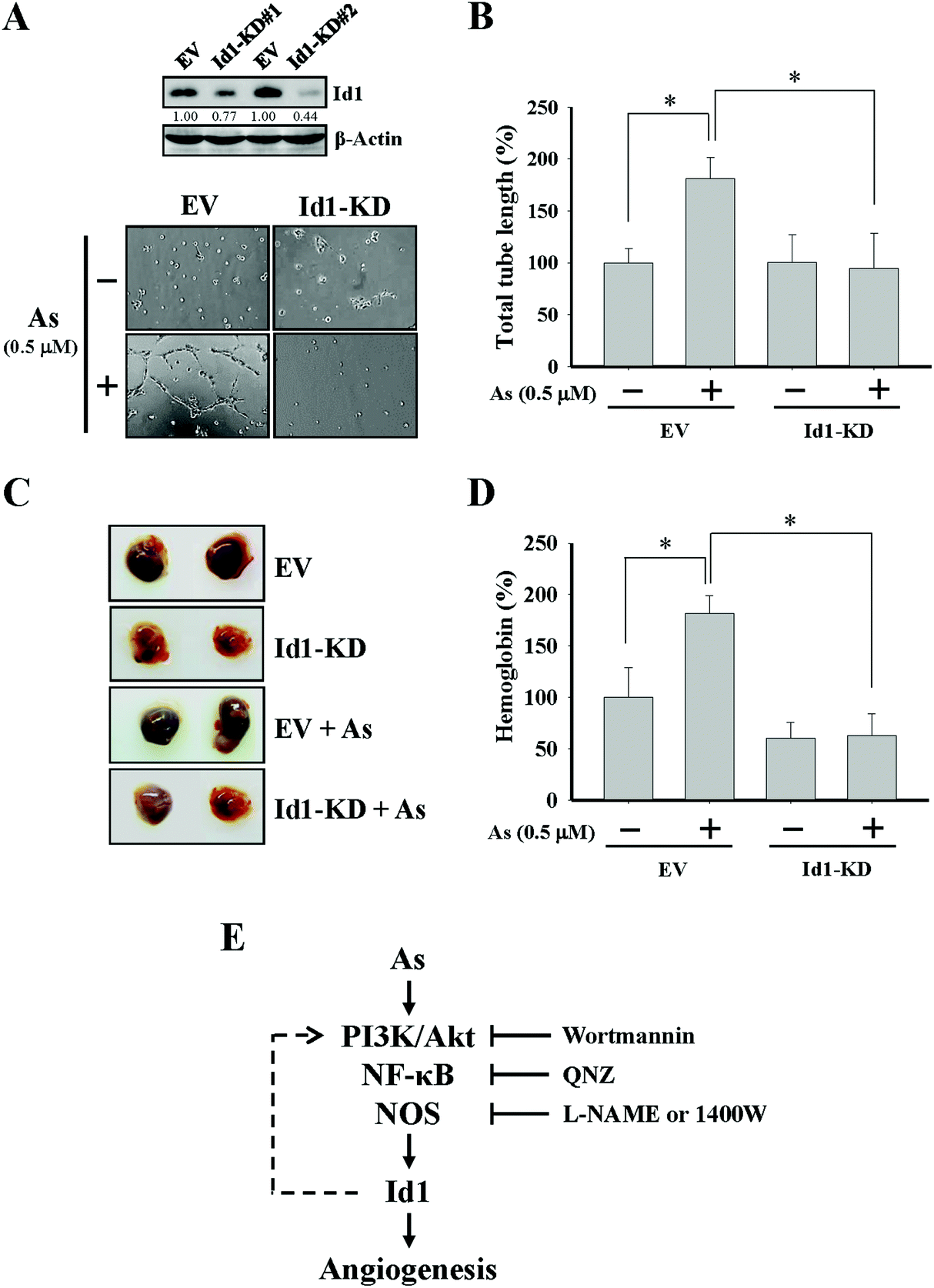 | ||
| Fig. 4 Involvement of Id1 in the arsenic-promoted angiogenesis. (A) Protein expression levels of Id1 in the Id1-knocked down (Id1-KD#1 and Id1-KD#2) and empty vector control (EV) MS1 cells were assessed by western blot. As a result, Id1-KD#2 MS1 cells were chosen for the experiments in (B)–(D). (A, B) Id1-KD and EV MS1 cells were treated with or without arsenic (0.5 μM) for 24 h, followed by the in vitro tube formation assay. (C, D) Id1-KD or EV MS1 cells were mixed with the Matrigel containing bovine FGF (1 ng mL−1), heparin (10 U mL−1), and either with or without arsenic (0.5 μM), followed by subcutaneous injection into immunodeficient mice for the Matrigel plug assay. (E) Schematic representation of a summary for the current work. The flow chart shows that arsenic-induced angiogenesis is mediated through Id1 expression regulated via PI3K/Akt, NF-κB and nitric oxide signaling. The dashed line suggests that a mutual regulation between Id1 and PI3K/Akt may possibly exist in this process (see the Discussion section). Wortmannin, PI3K/Akt inhibitor; QNZ, NF-κB inhibitor; L-NAME and 1400W, nitric oxide synthase inhibitors. *Significant difference of P < 0.05 by one-way ANOVA with post-hoc Dunnett's test. | ||
Discussion
Previous studies showed that exposure to arsenic was correlated with increasing risk of various carcinogenesis.8,24,41–45 In addition to the tumorigenic effect, the angiogenic activity of arsenic was also reported. For example, it was reported that low levels of arsenic promoted neovascularization and blood vessel remodeling.24,25 Treatment with arsenic also stimulated cell migration and tube formation of human microvascular endothelial cells.46 Moreover, the level of tumor angiogenesis was elevated by arsenic in mice xenografted with human adenocarcinoma cells.23 In the present study, we consistently showed that low arsenic promoted endothelial cell viability and angiogenesis (ESI Fig. 1†), whereas the opposite effect of arsenic at a higher concentration which caused the reduction of angiogenesis may be explained by its cytotoxic effect at such amounts of arsenic.35,36,47,48It was reported that PI3K/Akt signaling played an important role in arsenic-induced angiogenesis.49–51 Exposure to arsenic stimulated the PI3K/Akt phosphorylation cascade and resulted in cellular transformation characterized by increases in proliferation and anchorage-independent growth.52 Arsenic activated MAPK and PI3K/Akt pathways that were required for the arsenic-induced expression of COX-2, HIF-1α and VEGF.53,54 Our results suggested a novel regulatory role of PI3K/Akt in the arsenic-induced Id1 expression and tube formation (Fig. 2C and E).
The possible involvement of NF-κB, a downstream effector of PI3K/Akt in endothelial cells,55–57 was further investigated in this study. We found that arsenic was able to trigger NF-κB activation (Fig. 2A) as reported previously.47,58 Notably, the arsenic-enhanced Id1 expression and tube formation were suppressed in the presence of QNZ (Fig. 2D and F). We noticed that angiogenesis-related genes driven by NF-κB, such as IL-8 and Col1, were reportedly unnecessary for the arsenic-induced tube formation.59 Therefore, it would be worthwhile to determine whether there are specific NF-κB-regulated genes in the Id1-mediated angiogenesis induced by arsenic.
A previous report showed that the level of in vivo angiogenesis was reduced to a lesser degree in Id1−/− mice in contrast to that in Id1+/+ mice.28 In addition, the Id1−/− mice xenografted with human lung carcinoma cells had defects of microvascular formation on the implanted site, suggesting that Id1 might be a major effector for the tumor angiogenic activity.28 Strong Id1 expression was also observed in the surrounding blood vessels of high-grade glioma tumor tissues, while only weak Id1 expression was observed in those of low-grade tumor tissues.27 Our current data unveiled an angiogenic role of Id1 in the vascular endothelial cells when treated with arsenic. For instance, the arsenic-induced in vitro tube formation was significantly reduced in the Id1-knocked down MS1 cells compared with the control cells (Fig. 4A and B). Furthermore, the arsenic-induced in vivo angiogenic activity was suppressed in the mice xenografted with Id1-KD cells versus the mice xenografted with control cells (Fig. 4C and D). As angiogenesis is an essential event for tumor growth and metastasis, the Id1-mediated tumor angiogenesis may be developed into therapeutic potential against cancer progression.15,20,60
While nitric oxide is a known factor involved in arsenic-induced angiogenesis,61,62 the role of nitric oxide in arsenic-induced Id1 expression has not been elucidated. Several lines of evidence in our current study suggested the possible involvement of nitric oxide signaling and Id1 in these arsenic-induced events, including (1) the level of nitric oxide formation was increased by arsenic (Fig. 3A), (2) Id1 expression and tube formation were enhanced in the presence of DETA-NONOate, a known NO donor (Fig. 3B and C), and (3) application of the NOS inhibitors L-NAME and 1400W showed an inhibitory effect on Id1 expression and tube formation induced by arsenic (Fig. 3E and F). It was reported that NOS activities were regulated downstream of PI3K/Akt and NF-κB signaling.63–66 Intriguingly, a mutual regulation might be present between Id1 and PI3K/Akt.51,67–69 Therefore, it will be valuable to investigate the complex connection of these signaling molecules involved in the arsenic-induced angiogenesis (Fig. 4E).
In summary, our data showed for the first time that Id1 mediated arsenic-promoted angiogenesis. In addition, PI3K/Akt, NF-κB and nitric oxide had regulatory roles in this process. The current results may hence further our understanding towards the role of Id1 in arsenic-associated angiogenesis and suggest its potential application as a target of antiangiogenesis therapy in arsenic-associated cancer.
Acknowledgements
This work was supported by health and welfare surcharge of tobacco products from the Ministry of Health and Welfare (MOHW103-TD-B-111-05, MOHW104-TDU-B-212-124-003), National Science Council (NSC102-3114-Y-492-076-023), National Health Research Institutes (NHRI-EX98-/99-/100-9829BI, NHRI-EX102-/103/104-10212BI), Kaohsiung Medical University Hospital (KMUH102-2T07, KMUHI102-2R25), and Kaohsiung Medical University (KMU-TP103D18, Aim for the Top Journals Grant KMU-DT103010) in Taiwan.References
- R. S. Oremland and J. F. Stolz, Science, 2003, 300, 939–944 CrossRef CAS PubMed.
- M. Argos, H. Ahsan and J. H. Graziano, Rev. Environ. Health, 2012, 27, 191–195 CAS.
- M. N. Bates, A. H. Smith and C. Hopenhayn-Rich, Am. J. Epidemiol., 1992, 135, 462–476 CAS.
- Q.-b. Zeng, Y.-y. Xu, X. Yu, J. Yang, F. Hong and A.-h. Zhang, Toxicol. Res., 2014, 3, 359 RSC.
- M. Unterberg, L. Leffers, F. Hübner, H.-U. Humpf, K. Lepikhov, J. Walter, F. Ebert and T. Schwerdtle, Toxicol. Res., 2014, 3, 456–464 RSC.
- S. F. Farzan, M. R. Karagas and Y. Chen, Toxicol. Appl. Pharmacol., 2013, 272, 384–390 CrossRef CAS PubMed.
- M. S. Golub, M. S. Macintosh and N. Baumrind, J. Toxicol. Environ. Health, Part B, 1998, 1, 199–241 CAS.
- M. P. Waalkes, J. M. Ward and B. A. Diwan, Carcinogenesis, 2004, 25, 133–141 CrossRef CAS PubMed.
- D. J. Peacock, M. L. Banquerigo and E. Brahn, J. Exp. Med., 1992, 175, 1135–1138 CrossRef CAS PubMed.
- P. Martin, Science, 1997, 276, 75–81 CrossRef CAS PubMed.
- D. A. Greenberg and K. Jin, Cell. Mol. Life Sci., 2013, 70, 1753–1761 CrossRef CAS PubMed.
- A. Grochot-Przeczek, J. Dulak and A. Jozkowicz, Gene, 2013, 525, 220–228 CrossRef CAS PubMed.
- M. H. Liu, Z. H. Tang, G. H. Li, S. L. Qu, Y. Zhang, Z. Ren, L. S. Liu and Z. S. Jiang, Atherosclerosis, 2013, 229, 10–17 CrossRef CAS PubMed.
- J. Folkman, N. Engl. J. Med., 1971, 285, 1182–1186 CrossRef CAS PubMed.
- J. Welti, S. Loges, S. Dimmeler and P. Carmeliet, J. Clin. Invest., 2013, 123, 3190–3200 CAS.
- S. Bellou, G. Pentheroudakis, C. Murphy and T. Fotsis, Cancer Lett., 2013, 338, 219–228 CrossRef CAS PubMed.
- R. Benezra, R. L. Davis, D. Lockshon, D. L. Turner and H. Weintraub, Cell, 1990, 61, 49–59 CrossRef CAS PubMed.
- Y. C. Wong, X. Wang and M. T. Ling, Apoptosis, 2004, 9, 279–289 CrossRef CAS PubMed.
- Y. J. Cheng, J. W. Tsai, K. C. Hsieh, Y. C. Yang, Y. J. Chen, M. S. Huang and S. S. Yuan, Cancer Lett., 2011, 307, 191–199 CrossRef CAS PubMed.
- S. Fong, R. J. Debs and P. Y. Desprez, Trends Mol. Med., 2004, 10, 387–392 CrossRef CAS PubMed.
- L. Soroceanu, R. Murase, C. Limbad, E. Singer, J. Allison, I. Adrados, R. Kawamura, A. Pakdel, Y. Fukuyo, D. Nguyen, S. Khan, R. Arauz, G. L. Yount, D. H. Moore, P. Y. Desprez and S. D. McAllister, Cancer Res., 2013, 73, 1559–1569 CrossRef CAS PubMed.
- A. Iavarone and A. Lasorella, Cancer Lett., 2004, 204, 189–196 CrossRef CAS PubMed.
- L. Z. Liu, Y. Jiang, R. L. Carpenter, Y. Jing, S. C. Peiper and B. H. Jiang, PLoS One, 2011, 6, e20858 CAS.
- N. V. Soucy, M. A. Ihnat, C. D. Kamat, L. Hess, M. J. Post, L. R. Klei, C. Clark and A. Barchowsky, Toxicol. Sci., 2003, 76, 271–279 CrossRef CAS PubMed.
- A. C. Straub, D. B. Stolz, H. Vin, M. A. Ross, N. V. Soucy, L. R. Klei and A. Barchowsky, Toxicol. Appl. Pharmacol., 2007, 222, 327–336 CrossRef CAS PubMed.
- D. Lyden, A. Z. Young, D. Zagzag, W. Yan, W. Gerald, R. O'Reilly, B. L. Bader, R. O. Hynes, Y. Zhuang, K. Manova and R. Benezra, Nature, 1999, 401, 670–677 CrossRef CAS PubMed.
- D. A. Vandeputte, D. Troost, S. Leenstra, H. Ijlst-Keizers, M. Ramkema, D. A. Bosch, F. Baas, N. K. Das and E. Aronica, Glia, 2002, 38, 329–338 CrossRef PubMed.
- O. V. Volpert, R. Pili, H. A. Sikder, T. Nelius, T. Zaichuk, C. Morris, C. B. Shiflett, M. K. Devlin, K. Conant and R. M. Alani, Cancer Cell, 2002, 2, 473–483 CrossRef CAS PubMed.
- Y. J. Chen, C. M. Hung, N. Kay, C. C. Chen, Y. H. Kao and S. S. Yuan, Cancer Lett., 2012, 319, 223–231 CrossRef CAS PubMed.
- Y. J. Chen, Y. J. Cheng, A. C. Hung, Y. C. Wu, M. F. Hou, Y. C. Tyan and S. S. Yuan, Gynecol. Oncol., 2013, 131, 734–743 CrossRef CAS PubMed.
- S. S. Yuan, M. F. Hou, Y. C. Hsieh, C. Y. Huang, Y. C. Lee, Y. J. Chen and S. Lo, J. Natl. Cancer Inst., 2012, 104, 1485–1502 CrossRef CAS PubMed.
- G. N. Serbedzija, E. Flynn and C. E. Willett, Angiogenesis, 1999, 3, 353–359 CrossRef CAS.
- T. W. Shiue, Y. H. Chen, C. M. Wu, G. Singh, H. Y. Chen, C. H. Hung, W. F. Liaw and Y. M. Wang, Inorg. Chem., 2012, 51, 5400–5408 CrossRef CAS PubMed.
- A. Barchowsky, R. R. Roussel, L. R. Klei, P. E. James, N. Ganju, K. R. Smith and E. J. Dudek, Toxicol. Appl. Pharmacol., 1999, 159, 65–75 CrossRef CAS PubMed.
- Y. H. Kao, C. L. Yu, L. W. Chang and H. S. Yu, Chem. Res. Toxicol., 2003, 16, 460–468 CrossRef CAS PubMed.
- G. J. Roboz, S. Dias, G. Lam, W. J. Lane, S. L. Soignet, R. P. Warrell Jr. and S. Rafii, Blood, 2000, 96, 1525–1530 CAS.
- C. W. McCollum, C. Hans, S. Shah, F. A. Merchant, J. A. Gustafsson and M. Bondesson, Aquat. Toxicol., 2014, 152, 152–163 CrossRef CAS PubMed.
- G. Niu, K. L. Wright, M. Huang, L. Song, E. Haura, J. Turkson, S. Zhang, T. Wang, D. Sinibaldi, D. Coppola, R. Heller, L. M. Ellis, J. Karras, J. Bromberg, D. Pardoll, R. Jove and H. Yu, Oncogene, 2002, 21, 2000–2008 CrossRef CAS PubMed.
- S. S. Yuan, M. F. Hou, H. L. Chang, T. F. Chan, Y. H. Wu, Y. C. Wu and J. H. Su, Toxicol. in Vitro, 2003, 17, 139–143 CrossRef CAS PubMed.
- P. Basu, R. N. Ghosh, L. E. Grove, L. Klei and A. Barchowsky, Environ. Health Perspect., 2008, 116, 520–523 CrossRef CAS PubMed.
- L. M. Knobeloch, K. M. Zierold and H. A. Anderson, J. Health Popul. Nutr., 2006, 24, 206–213 Search PubMed.
- J. Liu, Y. Xie, J. M. Ward, B. A. Diwan and M. P. Waalkes, Toxicol. Sci., 2004, 77, 249–257 CrossRef CAS PubMed.
- S. A. Navarro Silvera and T. E. Rohan, Cancer, Causes Control, 2007, 18, 7–27 CrossRef PubMed.
- S. Oberoi, A. Barchowsky and F. Wu, Cancer Epidemiol., Biomarkers Prev., 2014, 23, 1187–1194 CrossRef CAS PubMed.
- K. M. Hunt, R. K. Srivastava, C. A. Elmets and M. Athar, Cancer Lett., 2014, 354, 211–219 CrossRef CAS PubMed.
- D. Meng, X. Wang, Q. Chang, A. Hitron, Z. Zhang, M. Xu, G. Chen, J. Luo, B. Jiang, J. Fang and X. Shi, Toxicol. Appl. Pharmacol., 2010, 244, 291–299 CrossRef CAS PubMed.
- A. Barchowsky, E. J. Dudek, M. D. Treadwell and K. E. Wetterhahn, Free Radical Biol. Med., 1996, 21, 783–790 CrossRef CAS PubMed.
- Y. S. Lew, S. L. Brown, R. J. Griffin, C. W. Song and J. H. Kim, Cancer Res., 1999, 59, 6033–6037 CAS.
- R. L. Carpenter, Y. Jiang, Y. Jing, J. He, Y. Rojanasakul, L. Z. Liu and B. H. Jiang, Biochem. Biophys. Res. Commun., 2011, 414, 533–538 CrossRef CAS PubMed.
- Y. Ma, J. Wang, L. Liu, H. Zhu, X. Chen, S. Pan, X. Sun and H. Jiang, Cancer Lett., 2011, 301, 75–84 CrossRef CAS PubMed.
- Y. Su, L. Gao, L. Teng, Y. Wang, J. Cui, S. Peng and S. Fu, J. Transl. Med., 2013, 11, 132 CrossRef CAS PubMed.
- R. Hubaux, D. D. Becker-Santos, K. S. Enfield, D. Rowbotham, S. Lam, W. L. Lam and V. D. Martinez, Mol. Cancer, 2013, 12, 20 CrossRef CAS PubMed.
- F. Wang, C. Wang, S. Liu, H. Wang, S. Xi and G. Sun, Toxicol. Res., 2015 10.1039/C1035TX00196J , in press.
- F. Wang, S. Liu, S. Xi, L. Yan, H. Wang, Y. Song and G. Sun, Toxicol. Lett., 2013, 222, 303–311 CrossRef CAS PubMed.
- T. H. Lee, H. Jung, K. H. Park, M. H. Bang, N. I. Baek and J. Kim, Exp. Biol. Med., 2014, 239, 1325–1334 CrossRef PubMed.
- M. Zhou, X. Song, Y. Huang, L. Wei, Z. Li, Q. You, Q. Guo and N. Lu, Vasc. Pharmacol., 2014, 60, 110–119 CrossRef CAS PubMed.
- G. Guan, H. Han, Y. Yang, Y. Jin, X. Wang and X. Liu, Endocrine, 2014, 47, 764–771 CrossRef CAS PubMed.
- E. Hossain, A. Ota, S. Karnan, L. Damdindorj, M. Takahashi, Y. Konishi, H. Konishi and Y. Hosokawa, Toxicol. Appl. Pharmacol., 2013, 273, 651–658 CrossRef CAS PubMed.
- L. R. Klei and A. Barchowsky, Toxicol. Sci., 2008, 102, 319–327 CrossRef CAS PubMed.
- S. M. Weis and D. A. Cheresh, Nat. Med., 2011, 17, 1359–1370 CrossRef CAS PubMed.
- P. Balakumar and J. Kaur, Cardiovasc. Toxicol., 2009, 9, 169–176 CrossRef CAS PubMed.
- Y. Kumagai and J. Pi, Toxicol. Appl. Pharmacol., 2004, 198, 450–457 CrossRef CAS PubMed.
- B. H. Chung, J. D. Kim, C. K. Kim, J. H. Kim, M. H. Won, H. S. Lee, M. S. Dong, K. S. Ha, Y. G. Kwon and Y. M. Kim, Biochem. Biophys. Res. Commun., 2008, 376, 404–408 CrossRef CAS PubMed.
- A. Hashimoto, G. Miyakoda, Y. Hirose and T. Mori, Atherosclerosis, 2006, 189, 350–357 CrossRef CAS PubMed.
- S. Yu, S. L. Wong, C. W. Lau, Y. Huang and C. M. Yu, Biochem. Biophys. Res. Commun., 2011, 407, 44–48 CrossRef CAS PubMed.
- C. Banerjee, A. Singh, R. Ramanb and S. Mazumder, Toxicol. Res., 2013, 2, 413–426 RSC.
- W. Li, H. Wang, C. Y. Kuang, J. K. Zhu, Y. Yu, Z. X. Qin, J. Liu and L. Huang, Mol. Cell. Biochem., 2012, 363, 135–145 CrossRef CAS PubMed.
- M. T. Ling, X. Wang, X. S. Ouyang, K. Xu, S. W. Tsao and Y. C. Wong, Oncogene, 2003, 22, 4498–4508 CrossRef CAS PubMed.
- Y. Su, L. Zheng, Q. Wang, J. Bao, Z. Cai and A. Liu, BMC Cancer, 2010, 10, 459 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5tx00280j |
| ‡ Equal contribution to this work. |
| This journal is © The Royal Society of Chemistry 2016 |