
HIF-2α, acting via miR-191, is involved in angiogenesis and metastasis of arsenite-transformed HBE cells†
Wenchao
Xu‡
ab,
Fei
Luo‡
ab,
Baofei
Sun‡
c,
Hua
Ye
d,
Jun
Li
c,
Le
Shi
ab,
Yi
Liu
ab,
Xiaolin
Lu
ab,
Bairu
Wang
ab,
Qingling
Wang
c,
Qizhan
Liu
*ab and
Aihua
Zhang
*c
aInstitute of Toxicology, School of Public Health, Nanjing Medical University, Nanjing 211166, Jiangsu, People's Republic of China
bThe Key Laboratory of Modern Toxicology, Ministry of Education, School of Public Health, Nanjing Medical University, Nanjing 211166, Jiangsu, People's Republic of China. E-mail: drqzliu@hotmail.com; Fax: +86-25-8686-8499; Tel: +86-25-8686-8424
cThe Key Laboratory of Environmental Pollution Monitoring and Disease Control, Ministry of Education, School of Public Health, Guiyang Medical University, Guiyang 550025, Guizhou, People's Republic of China. E-mail: aihuagy@aliyun.com; Fax: +86-851-8841-6175; Tel: +86-851-8841-6171
dSchool of Medicine, Yangzhou University, Yangzhou 225009, Jiangsu, People's Republic of China
First published on 6th November 2015
Abstract
Arsenic is a well established human carcinogen that causes diseases of the lung. Some studies have suggested that hypoxia-inducible factors (HIFs) and microRNAs (miRNAs) are involved in human lung cancer; however, their molecular mechanisms that causally contribute to arsenite-caused malignant transformation of cells remain unclear. To elucidate the mechanisms of angiogenesis and metastasis of lung cancer caused by arsenite, we investigated the role of HIF-2α regulation of miRNA-191 (miR-191) in the angiogenic and metastatic properties of human bronchial epithelial (HBE) cells transformed by arsenite. In HBE cells, HIF-2α binds to the hypoxia response element (HRE) in the promoter region of miR-191 and initiates transcription of miR-191. Blocking of HIF-2α with siRNA inhibited the up-regulation of miR-191, Wilms’ tumor 1 (WT1) protein, matrix metalloproteinase 9 (MMP-9), vascular endothelial growth factor (VEGF), and the down-regulation of brain acid-soluble protein 1 (BASP1). In arsenite-transformed HBE (T-HBE) cells, down-regulation of HIF-2α by siRNA blocked the process of angiogenesis and decreased their neoplastic properties and metastatic capacity, which were reversed by over-expression of miR-191 or by up-regulating WT1. Thus, HIF-2α up-regulates WT1 via miR-191, both of which are involved in the angiogenesis and metastasis of T-HBE cells. The results present a better understanding of the processes involved in lung cancer caused by arsenite exposure.
Introduction
Inorganic arsenic is a potent human carcinogen that causes cancers of the lung, skin, kidney, urinary bladder, and liver.1,2 Lung is considered to be the most sensitive site for arsenic toxicity, and chronic exposure to arsenite is associated with lung tumors.3Lung cancer remains the most common cancer and the most common cause of cancer-related death worldwide.4 The disease has a low rate of early diagnosis; metastases are detected in 75% of the cases at the time of diagnosis. The patients usually do not die of primary loci-induced symptoms, but of complications caused by metastatic loci, such as brain metastasis. Thus, an understanding of the mechanisms underlying lung cancer development and metastasis induced is essential for improving the diagnosis, prevention, and treatment of this disease.
Angiogenesis is a process of remodeling involving sprouting and growth of blood vessels5 that is essential to supply nutrients, oxygen, and growth factors that maintain cell function and survival. Angiogenic signals lead to the preferential differentiation of certain endothelial cells into so-called tip cells, which start to migrate and exist at the leading front of the growing vessels.6 Therefore, angiogenesis is associated with tumor metastasis which is the major cause of death among cancer patients. MicroRNAs (miRNAs) are 21–24 nucleotide, non-coding RNAs that affect RNA stability and translation and thereby regulate gene expression7 and various biological processes, including differentiation, proliferation, angiogenesis, and metastasis.8 One miRNA, miR-191, which is involved in human lung, breast, liver, pancreatic, and gastric cancer,9–13 is thought to be an emerging player in disease biology.14 Our previous study showed that, in transformed cells, miR-191 regulates the levels of brain acid-soluble protein 1 (BASP1) and Wilms’ tumor 1 (WT1) protein to modulate the epithelial–mesenchymal transition (EMT) and cancer stem cell (CSC)-like properties and that it is an onco-miR involved in the neoplastic and metastatic processes of these cells.15 Despite documented links between miR-191 and cancer, however, little is known about the factors that lead to its deregulation in tumor cells.
The regulators of hypoxic responses in cells are transcription factors called hypoxia-inducible factors (HIFs), which are heterodimers composed of an O2-labile α subunit (HIF-1α, HIF-2α, or the less characterized HIF-3α) and a stable β subunit (HIF-1β).16 HIFs are involved in tumor angiogenesis and metastasis.17–19 Both HIF-1α and HIF-2α are up-regulated in cancer cells and control tumor progression and sensitivity to therapy.20,21 HIF-2α, in contrast to the widely expressed HIF-1α, is expressed only in certain normal tissues, such as lung, intestine, liver, carotid body, and endothelium.22 Nevertheless, HIF-2α is detected in most types of solid tumors. In particular, it is associated with cancers of the digestive system.23 Our previous studies have demonstrated that HIF-2α, but not HIF-1α, is up-regulated by arsenite and that it is involved in the arsenite-induced malignant transformation of human bronchial epithelial (HBE) cells.24
miRNAs affect the expression of HIFs25,26 and are induced by HIFs under hypoxic conditions.27 It has not been determined, however, whether HIF-2α regulates miR-191 expression and its target genes, which are involved in the angiogenic and metastatic properties of T-HBE cells. Further, the biological functions of HIF-2α in the regulation of angiogenesis in T-HBE cells remain largely uninvestigated.
In the present study, the correlation of HIF-2α and miR-191 expression, the interaction and mechanism of HIF-2α in regulating the expression of miR-191, and the functions of HIF-2α in angiogenesis and metastasis were examined by the use of T-HBE cells. The results show that HIF-2α up-regulates WT1 via miR-191, which is involved in the angiogenic and metastatic properties of these cells.
Materials and methods
Cell culture and reagents
HBE cells, a SV40-transformed human bronchial epithelial cell line, are nontumorigenic and retain features of human bronchial epithelial cells. They are useful for studies of multistage bronchial epithelial carcinogenesis.28 Primary human umbilical vein endothelial cells (HUVECs) are used for angiogenesis assays. HBE and HUVEC cell lines, obtained from the Shanghai Institute of Cell Biology, Chinese Academy of Sciences (Shanghai, China), were maintained in 5% CO2 at 37 °C in Eagle's Minimum Essential Medium (MEM) and endothelial cell growth medium-2 (EGM-2), respectively, each supplemented with 10% fetal bovine serum (FBS, Life Technologies/Gibco, Grand Island, NY), 100 U per ml penicillin, and 100 μg per ml streptomycin (Life Technologies/Gibco, Gaithersburg, MD).We previously established the model of arsenite-transformed HBE cells.29 For chronic exposure, 1 × 106 cells were seeded into 10 cm (diameter) dishes for 24 h and maintained in 0.0 or 1.0 μM sodium arsenite (NaAsO2, Sigma, St Louis, MO, purity: 99.0%) for 48–72 h per passage. This process was continued for about 15 weeks (30 passages). All other reagents used were of analytical grade or the highest grade available.
Enzyme-linked immunosorbent assay (ELISA)
After transfection, transformed cells grew to 90–100% confluence, and the conditioned medium was collected. VEGF levels in the medium were measured using an ELISA kit from Beijing 4A Biotech Co., Ltd (Beijing) following the manufacturer's instructions and normalized to the cell numbers. All assays were performed in duplicate and repeated three times. The lower limit of detection of VEGF was 2 pg mL−1.Tube formation assay
An assay for endothelial cell tube formation in three-dimensional cultures was used to assess angiogenic potential. To perform the tube formation assay, conditioned medium (CM) from transfected cells was obtained by incubating 60–80% confluence with MEM. After a 24 h incubation, the CM was collected in a centrifugal tube and centrifuged at 3000g for 5 min to remove cell debris. Fifty microliters of Matrigel (BD Biosciences, Franklin Lakes, NJ, USA) were dispensed per well into 48-well plates using a cold pipette to avoid the formation of bubbles. The Matrigel was polymerized for 30 min at 37 °C with 5% CO2. HUVECs were starved in serum-free medium for 6 h and trypsinized. 5 × 106 cells were resuspended and plated onto Matrigel in the presence of 100 μL CM containing 1% FCS and incubated for 6 h at 37 °C with 5% CO2. The detailed experimental procedure was also described previously.30 Tube formation was examined in photographs taken under a microscope. The lengths of blood vessels, as an index of angiogenesis, were obtained by measuring the branches of blood vessels in 2 square millimeters using Olympus CellSens Standard digital imaging software. The numbers were normalized to values of the control groups.Quantitative real-time PCR
Total cellular RNA was isolated by the use of TRIzol (Invitrogen) according to the manufacturer's recommendations. For the detection of HIF-2α, total RNA (2 μg) was transcribed into cDNA by the use of AMV Reverse Transcriptase (Promega, Madison, Wisconsin, USA). Primers for HIF-2α were as follows: forward, 5′-CACCAAGGGTCAGGTAGTAAG-3′; and reverse, 5′-GGTTGCGAGGGTTGTAGAT-3′. For the detection of mature miRNAs, 2 μg of total RNA, miRNA-specific stem-loop RT primers, and MMLV reverse transcriptase (Promega Corp., Madison, WI) were used in reverse transcription following the manufacturer's protocol. The RT primers for miRNAs and U6 small nuclear RNA (snRNA) are listed in ESI Table 1.† The sequences of mature miRNAs were from Sanger miRBase (http://microrna.sanger.ac.uk/sequences/). qRT-PCR was performed using the Power SYBR Green Master Mix (Applied Biosystems, Foster City, CA, USA) and an ABI 7300 real-time PCR detection system (Applied Biosystems). Forward (F) and reverse (R) primers are listed in Table 1. All of the primers were synthesized by Invitrogen. U6 snRNA was used as an internal control. Fold changes in the expression of each gene were calculated by a comparative threshold cycle (Ct) method using the formula 2−(ΔΔCt).31| U6 | RT: 5-AAAATATGGAACGCTTCACG-3 |
| F: 5-CGCTTCGGCAGCACATATACTAAAATTGGAAC-3 | |
| R: 5-GCTTCACGAATTTGCGTGTCATCCTTGC-3 | |
| MiR-191 | RT: 5-CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGCAGCTGCT-3 |
| F: 5-ACACTCCAGCTGGGCAACGGAATCCCAAAAG-3 | |
| R: 5-TGGTGTCGTGGAGTCG-3 | |
| ChIP1 (HRE1) | F: 5-GCTGTGATAGTGTTTGGGTGG-3 |
| R: 5-GAGTGAACGAGGACAAGGTGA-3 | |
| ChIP2 (HRE2 and HRE3) | F: 5-CGTGGATTGGCTTCTCGGGTTC-3 |
| R: 5-CTTGCCCTTCCGCTTGCTCTGG-3 | |
| ChIP3 (HRE4) | F: 5-GTTTCTCCGGCTAGACGCG-3 |
| R: 5-AACCCGAGAAGCCAATCCAC-3 | |
| ChIP4 (HRE5) | F: 5-CAGTCTTAGGAGGCCGAGTTCC-3 |
| R: 5-GCCACGCTGCCAGGTGTT-3 | |
| ChIP5 (HRE6) | F: 5-CACCAGGGAAGCTCAACGG-3 |
| R: 5-GGGAACTCGGCCTCCTAAGACT-3 |
Western blots
Cells were washed twice with ice-cold phosphate-buffered saline (PBS), scraped off into 0.2 ml of lysis buffer (Beyotime), and incubated on ice for 30 min, followed by centrifugation at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 20 min. Protein concentrations were measured using the BCA protein assay (Beyotime). Afterwards, proteins were diluted to equal concentrations (20 μg), boiled for 10 min, separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and transferred to polyvinylidene fluoride membranes (Millipore, Billerica, MA, USA). The immune complexes were detected by enhanced chemiluminescence (Cell Signaling Technology). The antibodies used were those for HIF-1α, HIF-2α (Abcam); BASP1 and WT1 (Santa Cruz Biotechnology); matrix metalloproteinase 9 (MMP-9) and vascular endothelial growth factor (VEGF, Cell Signaling Technology); and glyceraldehyde 3-phosphate dehydrogenase (GAPDH, Sigma). After an additional incubation with a 1:1000 dilution of an anti-immunoglobin horseradish peroxidase-linked antibody for 1 h, the immune complexes were detected by enhanced chemiluminescence (Cell Signaling Technology). Blots were quantified by densitometry and normalized by the use of GAPDH to correct for differences in loading of the proteins. For densitometric analyses, the bands on the blots were measured by using an Eagle Eye II imaging system.
000 rpm for 20 min. Protein concentrations were measured using the BCA protein assay (Beyotime). Afterwards, proteins were diluted to equal concentrations (20 μg), boiled for 10 min, separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and transferred to polyvinylidene fluoride membranes (Millipore, Billerica, MA, USA). The immune complexes were detected by enhanced chemiluminescence (Cell Signaling Technology). The antibodies used were those for HIF-1α, HIF-2α (Abcam); BASP1 and WT1 (Santa Cruz Biotechnology); matrix metalloproteinase 9 (MMP-9) and vascular endothelial growth factor (VEGF, Cell Signaling Technology); and glyceraldehyde 3-phosphate dehydrogenase (GAPDH, Sigma). After an additional incubation with a 1:1000 dilution of an anti-immunoglobin horseradish peroxidase-linked antibody for 1 h, the immune complexes were detected by enhanced chemiluminescence (Cell Signaling Technology). Blots were quantified by densitometry and normalized by the use of GAPDH to correct for differences in loading of the proteins. For densitometric analyses, the bands on the blots were measured by using an Eagle Eye II imaging system.
RNA interference
Control siRNA and HIF-2α siRNA were purchased from Santa Cruz Biotechnology. Transfections were performed with an N-TER™ Nanoparticle siRNA Transfection System (Sigma). Briefly, 7 × 105 cells were seeded into each well of 6-well plates for 24 h, and then a nanoparticle formation solution containing 25 nM target siRNA was transferred to each well for another 24 h. The transient transfection system was also described previously.32 Cells were harvested and used for experiments.Plasmids and cell transfection
The pEGFP-N1-plasmid for overexpressing WT1 and its negative control were purchased from GeneChem (Shanghai, China). miR-191-mimic and miRNA negative control mimic were synthesized by RiBoBio (Guangzhou, China). Cells were transiently transfected using the Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer's protocol. At 24 h after transfection, cells were harvested and used for experiments.Structure analysis of the miR-191 promoter
Both HIF-1α and HIF-2α bind to the same hypoxia responsive element (HRE) at the target gene loci.16 By the use of the UCSC Genome Browser, 6 HREs that can bind to HIF-2α were found in the promoter of the miR-191 5′ flanking region. These results were consistent with others.12Chromatin immunoprecipitation assays
Chromatin immunoprecipitation (ChIP) assays were performed by the use of a Magna ChIP™A/G Chromatin Immunoprecipitation kit (Millipore) following the manufacturer's protocol. Briefly, cells (1 × 107) were treated with or without arsenite for 24 h and cross-linked in 1% formaldehyde for 10 min. After cell lysis, the chromatin was fragmented to an average size of 500 bp and enriched with a magnetic Dynal bead (Invitrogen)-coupled antibody against HIF-2α (Millipore), or isotype IgG at 4 °C overnight. The cross-links for the enriched and the input DNA were then reversed, and the DNA was cleaned by RNase A (0.2 mg mL−1) and proteinase K (2 μg mL−1) before phenol/chloroform purification. Primers were directed to the HREs of the 2000 bp upstream sequence at the 5′-end region of miR-191. There were 6 HREs in the 5′-end region; they were named as HRE1–HRE6 based on the distance from the pre-miR-191. The ChIP primers for HREs are listed in ESI Table 1.† Amplification conditions were as follows: pre-denaturing at 94 °C for 3 min, 94 °C for 30 s, 58 °C for 45 s, and 72 °C for 45 s (40 cycles) and extending at 72 °C for 10 min.Anchorage-independent growth
Soft agar plates were prepared in 10 cm (diameter) dishes with under-layers of 0.70% agarose in MEM supplemented with 10% FBS. To test their capacity for colony growth in soft agar, cells were plated in triplicate at a density of 1 × 104 in 2 ml of 0.35% agarose over the agar base. Cultures were fed every three days; after 14 days, colonies with >30 cells were counted.Transwell assays
Migration of T-HBE cells was evaluated by the use of Transwell™ chambers with 8 μm pore filters (Corning Inc., Corning, NY, USA). At 24 h after transfection, cells transfected with control siRNA, HIF-2α siRNA, WT1 siRNA, miR-191-mimic, or the negative control mimic were trypsinized, and 5 × 104 per 100 μL cells were plated in the upper chambers in serum-free medium. MEM containing 10% FBS was added to the lower chamber as a chemoattractant. After incubation for 24 h at 37 °C, non-migrating cells were removed with cotton swabs. Cells that migrated to the bottom of the membrane were fixed with 4% paraformaldehyde, stained with crystal violet solution for 30 min, and washed twice with PBS. Stained cells were visualized under a microscope (high-power field), and the number of cells counted in five random fields were averaged. To assess the capacity for invasion of T-HBE cells, 5 × 104 per 100 μL cells were added to the upper chambers that had been coated with 35 μl of Matrigel. MEM containing 10% FBS was added to the lower chambers. Cells were incubated for 24 h at 37 °C, and then non-invading cells were removed with cotton swabs. Invading cells were fixed, stained, and counted.Statistical analyses
Derived values are presented as the means ± SD. Comparison of the mean data among multiple groups was analyzed by one-way analysis of variance (ANOVA), and a multiple-range least significant difference was used for inter-group comparisons. P values < 0.05 were considered statistically significant. All statistical analyses were performed with SPSS 16.0.Results
Exposure of HBE cells to arsenite increases the expression of HIF-2α, MMP-9, and VEGF
HIFs are involved in carcinogenesis and tumor progression.33 There are three isoforms of the HIF-α subunit (HIF-1α, HIF-2α, and HIF-3α). Of these, HIF-1α and HIF-2α are the best understood. Matrix metalloproteinase 9 (MMP-9) is involved in the progression of several types of cancer by increasing tumor growth, migration, invasion, and metastasis.34 Vascular endothelial growth factor (VEGF) is a secreted, endothelial-specific growth factor that induces angiogenesis.35 MMP-9 and VEGF are targets of WT1.36 With long-term exposure to arsenite, the expression of HIF-1α was not appreciably changed, but those of HIF-2α, MMP-9, and VEGF were increased; such changes did not occur in control cells (Fig. 1A and B). To determine whether arsenite is involved in the expression of HIF-1α, HIF-2α, MMP-9, and VEGF, HBE cells were exposed to 1.0 mM arsenite for 0, 3, 6, 12, or 24 h. In this experiment, arsenite enhanced the expression of HIF-2α and VEGF but not HIF-1α or MMP-9 (Fig. 1C and D).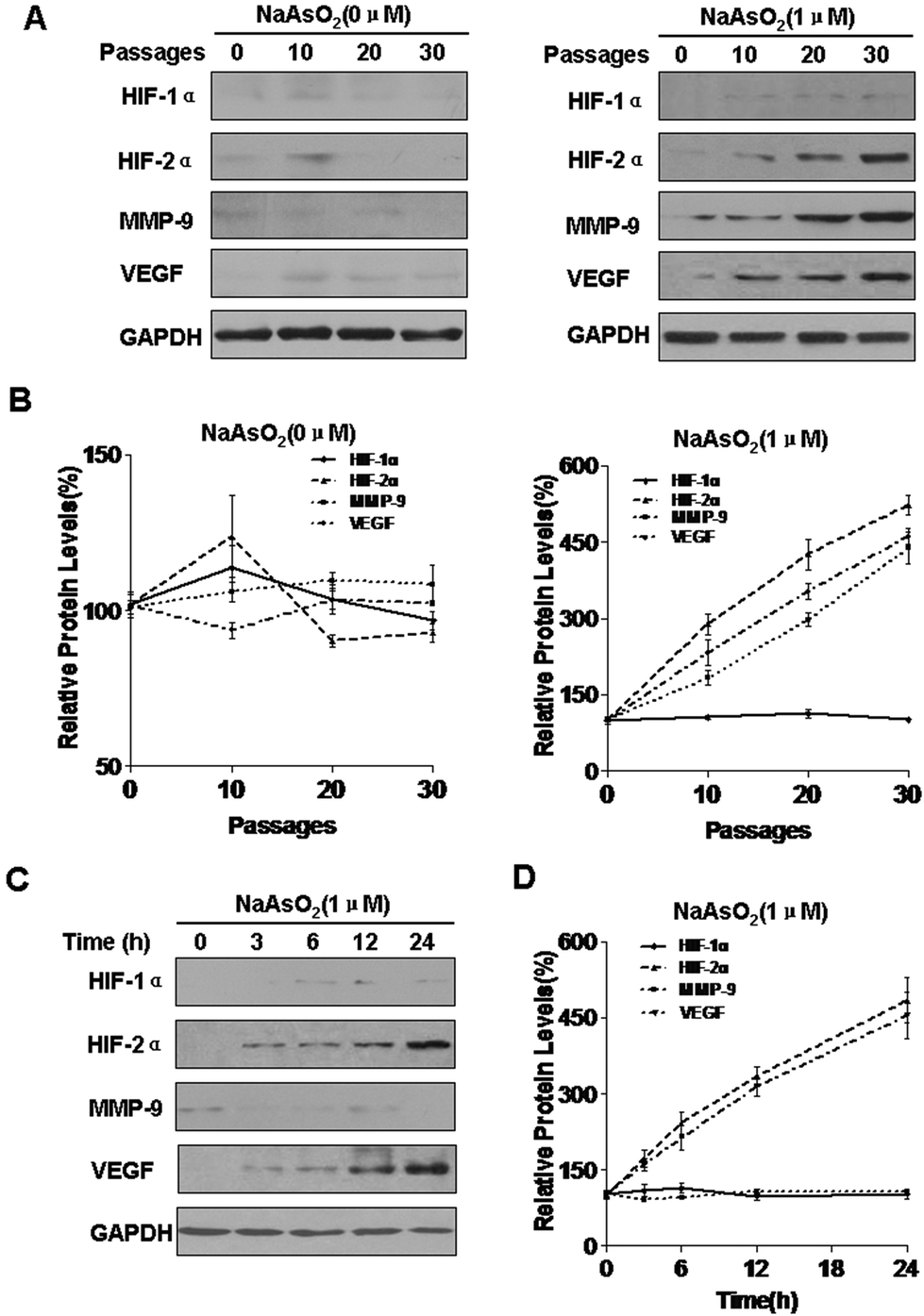 | ||
| Fig. 1 Exposure of HBE cells to arsenite increases the expression of HIF-2α, MMP-9, and VEGF. Densities of bands were quantified by using Eagle Eye II software. GAPDH levels, measured in parallel, served as controls. HBE cells were exposed to 0.0 or 1.0 μM arsenite for 0, 10, 20, or 30 passages. (A) Western blots and (B) relative protein levels (means ± SD, n = 3) of HIF-1α, HIF-2α, MMP-9, and VEGF. HBE cells were exposed to 1.0 μM arsenite for 0, 3, 6, 12, or 24 h. (C) Western blots and (D) relative protein levels (means ± SD, n = 3) of HIF-1α, HIF-2α, MMP-9, and VEGF. | ||
HIF-2α is involved in the increases of miR-191 and regulation of its target proteins induced by arsenite
miRNAs, a class of endogenous, small non-coding RNAs that are highly conserved among species, are aberrantly expressed in human malignancies and can act as onco-miRs or tumor suppressor miRs.37 We previously reported that exposure of HBE cells to arsenite increases the expression of miR-191, which is involved in the neoplastic and metastatic processes of transformed cells.15 However, the mechanisms by which this occurs are unclear. In pancreatic cancers, HIF-1α binds to HRE5 in the 5′-flanking region of miR-191 and initiates its transcription.12 We have reported that HIF-2α, but not HIF-1α, is up-regulated by arsenite and is involved in the arsenite-induced malignant transformation of HBE cells. Since HIF-2α has the same HRE binding sites as HIF-1α,16 we hypothesized that HIF-2α transcription regulates miR-191 expression in HBE cells. To assess the effects of HIF-2α on arsenite-induced increases of miR-191 expression, blockage of HIF-2α was accomplished with HIF-2α siRNA (Fig. S1A and B†) and with the HIF inhibitor, topotecan (Fig. S2A and B†). In arsenite-treated cells, reduction of HIF-2α reduced miR-191 expression (Fig. 2A and S2C†). BASP1, a target of miR-191, regulates WT1 transcriptional activity38 and induces its expression.15 Further, HIF-2α siRNA treatment restored the levels of BASP1 and reduced the levels of WT1 and VEGF (Fig. 2B and C). MMP-9 was not detectable in normal HBE cells (Fig. 2B). Thus, in HBE cells exposed to arsenite, HIF-2α is involved in the increases of miR-191 and in the regulation of its target proteins.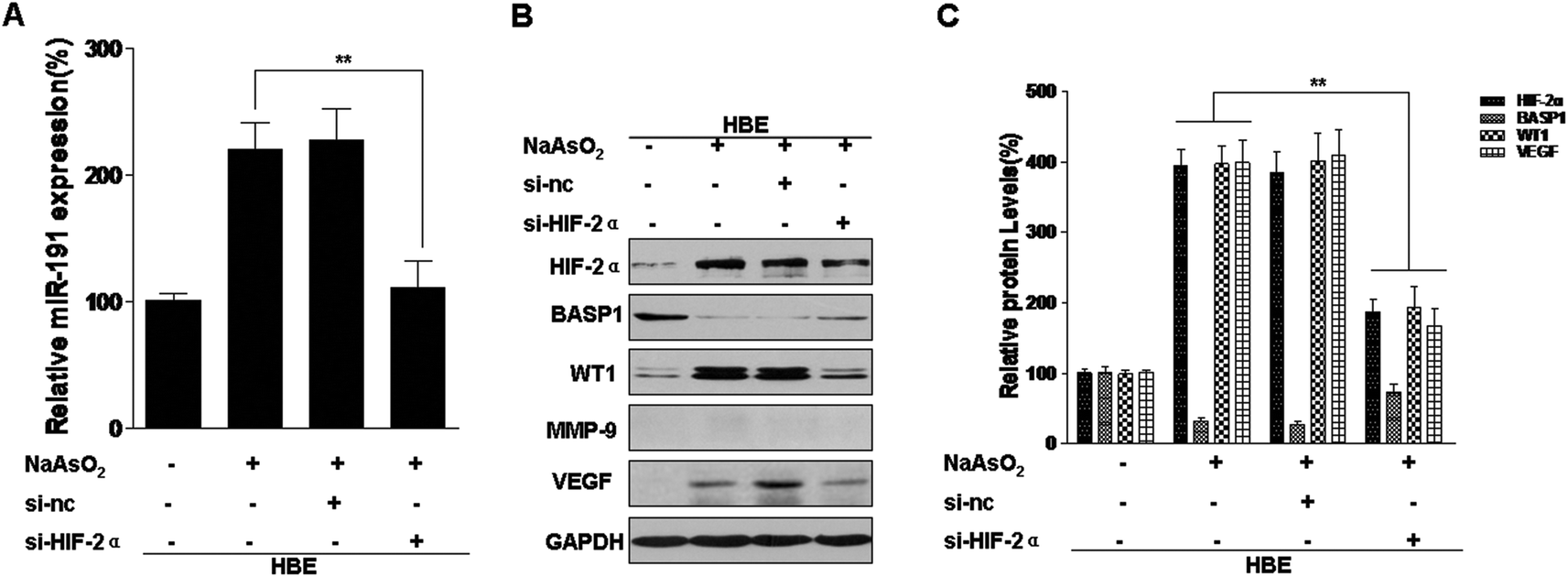 | ||
| Fig. 2 HIF-2α is involved in arsenite-induced increases of miR-191 levels and the regulation of its target proteins. Abbreviations: HBE, passage control HBE cells. Densities of bands were quantified by using Eagle Eye II software. GAPDH levels, measured in parallel, served as controls. After HBE cells were transfected with 25 nM control siRNA or 25 nM HIF-2α siRNA for 24 h, they were exposed to 0.0 or 1.0 μM arsenite for 24 h. (A) Levels of miR-191 were determined by qRT-PCR assays (means ± SD, n = 3). (B) Western blots were performed, and (C) levels of HIF-2α, BASP1, WT1, and VEGF were determined (means ± SD, n = 3). **P < 0.05 difference from arsenite-treated cells in the absence of HIF-2α siRNA. | ||
miR-191 is a transcriptional target of HIF-2α
HIFs were originally considered to be targets of miRNAs,26,39,40 but they can also induce the expression of miRNAs.27,41 Analysis of the promoter region of miR-191 by using the UCSC Genome Browser revealed that there were six putative HRE binding sites (Fig. 3A). HBE cells were exposed to 0 or 1.0 μM arsenite for 24 h, and the binding of HIF-2α to the miR-191 promoter was analyzed by the use of a ChIP assay. The results showed that, in HBE cells treated with arsenite, protein/DNA complexes immunoprecipitated with the anti-HIF-2α antibody resulted in a specific PCR product brighter in ChIP2 (HRE2 and HRE3) (Fig. 3B). To eliminate interference, the trials were repeated with an input positive control and an IgG negative control. The results were the same (Fig. 3C). Then, the effect of HIF-2α on miR-191 promoter activity was assessed. After HBE cells were co-transfected with miR-191 promoter plasmid, si-nc, or si-HIF-2α, they were exposed to 0 or 1.0 μM arsenite for 24 h. Luciferase assays indicated that, in HBE cells treated with arsenite, the activity of the miR-191 promoter was higher than in control HBE cells (Fig. 3D). Moreover, down-regulation of HIF-2α reduced luciferase activity, an effect similar to that caused by arsenite. Overall, these results suggest that HIF-2α increases miR-191 expression via binding to the HRE on the miR-191 promoter.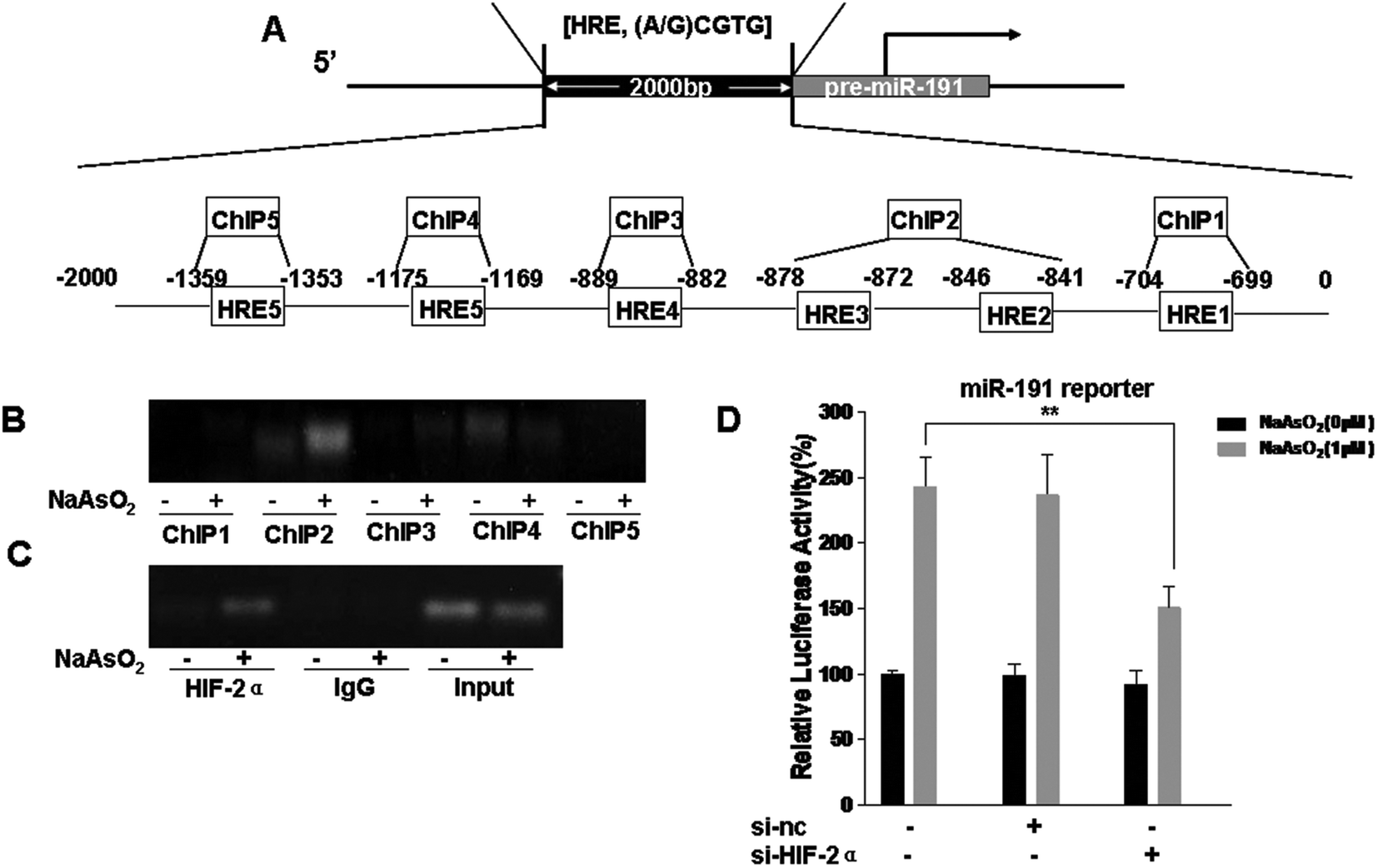 | ||
| Fig. 3 miR-191 is a transcriptional target of HIF-2α. (A) Schematic representation of 6 putative HRE sites in the promoter region of miR-191 that could bind HIF-2α. After HBE cells were co-transfected with luciferase reporters (containing the miR-191 promoter region) and 25 nM control siRNA or 25 nM HIF-2α siRNA for 24 h, they were exposed to 0 or 1.0 μM arsenite for 24 h. (B) ChIP assays using antibodies of HIF-2α for HRE1, HRE2 and HRE3, HRE4, HRE5, and HRE6 of the miR-191 promoter. (C) ChIP assays using antibodies of HIF-2α for the HRE2 and HRE3 of the miR-191 promoter in the input positive control and IgG negative control. (D) Luciferase activity was measured and normalized according to Renilla luciferase activity (means ± SD, n = 3); **P < 0.05 difference from arsenite-treated cells in the absence of HIF-2α siRNA. | ||
Involvement of HIF-2α in the decreases of BASP1 and the increases of WT1, MMP9, and VEGF levels in T-HBE cells via miR-191
To elucidate the effects of HIF-2α regulation of BASP1 and its downstream genes via miR-191, T-HBE cells were treated with HIF-2α siRNA, which reduced the levels of HIF-2α (Fig. 4A and B). The levels of BASP1 were restored by si-HIF-2α, but the levels of WT1, MMP-9, and VEGF were reduced (Fig. 4A). In T-HBE cells co-transfected with HIF-2α siRNA and the miR-191 mimic, BASP1 levels were decreased and those for WT1, MMP-9, and VEGF were increased (Fig. 4A and B). A similar miR-191 expression was found by a qRT-PCR assay (Fig. 4C). | ||
| Fig. 4 HIF-2α, acting via miR-191, is involved in the decreases of BASP1 and the increases of WT1, MMP9, and VEGF levels in T-HBE cells. Abbreviations: C-HBE, passage control HBE cells; T-HBE, arsenite-transformed HBE cells. Densities of bands were quantified by using Eagle Eye II software. GAPDH levels, measured in parallel, served as controls. After T-HBE cells were transfected with 25 nM control siRNA or 25 nM HIF-2α siRNA for 24 h, they were transfected with 80 nM miR-mimic-nc or 80 nM miR-191 mimic for 24 h. (A) Western blots and (B) relative protein levels of HIF-2α, BASP1, WT1, MMP-9, and VEGF were determined (means ± SD, n = 3). (C) Levels of miR-191 were determined by qRT-PCR assays (means ± SD, n = 3). **P < 0.05 difference from T-HBE cells. ##P < 0.05 difference from T-HBE cells transfected with HIF-2α siRNA. | ||
Involvement of HIF-2α in the secretion of VEGF and angiogenesis in T-HBE cells via miR-191
HIF-2α is involved in various processes associated with cancer, including tumorigenesis, angiogenesis, and metastasis.23 Our previous study confirmed that conditioned medium from arsenite-transformed cells cultured in the absence of arsenite increased tube formation by HUVECs.42 To determine whether HIF-2α affects the angiogenesis of arsenite-induced T-HBE cells via miR-191, appropriate assays were performed. Down-regulation of HIF-2α inhibited the secretion of VEGF into the medium (Fig. 5A) and tube formation in HUVECs (Fig. 5B and C). The transformed cells were co-transfected with HIF-2α siRNA and the miR-191 mimic. The effects of HIF-2α inhibition on decreasing VEGF secretion and tube formation were reversed by the miR-191 mimic (Fig. 5 A–C), demonstrating that, in transformed cells, miR-191 is required for HIF-2α-dependent angiogenesis. In summary, these results indicate that HIF-2α is involved in the secretion of VEGF and angiogenesis in T-HBE cells via miR-191.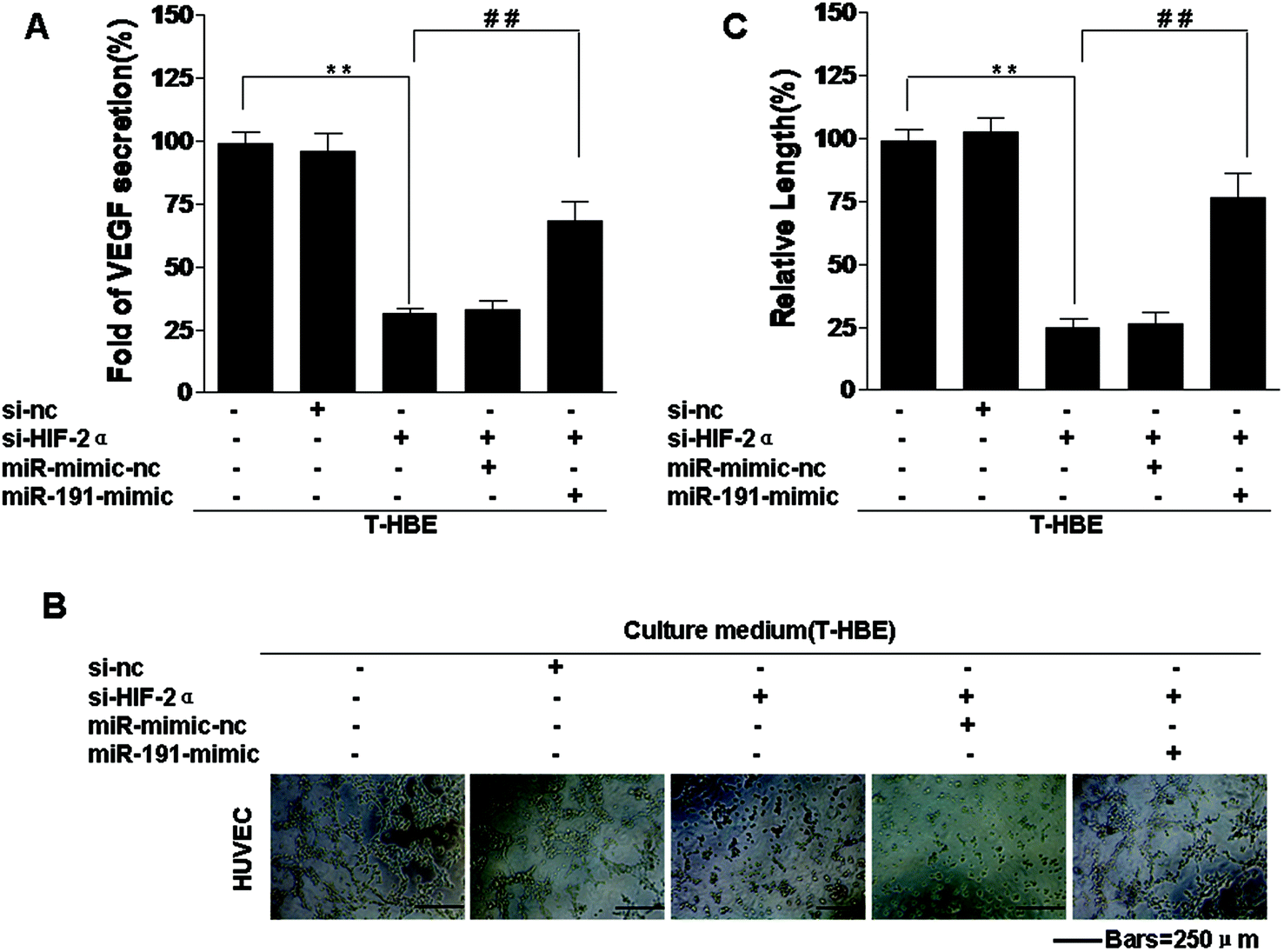 | ||
| Fig. 5 Involvement of HIF-2α in the increase of VEGF secretion and angiogenesis in T-HBE cells via miR-191. Abbreviations: C-HBE, passage control HBE cells; T-HBE, arsenite-transformed HBE cells. After T-HBE cells were transfected with 25 nM control siRNA or 25 nM HIF-2α siRNA for 24 h, they were transfected with 80 nM miR-mimic-nc or 80 nM miR-191 mimic for 24 h. (A) VEGF secretion, measured by ELISA, in the culture medium. HUVEC cells, treated for 6 h with conditioned media from cells in the upper chamber, were seeded onto Matrigel for 12 h. (B) Tube formation of HUVEC cells (bars = 250 μm) and (C) relative tube lengths of HUVEC cells. **P < 0.05 difference from T-HBE cells. ##P < 0.05 difference from T-HBE cells transfected with HIF-2α siRNA. | ||
Involvement of HIF-2α in the neoplastic and metastatic properties of T-HBE cells via miR-191
Previous studies have established that HIF-2α and miR-191 are associated with the malignant transformation of HBE cells.15,24 Since miR-191 is induced by HIF-2α (Fig. 2A), we determined whether HIF-2α affected the neoplastic and metastatic capacities of T-HBE cells via miR-191. Down-regulation of HIF-2α inhibited their capacities for colony formation (Fig. 6A and B) and migration and invasion (Fig. 6C and D). The transformed cells were co-transfected with HIF-2α siRNA and the miR-191-mimic and examined for their colony formation, migration, and invasion capacities. The effects of HIF-2α inhibition on decreasing colony formation (Fig. 6A and B), migration, and invasion (Fig. 6C and D) were reversed by the presence of the miR-191-mimic, indicating that miR-191 is required for these HIF-2α-dependent properties of transformed cells. These results indicate the involvement of HIF-2α in the neoplastic and metastatic properties of T-HBE cells via miR-191.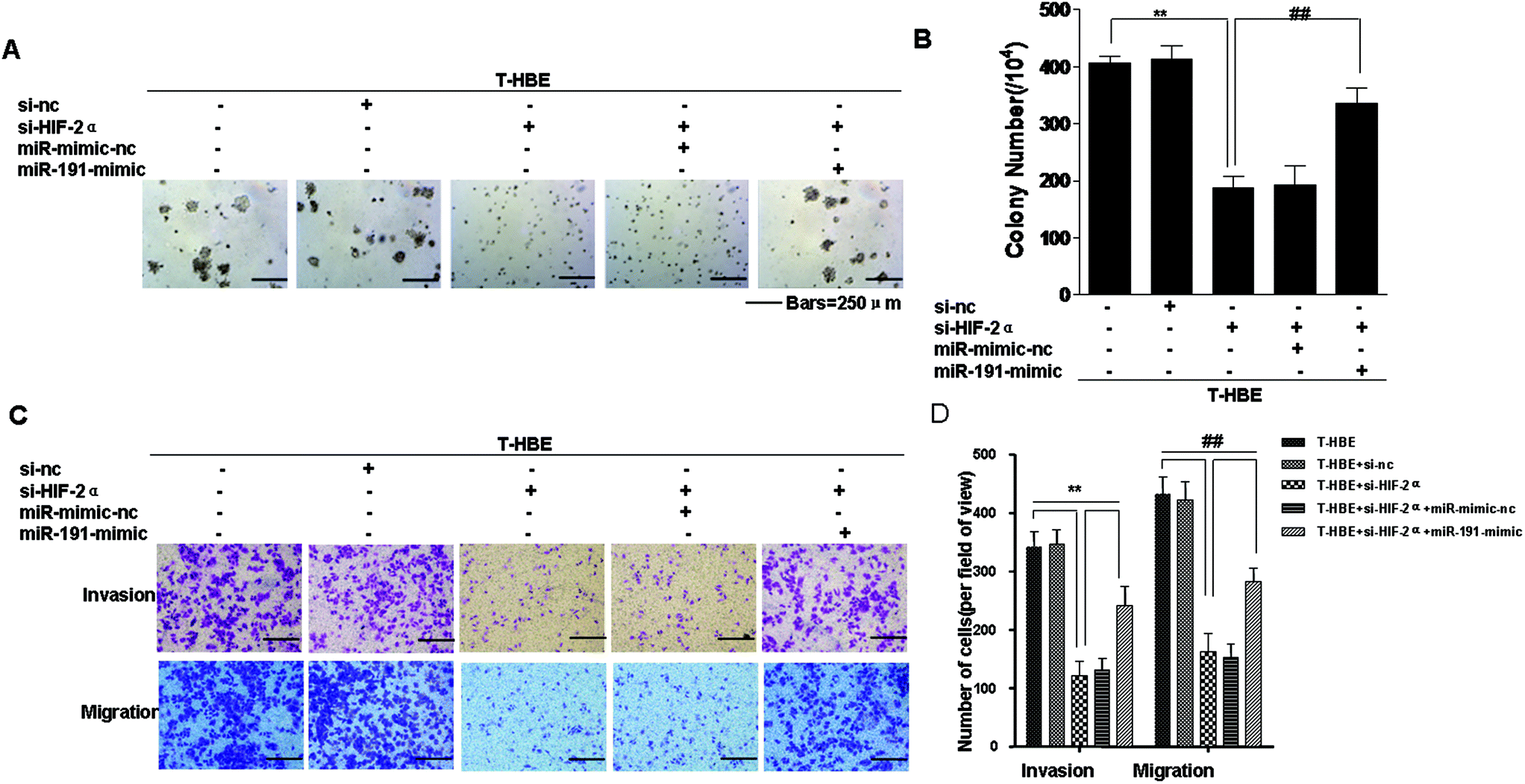 | ||
| Fig. 6 Involvement of HIF-2α in the neoplastic and metastatic properties of T-HBE cells via miR-191. Abbreviations: C-HBE, passage control HBE cells; T-HBE, arsenite-transformed HBE cells. After T-HBE cells were transfected with 25 nM control siRNA or 25 nM HIF-2α siRNA for 24 h, they were transfected with 80 nM miR-mimic-nc or 80 nM miR-191 mimic for 24 h. (A) Colony formation was assessed in soft agar (bars = 250 μm), and (B) their numbers (means ± SD, n = 3) were quantified. (C) Representative images of cell invasion and migration in Transwell assays (bars = 250 μm). (D) Relative levels of cell migratory and invasive capacities were determined. **P < 0.05 difference from T-HBE cells. ##P < 0.05 difference from T-HBE cells transfected with HIF-2α siRNA. | ||
Involvement of HIF-2α in the regulation of MMP-9 and VEGF levels and the angiogenesis of T-HBE cells via WT1
The WT1 gene was initially identified as a tumor suppressor due to its inactivation in Wilms’ tumor (nephroblastoma), the most common pediatric kidney tumor.43 However, WT1 also acts as an oncogene in lung cancer and other tumors.44 MMP-9 and VEGF are targets of WT1,36 and WT1 is a regulator of tumor angiogenesis and metastasis.45 However, the function of WT1 in the malignant transformation of HBE cells has not been established. To determine whether WT1 is involved in the angiogenesis of T-HBE cells, plasmids were used. Transformed cells, co-transfected with HIF-2α siRNA and pEGFP-WT1, were examined for VEGF secretion and tube formation. HIF-2α and BASP1 levels were decreased, and WT1, MMP-9, and VEGF levels were increased (Fig. 7A and B). In transformed cells, the effect of HIF-2α inhibition on decreasing VEGF secretion and tube formation was reversed by WT1 over-expression (Fig. 7C–E), indicating that WT1 is required for HIF-2α-dependent angiogenesis. These results show that, in T-HBE cells, HIF-2α is involved in the secretion of VEGF and angiogenesis via WT1.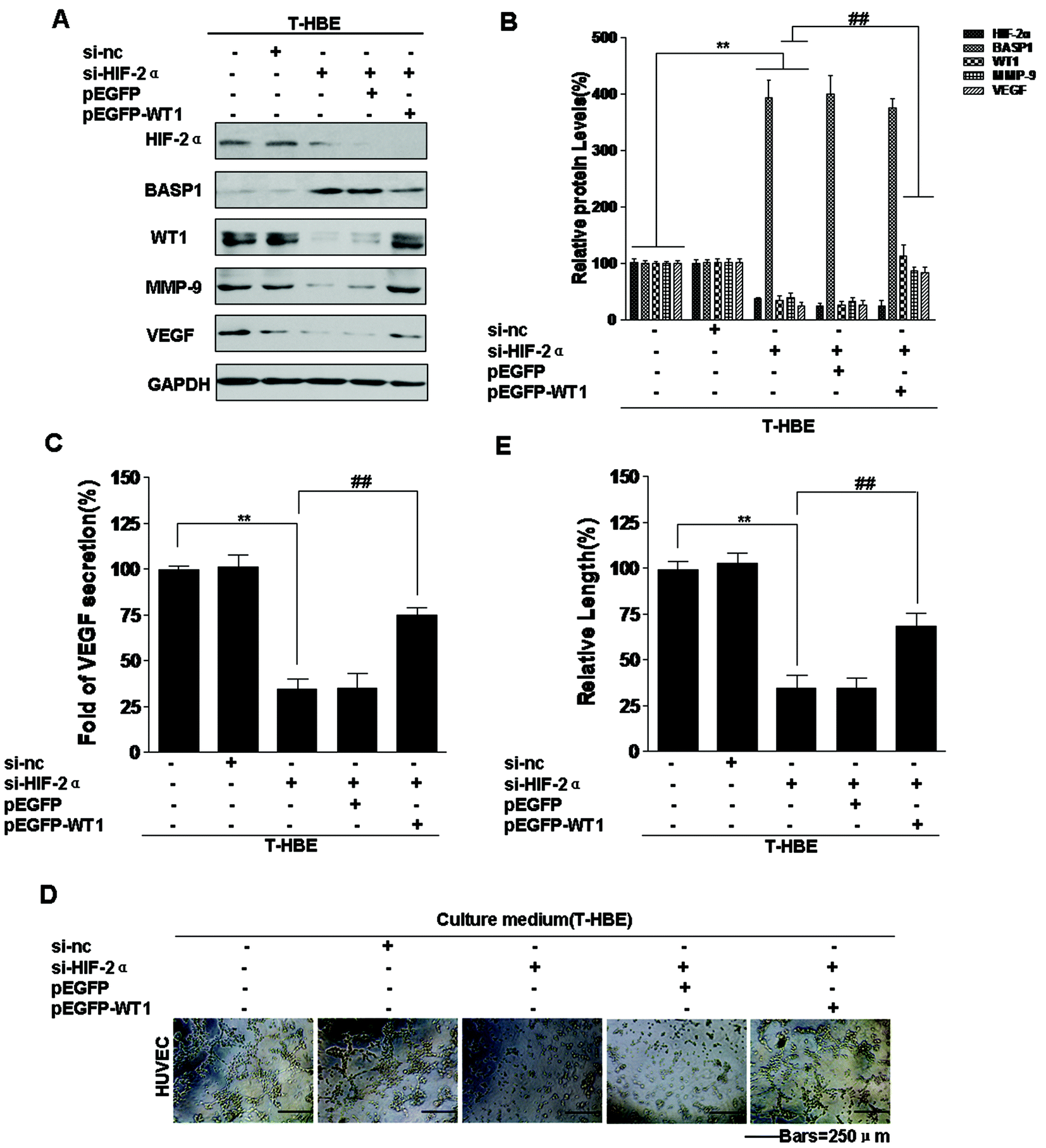 | ||
| Fig. 7 Involvement of HIF-2α in the increases of MMP-9 and VEGF levels and angiogenesis of T-HBE cells via WT1. Abbreviations: C-HBE, passage control HBE cells; T-HBE, arsenite-transformed HBE cells. Densities of bands were quantified by using Eagle Eye II software. GAPDH levels, measured in parallel, served as controls. After T-HBE cells were transfected with 25 nM control siRNA or 25 nM HIF-2α siRNA for 24 h, they were transfected with 15 μg of pEGFP vector plasmid or pEGFP-WT1 plasmid for 24 h. (A) Western blots and (B) relative protein levels of HIF-2α, BASP1, WT1, MMP-9, and VEGF were determined (means ± SD, n = 3). (C) ELISA analysis of VEGF secreted into the culture medium. HUVEC cells, which were treated with conditioned medium from cells in the upper chamber for 6 h, were seeded onto Matrigel for 12 h. (D) Tube formation of HUVEC cells (bars = 250 μm) and (E) the relative tube lengths of HUVEC cells. **P < 0.05 difference from T-HBE cells. ##P < 0.05 difference from T-HBE cells transfected with HIF-2α siRNA. | ||
Involvement of HIF-2α in the neoplastic and metastatic properties of T-HBE cells via WT1
Since our previous study established that miR-191 is involved in the neoplastic and metastatic properties of transformed cells and that miR-191 regulates the levels of WT1 by BASP1,15 we determined whether the expression of HIF-2α affected the neoplastic capacity and metastatic properties of T-HBE cells via WT1. Transformed cells were co-transfected with HIF-2α siRNA and pEGFP-WT1, and their capacity for colony formation was evaluated. The effects of HIF-2α inhibition on decreasing colony formation (Fig. 8A and B) and on migration and invasion were reversed by over-expression of WT1 (Fig. 8C and D), indicating that WT1 is required for these HIF-2α-dependent properties. The results demonstrate that HIF-2α and WT1 are involved in the neoplastic and metastatic properties of T-HBE cells. | ||
| Fig. 8 Involvement of HIF-2α in the neoplastic and metastatic properties of T-HBE cells via WT1. Abbreviations: C-HBE, passage control HBE cells; T-HBE, arsenite-transformed HBE cells. After T-HBE cells were transfected with 25 nM control siRNA or 25 nM HIF-2α siRNA for 24 h, they were transfected with 15 μg of pEGFP vector plasmid or pEGFP-WT1 plasmid for 24 h. (A) Colony formation was assessed in soft agar (bars = 250 μm), and (B) their numbers (means ± SD, n = 3) were quantified. (C) Representative images of cell invasion and cell migration in Transwell assays (bars = 250 μm) and (D) relative levels of cell migratory and invasive capacities were determined. **P < 0.05 difference from T-HBE cells. ##P < 0.05 difference from T-HBE cells transfected with HIF-2α siRNA. | ||
Discussion
Inorganic arsenite is a widely distributed, naturally occurring environmental contaminant affecting tens of millions of people worldwide.46 A close association exists between arsenite exposure and increased incidence of various forms of cancer, as documented from the studies of different arsenite-endemic areas in the world.47 Although there is evidence for the lung carcinogenicity of inorganic arsenic compounds in humans, the molecular mechanisms remain incompletely defined.miRNAs are non-protein-encoding RNA oligonucleotides that regulate gene expression.48 Abnormal expression of miRNAs is associated with cancer metastasis.49 miR-191 is over-expressed in malignancies, including breast,11 lung,9 and prostate cancers,50 but is decreased in chronic lymphocytic leukemia.51 Although miR-191 is linked to cancer, little is known about the factors that lead to its deregulation in tumor cells. We previously demonstrated that exposure of HBE cells to arsenite increases the expression of miR-191,15 and in that study we have performed miRNA array analyses in normal HBE cells and T-HBE cells to acquire miRNA expression profiles. Relative to normal HBE cells, the expression of 30 miRNAs was up-regulated in T-HBE cells. To validate their fold change in the miRNA array, miR-191 and other miRNAs were examined in normal HBE cells and T-HBE cells by qRT-PCR. The results showed that the miR-191 levels were up-regulated at the highest fold change; thus miR-191 was selected for further study. However, the mechanisms by which this occurs are unclear. In the present investigation, we provide evidence that miR-191 is a transcriptional target of HIF-2α and contributes to the angiogenic and metastatic properties of T-HBE cells.
HIF-2α is increased in a variety of human tumors and correlates with tumor angiogenesis and patient mortality.26 Although it can be induced by non-hypoxic mechanisms involving growth factor-induced or oncogenic activation of translational and transcriptional pathways, our previous study established that a low level of arsenite up-regulates the stabilization and transactivation of HIF-2α, but not that of HIF-1α, by inhibiting the ubiquitin-proteasome pathway in normoxia.24 The present results show that, with long-term exposure of HBE cells to arsenite, the expression of HIF-1α is not appreciably changed, the results that are consistent with our previous study,24 and that HIF-2α, MMP-9, and VEGF are increased. Since MMP-9 activity is an excellent marker for arsenic-induced malignant transformation,52 the MMP-9 levels of HBE cells were very low, which is consistent with their weak neoplastic properties, in an experiment involving acute exposure. According to HIF-1α regulating the expression of some miRNAs,39,53 we speculated that there is a relationship between HIF-2α and miR-191 and assessed the effects of HIF-2α on arsenite-induced increases of miR-191 expression. Blockage of HIF-2α was accomplished with HIF-2α siRNA and the HIF inhibitor, topotecan. In arsenite-treated cells, reduction of HIF-2α reduced miR-191 expression and restored the levels of BASP1, a target of miR-191.15 Moreover, WT1, MMP-9, and VEGF, which are downstream genes of BASP1, are also regulated by HIF-2α. Thus, we hypothesized that the arsenite-induced increase of miR-191 expression was via HIF-2α.
To determine whether HIF-2α regulates miR-191 transcription, we searched the promoter regions of miR-191 by the use of the UCSC Genome Browser and found 6 putative HRE sites that can bind HIF-2α. HBE cells were then exposed to 0 or 1.0 μM arsenite for 24 h, and the binding of HIF-2α to the miR-191 promoter was analyzed by the use of a ChIP assay. The results showed that, in HBE cells treated with arsenite, protein/DNA complexes immunoprecipitated with the anti-HIF-2α antibody resulted in a specific PCR product brighter in ChIP2, which suggested that HIF-2α binds to HRE2 and HRE3. Song Z et al. demonstrated that HIF-1α binds to HRE5 in hypoxia to initiate the transcription of miR-191.12 The reason for these differences between our data and their findings may be because HIF-1α and HIF-2α are similar, but not identical, and because the cell lines chosen for the study were different.
In addition, we evaluated the effect of HIF-2α on miR-191 promoter activity by luciferase assays. These activities in HBE cells transfected with the miR-191 promoter Luc construct and exposed to arsenite were lower than those in control HBE cells. Moreover, down-regulation of HIF-2α by transfection with HIF-2α siRNA reduced the luciferase activities of cells co-transfected with the miR-191 promoter Luc construct, an effect that was blocked by arsenite. These results indicate that miR-191 is a transcriptional target of HIF-2α.
Metastasis is a multistep process made up of a cascade of sequential steps involving changes in cellular invasion, migration, adhesion, movement of cancer cells through the circulatory system, and their re-implantation within an organ located at a distant site in the body, followed by colonization, tumor growth, and the associated formation of new capillaries.54 Metastasis is the major cause of death among cancer patients.55 Thus, an understanding of the mechanisms underlying metastasis development and progression is essential for improving the treatment of lung cancer.
Angiogenesis is characterized by the formation of capillary blood vessels that originate from pre-existing vasculature. This process is essential to provide nutrients, oxygen, and growth factors that maintain cell function and survival.56 Therefore, angiogenesis is associated with tumor growth and metastasis.57,58 Although previous studies showed that short-term arsenite treatment of HUVECs stimulated angiogenesis,42 it remains to be determined whether media of T-HBE cells have an angiogenic effect. In humans, HIF-2α is commonly increased in a variety of human tumors and correlates with tumor angiogenesis and patient mortality.59 Our previous studies have established that HIF-2α expression elevates arsenite-induced proliferation and inhibits p53 activation, processes that are involved in malignant transformation.24 However, the role of HIF-2α in the angiogenesis of T-HBE cells is not known. In the present study, after HIF-2α was knocked down in T-HBE cells by siRNA, the levels of BASP1 were restored, and the levels of WT1, MMP-9, and VEGF were reduced. In cells co-transfected with HIF-2α siRNA and the miR-191 mimic, BASP1 levels were decreased and WT1, MMP-9, and VEGF levels were increased.
VEGF, a secreted protein, is involved in angiogenesis.60 Down-regulation of HIF-2α inhibited the secretion of VEGF and tube formation in HUVECs. When transformed cells were co-transfected with miR-HIF-2α siRNA and the miR-191 mimic, the effect of HIF-2α inhibition on decreasing VEGF secretion and tube formation was reversed by the miR-191 mimic. Thus, these results indicate that, in T-HBE cells, HIF-2α is involved in the secretion of VEGF and angiogenesis via miR-191.
HIF-2α and miR-191 are associated with the malignant transformation of HBE cells.15,24 Since miR-191 is induced by HIF-2α, we determined whether HIF-2α affects the neoplastic and metastatic capacities of T-HBE cells via miR-191. The results were consistent with our hypothesis. Knockdown of HIF-2α inhibited the capacity of transformed cells for colony formation, migration, and invasion. Transformed cells were co-transfected with HIF-2α siRNAs and the miR-191-mimic and examined their capacities for colony formation, migration, and invasion. The effects of HIF-2α inhibition on decreasing these properties were reversed by the miR-191-mimic, indicating that miR-191 is required for HIF-2α-dependent colony formation, migration, and invasion.
WT1, a transcriptional regulatory protein, is over-expressed in a variety of tumor types, including leukemia, breast cancer, and sarcomas.61–64 Although WT1 has been implicated in the regulation of angiogenesis and metastasis,45,65 its role in T-HBE cells remains unclear. BASP1 binds to the suppression domain of WT1 (residues 71–101) and inhibits its transcriptional activation.38 Our previous study confirmed that miR-191 targets BASP1 to up-regulate the expression of WT1.15 Our present results show that, in T-HBE cells, up-regulation of WT1 levels increases MMP-9 and VEGF. The effect of HIF-2α inhibition on decreasing VEGF secretion and tube formation was reversed by over-expression of WT1. Thus, WT1 is involved in the regulation of MMP-9 and VEGF levels and in the angiogenesis of T-HBE cells.
Since our previous study established that miR-191 is involved in the neoplastic and metastatic properties of transformed cells and that miR-191 regulates the levels of WT1 by BASP1,15 we determined whether the expression of WT1 affected these characteristics. The effect of HIF-2α inhibition on decreasing colony formation, migration, and invasion was reversed by WT1 over-expression, indicating that WT1 is required for these HIF-2α-dependent properties.
In summary, during arsenite-induced malignant transformation of HBE cells, HIF-2α increases WT1 levels via miR-191. HIF-2α binds to the HRE in the promoter region of miR-191 and initiates its transcription. The results demonstrate that HIF-2α up-regulates WT1 via miR-191, which is also involved in the angiogenic, neoplastic, and metastatic properties of transformed cells. The results elucidate the mechanisms of arsenite-induced angiogenesis and transformation of HBE cells and provide a link between HIFs, miRNAs, and malignant transformation, thereby contributing to an understanding of the processes involved in lung cancer caused by arsenite exposure and providing a new approach to the development of treatments for lung cancer.
Competing interests declaration
The authors declare that they have no competing financial interests.Acknowledgements
The authors wish to thank Donald L. Hill (University of Alabama at Birmingham, USA) for editing. This work was supported by the Natural Science Foundation of China (81273114 and 81430077), the Postgraduate Innovation Project of Jiangsu province (CXZZ14_0421, CXZZ14_0951, CXZZ13_0600, and KYLX15_0974), and the Priority Academic Program Development of Jiangsu Higher Education Institutions (2010).References
- World Health Organization, IARC monographs on the evaluation of carcinogenic risks to humans, International Agency for Research on Cancer, 2012, vol. 100, 11–465 Search PubMed.
- IARC monographs on the evaluation of the carcinogenic risk of chemicals to humans, 1980, 23, 39–141.
- C. L. Chen, L. I. Hsu, H. Y. Chiou, Y. M. Hsueh, S. Y. Chen, M. M. Wu and C. J. Chen, JAMA, 2004, 292, 2984–2990 CrossRef CAS PubMed.
- A. Jemal, F. Bray, M. M. Center, J. Ferlay, E. Ward and D. Forman, Ca-Cancer J. Clin., 2011, 61, 69–90 CrossRef.
- J. Folkman and Y. Shing, J. Biol. Chem., 1992, 267, 10931–10934 CAS.
- Z. Wang, C. Dabrosin, X. Yin, M. M. Fuster, A. Arreola, W. K. Rathmell, D. Generali, G. P. Nagaraju, B. El-Rayes, D. Ribatti, Y. C. Chen, K. Honoki, H. Fujii, A. G. Georgakilas, S. Nowsheen, A. Amedei, E. Niccolai, A. Amin, S. S. Ashraf, B. Helferich, X. Yang, G. Guha, D. Bhakta, M. R. Ciriolo, K. Aquilano, S. Chen, D. Halicka, S. I. Mohammed, A. Azmi, A. Bilsland, N. Keith and L. D. Jensen, Semin. Cancer Biol., 2015 DOI:10.1016/j.semcancer.2015.01.001.
- E. van Rooij, Circ. Res., 2011, 108, 219–234 CrossRef CAS.
- W. Sun, Y. S. Julie Li, H. D. Huang, J. Y. Shyy and S. Chien, Annu. Rev. Biomed. Eng., 2010, 12, 1–27 CrossRef CAS PubMed.
- S. K. Patnaik, E. Kannisto and S. Yendamuri, PLoS One, 2010, 5, e9219 Search PubMed.
- Y. He, Y. Cui, W. Wang, J. Gu, S. Guo, K. Ma and X. Luo, Neoplasia, 2011, 13, 841–853 CrossRef CAS PubMed.
- B. Wang, J. Li, M. Sun, L. Sun and X. Zhang, IUBMB Life, 2014, 66, 371–377 CrossRef CAS PubMed.
- Z. Song, H. Ren, S. Gao, X. Zhao, H. Zhang and J. Hao, J. Int. Soc. Oncodev. Biol. Med., 2014, 35, 11319–11328 CrossRef CAS.
- W. Z. Peng, R. Ma, F. Wang, J. Yu and Z. B. Liu, Int. J. Mol. Sci., 2014, 15, 4031–4048 CrossRef CAS.
- N. Nagpal and R. Kulshreshtha, Front. Genet., 2014, 5, 99 CrossRef.
- W. Xu, J. Ji, Y. Xu, Y. Liu, L. Shi, X. Lu, Y. Zhao, F. Luo, B. Wang, R. Jiang, J. Zhang and Q. Liu, Mol. Carcinog., 2015, 54(Suppl. 1), E148–E161 CrossRef CAS PubMed.
- A. Loboda, A. Jozkowicz and J. Dulak, Mol. Cells, 2010, 29, 435–442 CrossRef CAS PubMed.
- G. L. Semenza, Trends Mol. Med., 2012, 18, 534–543 CrossRef CAS.
- C. Martinengo, T. Poggio, M. Menotti, M. S. Scalzo, C. Mastini, C. Ambrogio, E. Pellegrino, L. Riera, R. Piva, D. Ribatti, F. Pastorino, P. Perri, M. Ponzoni, Q. Wang, C. Voena and R. Chiarle, Cancer Res., 2014, 74, 6094–6106 CrossRef CAS PubMed.
- D. Zhao, B. Zhai, C. He, G. Tan, X. Jiang, S. Pan, X. Dong, Z. Wei, L. Ma, H. Qiao, H. Jiang and X. Sun, Cell. Signalling, 2014, 26, 1030–1039 CrossRef CAS PubMed.
- C. J. Hu, L. Y. Wang, L. A. Chodosh, B. Keith and M. C. Simon, Mol. Cell. Biol., 2003, 23, 9361–9374 CrossRef CAS PubMed.
- H. Menrad, C. Werno, T. Schmid, E. Copanaki, T. Deller, N. Dehne and B. Brune, Hepatology, 2010, 51, 2183–2192 CrossRef CAS PubMed.
- B. Keith, R. S. Johnson and M. C. Simon, Nat. Rev. Cancer, 2012, 12, 9–22 CAS.
- J. Zhao, F. Du, G. Shen, F. Zheng and B. Xu, Cell Death Dis., 2015, 6, e1600 CrossRef CAS PubMed.
- Y. Xu, Y. Li, Y. Pang, M. Ling, L. Shen, R. Jiang, Y. Zhao, J. Zhou, X. Wang and Q. Liu, Arch. Toxicol., 2012, 86, 947–959 CrossRef CAS PubMed.
- H. Xu, L. Zhao, Q. Fang, J. Sun, S. Zhang, C. Zhan, S. Liu and Y. Zhang, PLoS One, 2014, 9, e115565 Search PubMed.
- H. Zhang, J. Pu, T. Qi, M. Qi, C. Yang, S. Li, K. Huang, L. Zheng and Q. Tong, Oncogene, 2014, 33, 387–397 CrossRef CAS PubMed.
- S. Y. Chan and J. Loscalzo, Cell Cycle, 2010, 9, 1072–1083 CrossRef CAS.
- R. R. Reddel, Y. Ke, B. I. Gerwin, M. G. McMenamin, J. F. Lechner, R. T. Su, D. E. Brash, J. B. Park, J. S. Rhim and C. C. Harris, Cancer Res., 1988, 48, 1904–1909 CAS.
- Y. Xu, Y. Li, Y. Pang, M. Ling, L. Shen, X. Yang, J. Zhang, J. Zhou, X. Wang and Q. Liu, PLoS One, 2012, 7, e37765 CAS.
- H. Zhang, L. Yang, X. Teng, Z. Liu, C. Liu and L. Zhang, J. Int. Soc. Oncodev. Biol. Med., 2015 DOI:10.1007/s13277-015-4051-5.
- K. J. Livak and T. D. Schmittgen, Methods, 2001, 25, 402–408 CrossRef CAS PubMed.
- W. Guo, Z. Qiu, Z. Wang, Q. Wang, N. Tan, T. Chen, Z. Chen, S. Huang, J. Gu, J. Li, M. Yao, Y. Zhao and X. He, Hepatology, 2015, 62, 1132–1144 CrossRef CAS PubMed.
- B. Philip, K. Ito, R. Moreno-Sanchez and S. J. Ralph, Carcinogenesis, 2013, 34, 1699–1707 CrossRef CAS PubMed.
- H. Li, K. Zhang, L. H. Liu, Y. Ouyang, J. Bu, H. B. Guo and T. Xiao, J. Int. Soc. Oncodev. Biol. Med., 2014, 35, 5487–5491 CrossRef CAS PubMed.
- N. Ferrara and S. Bunting, Curr. Opin. Nephrol. Hypertens., 1996, 5, 35–44 CrossRef CAS PubMed.
- V. Katuri, S. Gerber, X. Qiu, G. McCarty, S. D. Goldstein, H. Hammers, E. Montgomery, A. R. Chen and D. M. Loeb, OncoTargets Ther., 2014, 5, 2436–2449 CrossRef.
- S. H. Chan and L. H. Wang, J. Biomed. Sci., 2015, 22, 9 CrossRef CAS PubMed.
- B. Carpenter, K. J. Hill, M. Charalambous, K. J. Wagner, D. Lahiri, D. I. James, J. S. Andersen, V. Schumacher, B. Royer-Pokora, M. Mann, A. Ward and S. G. Roberts, Mol. Cell. Biol., 2004, 24, 537–549 CrossRef CAS PubMed.
- Y. Liu, H. Nie, K. Zhang, D. Ma, G. Yang, Z. Zheng, K. Liu, B. Yu, C. Zhai and S. Yang, FEBS Lett., 2014, 588, 3137–3146 CrossRef CAS PubMed.
- T. Umezu, H. Tadokoro, K. Azuma, S. Yoshizawa, K. Ohyashiki and J. H. Ohyashiki, Blood, 2014, 124, 3748–3757 CrossRef CAS PubMed.
- J. K. Seok, S. H. Lee, M. J. Kim and Y. M. Lee, Nucleic Acids Res., 2014, 42, 8062–8072 CrossRef CAS PubMed.
- Y. Zhao, Y. Xu, F. Luo, W. Xu, B. Wang, Y. Pang, J. Zhou, X. Wang and Q. Liu, Toxicol. Lett., 2013, 223, 35–41 CrossRef CAS PubMed.
- K. M. Call, T. Glaser, C. Y. Ito, A. J. Buckler, J. Pelletier, D. A. Haber, E. A. Rose, A. Kral, H. Yeger and W. H. Lewis, et al. , Cell, 1990, 60, 509–520 CrossRef CAS PubMed.
- I. Lindstedt, M. A. Lindgren, E. Andersson and W. Engstrom, In Vivo, 2014, 28, 675–681 CAS.
- K. D. Wagner, J. Cherfils-Vicini, N. Hosen, P. Hohenstein, E. Gilson, N. D. Hastie, J. F. Michiels and N. Wagner, Nat. Commun., 2014, 5, 5852 CrossRef CAS.
- IARC, IARC monographs on the evaluation of carcinogenic risks to humans/World Health Organization, International Agency for Research on Cancer, 2004, vol. 84, 269–477 Search PubMed.
- K. P. Cantor and J. H. Lubin, Toxicol. Appl. Pharmacol., 2007, 222, 252–257 CrossRef CAS PubMed.
- C. Augello, V. Vaira, L. Caruso, A. Destro, M. Maggioni, Y. N. Park, M. Montorsi, R. Santambrogio, M. Roncalli and S. Bosari, Liver Int., 2012, 32, 772–782 CrossRef CAS.
- Y. Wang, S. Kim and I. M. Kim, Front. Radiat. Oncol., 2014, 4, 143 Search PubMed.
- S. Stegeman, L. Moya, L. A. Selth, A. B. Spurdle, J. Clements and J. Batra, Endocr.-Relat. Cancer, 2015, 22(2), 265–276 CrossRef CAS PubMed.
- G. A. Calin, C. G. Liu, C. Sevignani, M. Ferracin, N. Felli, C. D. Dumitru, M. Shimizu, A. Cimmino, S. Zupo, M. Dono, M. L. Dell'Aquila, H. Alder, L. Rassenti, T. J. Kipps, F. Bullrich, M. Negrini and C. M. Croce, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 11755–11760 CrossRef CAS PubMed.
- J. Pi, B. A. Diwan, Y. Sun, J. Liu, W. Qu, Y. He, M. Styblo and M. P. Waalkes, Free Radical Biol. Med., 2008, 45, 651–658 CrossRef CAS PubMed.
- L. Li, K. Huang, Y. You, X. Fu, L. Hu, L. Song and Y. Meng, Int. J. Oncol., 2014, 44, 2111–2120 CAS.
- X. Xiao, Z. Liu, R. Wang, J. Wang, S. Zhang, X. Cai, K. Wu, R. C. Bergan, L. Xu and D. Fan, OncoTargets Ther., 2015, 6(5), 3225–3239 CrossRef.
- Y. Goto, L. Zeng, C. J. Yeom, Y. Zhu, A. Morinibu, K. Shinomiya, M. Kobayashi, K. Hirota, S. Itasaka, M. Yoshimura, K. Tanimoto, M. Torii, T. Sowa, T. Menju, M. Sonobe, H. Kakeya, M. Toi, H. Date, E. M. Hammond, M. Hiraoka and H. Harada, Nat. Commun., 2015, 6, 6153 CrossRef CAS.
- R. G. Bonhin, V. B. Rocha, G. M. de Carvalho, A. C. Guimaraes, A. N. Crespo, C. T. Chone and E. M. Amstalden, Braz. J. Otorhinolaryngol., 2015, 81, 58–62 CrossRef PubMed.
- D. Nie and K. V. Honn, Semin. Thromb. Hemostasis, 2004, 30, 119–125 CrossRef CAS PubMed.
- K. Sappayatosok, Y. Maneerat, S. Swasdison, P. Viriyavejakul, K. Dhanuthai, J. Zwang and U. Chaisri, Med. Oral Patol. Oral Cir. Bucal, 2009, 14, E319–E324 Search PubMed.
- G. Xue, H. L. Yan, Y. Zhang, L. Q. Hao, X. T. Zhu, Q. Mei and S. H. Sun, Oncogene, 2015, 34, 1393–1406 CrossRef CAS.
- A. Sene, D. Chin-Yee and R. S. Apte, Trends Mol. Med., 2015, 21, 43–51 CrossRef CAS PubMed.
- D. F. Carpentieri, K. Nichols, P. M. Chou, M. Matthews, B. Pawel and D. Huff, Mod. Pathol., 2002, 15, 1080–1086 CrossRef CAS PubMed.
- K. Inoue, H. Ogawa, Y. Sonoda, T. Kimura, H. Sakabe, Y. Oka, S. Miyake, H. Tamaki, Y. Oji, T. Yamagami, T. Tatekawa, T. Soma, T. Kishimoto and H. Sugiyama, Blood, 1997, 89, 1405–1412 CAS.
- D. M. Loeb, E. Evron, C. B. Patel, P. M. Sharma, B. Niranjan, L. Buluwela, S. A. Weitzman, D. Korz and S. Sukumar, Cancer Res., 2001, 61, 921–925 CAS.
- T. Merghoub, C. Gurrieri, F. Piazza and P. P. Pandolfi, Blood Cells, Mol., Dis., 2001, 27, 231–248 CrossRef CAS PubMed.
- Z. Liu, K. Yamanouchi, T. Ohtao, S. Matsumura, M. Seino, V. Shridhar, T. Takahashi, K. Takahashi and H. Kurachi, Anticancer Res., 2014, 34, 2331–2340 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5tx00225g |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2016 |