
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stability, electronic structures and thermoelectric properties of binary Zn–Sb materials
Xin
He
a,
Yuhao
Fu
a,
David J.
Singh
*ab and
Lijun
Zhang
*a
aCollege of Materials Science and Engineering and Key Laboratory of Automobile Materials of MOE, Jilin University, Changchun 130012, China. E-mail: lijun_zhang@jlu.edu.cn
bDepartment of Physics and Astronomy, University of Missouri, Columbia, MO 65211-7010, USA. E-mail: singhdj@missouri.edu
First published on 3rd November 2016
Abstract
We report first principles studies of the binary Zn–Sb phases in relation to thermoelectric properties and chemical stability. We identify the unknown structure of the Zn3Sb2 phase using particle swarm optimization, finding a tetragonal structure different from the hexagonal Mg3Sb2 and the hexagonal or cubic Ca3Sb2 phases. All the phases are found to be semiconducting with bandgaps in the range of 0.06–0.77 eV. This semiconducting behavior is understood in Zintl terms as a balance between the Zn![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Sb and Sb3−:½(Sb2)4− ratios in the stable crystal structures. With the exception of Zn3Sb2, which has a small gap, all the compounds have electronic properties favorable for thermoelectric performance.
:Sb and Sb3−:½(Sb2)4− ratios in the stable crystal structures. With the exception of Zn3Sb2, which has a small gap, all the compounds have electronic properties favorable for thermoelectric performance.
1 Introduction
Thermoelectric (TE) devices can directly convert between electrical and thermal energies for cooling or heating or to recover waste heat. These materials have traditionally had an important role in energy conversion, especially portable refrigeration devices, and power generation especially for space crafts.1–3 They are increasingly seen as having the potential to make important contributions to energy technology more generally3–5 and have additionally attracted fundamental interest due to the connection with topological insulators and the possibility of new types of thermoelectrics.5–9 Furthermore there has been interest in transient thermoelectric effects.10–15 A key challenge for enabling applications is to improve the TE properties of the active materials.Efficiency plays a central role in energy technology. In the case of TE, this is limited by a figure of merit ZT = σS2T/k, where σ is the electrical conductivity, S is the thermopower, k is the thermal conductivity, and T is the absolute temperature. A good TE material should have excellent electrical transport properties, measured by the TE power factor (PF = S2σ) and also a low thermal conductivity k.
Caillat et al. found a high ZT of 1.3 at 970 K in p-type β-Zn4Sb3 in 1997.16 The high performance combined with the relatively inexpensive, environmentally acceptable composition stimulated considerable interest. Four compounds in the narrow range of 50–60 atomic% of Zn were reported in the binary Zn–Sb phase diagram: ZnSb is a stoichiometric compound with a well characterised crystal structure (CdSb-type)17 and stable up to 822 K. Zn4Sb3 displays temperature dependent polymorphism, with a β–α transition at approximately 250 K.18,19 Two phases of Zn8Sb7 have been reported, α and β.20,21 Zn3Sb2 is metastable and has been reported without any crystal structure solution.22 Interestingly, the experimental values and temperature dependences of S and σ are very similar for ZnSb, β-Zn4Sb3 and β-Zn8Sb7. What makes the β-phase special is the exceptionally low thermal conductivity, which is as low as that expected for glass-like materials. The presence of the interstitial Zn atoms in the β-phase crystal structures is the key to the low lattice thermal conductivity.20,23 There have been several studies of electrical and transport properties for β-Zn4Sb3, α-Zn8Sb7 and ZnSb,24–32 including first principles studies of the electronic structures of these binaries26,30–32 and the related Zintl phases SrZn2Sb2 and YbZn2Sb2. However, the other phases have not been studied in similar detail. Here we report detailed first-principles calculations to establish the electronic structures of Zn–Sb binary system compounds, elucidate the structures and bonding, and to assess their thermoelectric potential.
2 Computational methodology
Full structure optimizations were performed using the projector augmented wave (PAW)33 method with generalized approximation (GGA) of Perdew–Burke–Ernzerhof (PBE) as implemented in the VASP code.34 Following a series of test calculations a plane-wave cutoff of 360 eV and k-point meshes of spacing 2π × 0.03 Å−1 or better were adopted. In all of the calculations the convergence limit was set to 1 × 10−4 eV Å−1 for the force and 1 × 10−5 eV Å−1 for the energy. Standard GGA methods underestimate the band gaps due to the presence of artificial self-interaction and the absence of the derivative discontinuity in the exchange–correlation potential.35,36 We used the modified Becke Johnson (MBJ)37 potential to obtain more accurate bandgaps and optical properties. This potential generally improves the accuracy of the gap values relative to experiments. For example, the band gaps calculated using MBJ potential for ZnSb and β-Zn4Sb3 (0.60 and 0.77 eV, respectively) are much closer to experimental values (0.61 and 0.8 eV38,39) than PBE values (0.15 and 0.4 eV).This is important for predicting the thermoelectric properties, as the thermopower in particular can be strongly reduced by spurious bipolar effects from underestimated band gaps. However, the MBJ potential does not have a corresponding energy functional and therefore cannot be used to predict the structures. Therefore we use the PBE functional, which is designed to produce reliable total energies and structures, but not band gaps, to obtain crystal structures, and then do calculations with the MBJ potential to obtain band structures and transport properties. We did not include the spin–orbit coupling (SOC) effect in transport calculations since it shows a negligible effect on the electronic structures of the studied system. The values of the gaps are given in Table 1 along with calculated transport coefficients at 400 K. The available experimental and calculated data are listed for comparison. The transport coefficients are discussed for each compound in the sections below.
| Compound | E g (indirect) | E g (direct) | S (400 K, 1020 cm−3) | σS 2/τ (400 K, 1020 cm−3) |
|---|---|---|---|---|
| ZnSb | 0.60 | 0.88 | 98 | 7.3 |
| 0.61(Exp.)38 | 90(Cal.)29 | |||
| 0.56(Cal.)31 | 135(Cal.)31 | |||
| α-Zn4Sb3 | 0.68 | 0.77 | 205 | 14.5 |
| 0.77 | 161 | |||
| β-Zn4Sb3 | 0.8(Exp.)39 | 0.81 | 110(Cal.)29 | 15.9 |
| 0.3(Cal.)30 | 180(Cal.)26 | |||
| α-Zn8Sb7 | 0.71 | 0.76 | 164 | 7.5 |
| 142(Cal.)29 | ||||
| β-Zn8Sb7 | 0.61 | 0.61 | 129 | 8.1 |
| Zn3Sb2 | 0.06 | 0.1 | 109 | 6.9 |
In order to demonstrate the validity of using the MBJ potential, we calculate the optical spectra shown in Fig. 1. The onset of absorption is at 0.85 eV. The experimental onset of strong absorption transition is at approximately 1.05 eV. We find noticeable anisotropy in the optical absorption, very close to the onset. It shows significantly stronger absorption for polarization along c, in accord with the experiment.38 We predict that this anisotropy switches at higher energy so that the strongest absorption is for polarization along a in the range 2–4 eV.
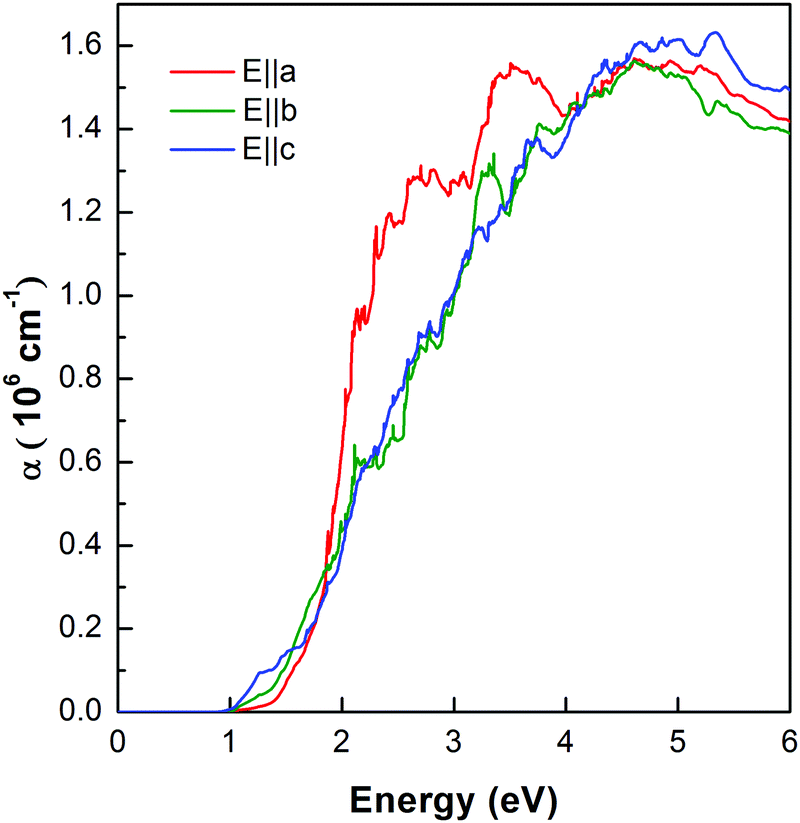 | ||
| Fig. 1 Optical spectra of ZnSb calculated by using the MBJ functional. | ||
The transport calculations were done using the BoltzTraP program.40 We used approximately 5000 k-points in the irreducible Brillouin Zone, with the constant relaxation time approximation. The necessary crystal structure and eigen-energies were obtained from VASP results by using MBJ potential. This approach uses a rigid band approximation. This is generally valid for semiconductors that have good conductivity when doped, as is the case for the Zn–Sb phases (note that strong band distortion upon doping is indicative of strong electronic scattering by dopants). It is also consistent with spectroscopic measurements for some of these phases in relation to prior density functional calculations.30–32 Going beyond the rigid band approximation would require knowledge of the specific doping elements and defects that occur in actual samples.
Previous studies on the phase diagram of the zinc-antimony system have reported compound Zn3Sb2.41,42 But it is metastable and crystals amorphize over time, which impedes its structure and property characterization. We performed a crystal structure prediction for Zn3Sb2via global minimization of free energy surfaces as implemented in the CALYPSO code, which searches for the stable structures of the given compounds. Using particle swarm optimization (PSO) the most stable structures based on the chemical composition can be found. Importantly, this approach is unbiased by already known structures.43,44 In this sense it differs from approaches such as those used in most materials genome projects, for example, the materials project,45 where known structures and structure types are generally relied upon. The advantage of the PSO approach is that it can find structures that were previously unknown, while on the other hand it requires calculations for many structures during the global optimization. Structure searching was performed at zero pressure with simulation cells consisting of 1–4 formula units (f.u.). This global optimization involved density functional relaxations of approximately 1500 distinct structures to find the ground state. The most stable predicted structure was then further optimized using VASP as described above. This calculation adds the structure and energy of Zn3Sb2 to the phase diagram.
3 Structure and phase diagram
We consider all Zn–Sb binary materials for which structures have been reported and Zn3Sb2 with a predicted structure. Orthorhombic ZnSb (space group Pbac) was reported in 1948.17 It may be regarded as an arrangement of Zn centered, vertex and corner sharing Sb4 tetrahedra (quasitetrahedral close-packing), as shown in Fig. 2a. The formal valence state of Sb is −2, and it has the appearance of a classical Zintl phase with Sb present as (Sb2)4− dimers. The ZnSb4 tetrahedra are linked by Sb–Sb bonds. We note that such Sb–Sb dumbbells have been implicated in the low thermal conductivity of the Zn4Sb3 phase.46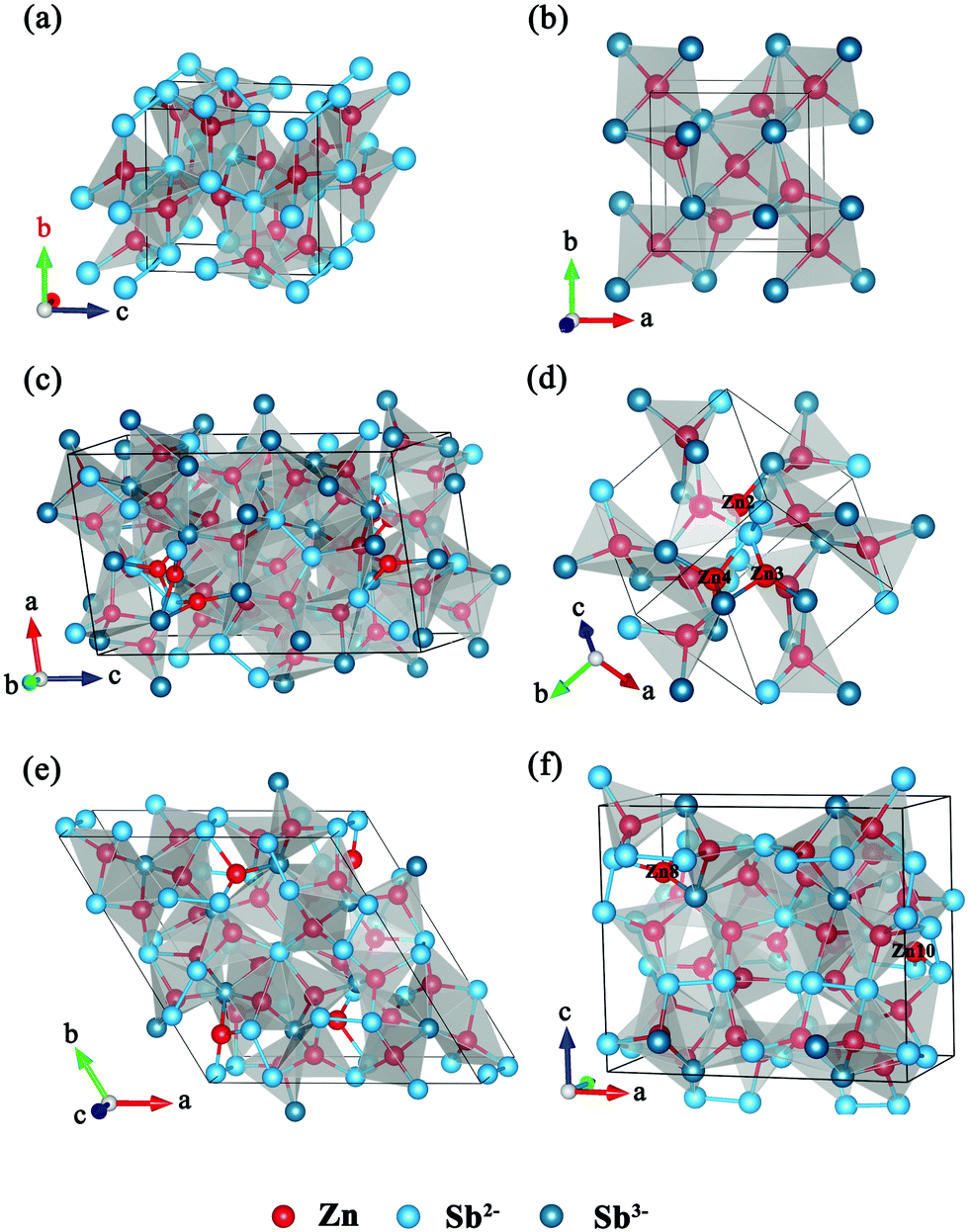 | ||
| Fig. 2 Crystal structure of (a) ZnSb, (b) predicted Zn3Sb2 structure with CALYPSO, (c) α-Zn4Sb3, (d) β-Zn4Sb3, Zn2, Zn3, and Zn4 are interstitial atoms, (e) α-Zn8Sb7, (f) β-Zn8Sb7, Zn8, Zn10 are interstitial atoms. | ||
The low temperature α-phase and high temperature β-phase of Zn4Sb3 were considered in this work. The unit cell of β-Zn4Sb3 is rhombohedral (space group R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) C) and contains three distinct atomic positions (36 Zn, 18 Sb1, and 12 Sb2 amounting to a Zn3.6Sb3 stoichiometry). The atoms on the Sb1 site are free Sb3− while those on the Sb2 site form (Sb2)4− dimers, according to the Zintl-Klemm concept.47 The problem posed by apparent deviation from the nominal composition was solved by Snyder et al.23 They found a high degree of disorder in the Zn substructure of β-Zn4Sb3, and in particular interstitial Zn atoms distributed on three partially occupied general sites in space group RC (36f). Additionally, the regular framework position (also 36f) displayed a considerable occupational deficiency with only 0.89–0.9 of the sites filled. Based on this, the framework of β-Zn4Sb3 for our calculations was constructed in the following way:48,49 first, a rhombohedral primitive cell was built with full occupancy of Zn1, Sb1 and Sb2 sites resulting in the Zn12Sb10 stoichiometry. Then we removed two Zn atoms from the main site (Zn1) and inserted three different Zn interstitials in the Zn2, Zn3 and Zn4 sites, respectively, while maximizing the distance between interstitial atoms to minimize the total energy.25 We considered all the possible configurations and chose the lowest total energy one. The final structure is shown in Fig. 2d. Below about 250 K β-Zn4Sb3 transforms into α-Zn4Sb3, which somewhat surprisingly has a completely ordered structure with a crystallographic composition of Zn13Sb10 for fully occupied sites.19 The structure of α-Zn4Sb3 is triclinic, but is often described with a C-centred cell (i.e. C
C) and contains three distinct atomic positions (36 Zn, 18 Sb1, and 12 Sb2 amounting to a Zn3.6Sb3 stoichiometry). The atoms on the Sb1 site are free Sb3− while those on the Sb2 site form (Sb2)4− dimers, according to the Zintl-Klemm concept.47 The problem posed by apparent deviation from the nominal composition was solved by Snyder et al.23 They found a high degree of disorder in the Zn substructure of β-Zn4Sb3, and in particular interstitial Zn atoms distributed on three partially occupied general sites in space group RC (36f). Additionally, the regular framework position (also 36f) displayed a considerable occupational deficiency with only 0.89–0.9 of the sites filled. Based on this, the framework of β-Zn4Sb3 for our calculations was constructed in the following way:48,49 first, a rhombohedral primitive cell was built with full occupancy of Zn1, Sb1 and Sb2 sites resulting in the Zn12Sb10 stoichiometry. Then we removed two Zn atoms from the main site (Zn1) and inserted three different Zn interstitials in the Zn2, Zn3 and Zn4 sites, respectively, while maximizing the distance between interstitial atoms to minimize the total energy.25 We considered all the possible configurations and chose the lowest total energy one. The final structure is shown in Fig. 2d. Below about 250 K β-Zn4Sb3 transforms into α-Zn4Sb3, which somewhat surprisingly has a completely ordered structure with a crystallographic composition of Zn13Sb10 for fully occupied sites.19 The structure of α-Zn4Sb3 is triclinic, but is often described with a C-centred cell (i.e. C![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ). Here, the interstitial Zn atoms are located on two fully occupied sites, and the main Zn site is also fully occupied (Fig. 2c).18
). Here, the interstitial Zn atoms are located on two fully occupied sites, and the main Zn site is also fully occupied (Fig. 2c).18
β-Zn8Sb7 is a new metastable compound whose synthesis was reported in 2015.20 It crystallizes in a noncentrosymmetric orthorhombic space group Pmn21. Twenty-four Zn atoms are distributed over 6 fully occupied crystallographic sites, while the remaining 8 Zn atoms are distributed over 4 sites with partial occupancies. It contains 10 (Sb2)4− dumbbells and 8 isolated Sb3− anions, which is similar to Zn13Sb10. Each Zn atom is surrounded by distorted tetrahedra with sharing of common edges. The distances of the reported Zn7–Zn8 and Zn9–Zn10 sites are in the range of 1.2–1.4 Å. This indicates that only one of two possible Zn atoms is present. Thus, we use one Zn8 atom to replace the nearest Zn7 atom, similarly for Zn9 and Zn10. The final crystal structure with the lowest total energy is shown in Fig. 2f. The α-Zn8Sb7 phase was reported as Zn1+δSb (with δ = 0.068) with a space group P.21 It has an ordered structure with an ideal stoichiometry of Zn8Sb7 and few Zn defects in it. We adopt its ideal full occupancy structure in our calculation (Fig. 2e).
We performed variable-cell prediction simulations using the PSO methodology for Zn3Sb2 at 0 GPa. The lowest energy structure was chosen, as shown in Fig. 2b. Its crystal structure is tetragonal (space group P![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 21m, Z = 2), with lattice parameters a = 7.090 Å and c = 4.777 Å. Sb occupies the 4e (1.134, 0.634, 0.738) sites, while Zn atoms are at the 4e sites (1.134, 0.634, 0.738) and 2d sites (0, 0, 0.5). All Sb in this structure are isolated Sb3− anions, with no bonds between Sb atoms. We note that this structure differs from the hexagonal and cubic structures of Mg3Sb2 and Ca3Sb2. Each Sb atom is exclusively surrounded by six Zn atoms (at distances between 2.65 and 2.82 Å, similar to the known structures for β-Zn4Sb3 2.7–2.8 Å, for α-Zn4Sb3 2.67–2.75 Å, for β-Zn8Sb7 2.64–2.87 Å, and 2.56–2.92 Å for the α-Zn8Sb7 phase). Each ZnSb4 tetrahedron shares a common Sb atom with another five tetrahedra.
21m, Z = 2), with lattice parameters a = 7.090 Å and c = 4.777 Å. Sb occupies the 4e (1.134, 0.634, 0.738) sites, while Zn atoms are at the 4e sites (1.134, 0.634, 0.738) and 2d sites (0, 0, 0.5). All Sb in this structure are isolated Sb3− anions, with no bonds between Sb atoms. We note that this structure differs from the hexagonal and cubic structures of Mg3Sb2 and Ca3Sb2. Each Sb atom is exclusively surrounded by six Zn atoms (at distances between 2.65 and 2.82 Å, similar to the known structures for β-Zn4Sb3 2.7–2.8 Å, for α-Zn4Sb3 2.67–2.75 Å, for β-Zn8Sb7 2.64–2.87 Å, and 2.56–2.92 Å for the α-Zn8Sb7 phase). Each ZnSb4 tetrahedron shares a common Sb atom with another five tetrahedra.
It is perhaps surprising that Zn3Sb2 has a different structure than hexagonal Mg3Sb2. However, we note that the electronegativity of Zn is significantly higher than Mg, which suggests chemical differences. We carefully checked the result by performing all-electron calculations for Zn3Sb2 in the fully relaxed Mg3Sb2 structure and our predicted tetragonal ground state structure. These calculations were done using the general potential linearized augmented planewave (LAPW) method50 as implemented in the WIEN2k code.51 We used highly converged basis sets based on the accurate LAPW augmentation, plus local orbitals to treat semicore states. We added additional local orbitals to improve the accuracy for the Zn d state.52 Spin–orbit was included in these calculations. We find that our predicted tetragonal structure is lower in energy by 0.25 eV per formula unit. This means that the tetragonal structure is strongly preferred over the Mg3Sb2 structure for Zn3Sb2.
We calculated the formation energy with respect to the elements for six configurations. As plotted in Fig. 3, the formation enthalpies of all the configurations are negative. In order to analyze phase stability at 0 K, we constructed the convex hull of formation enthalpy, as illustrated by the solid lines in Fig. 3. Here we included the SOC effect. ZnSb is the most stable system with a formation energy of approximately 27 meV per atom. The data shown for the interstitial β-phase structures are for the lowest formation energy structure out of all those that we built. A structure whose formation enthalpy lies above the convex hull, which can be considered as a global stability line, is metastable or unstable. The formation enthalpies of Zn4Sb3, Zn8Sb7 and Zn3Sb2 are above the convex hull. As expected, the interstitial frameworks of β-phases are less stable than those of the ordered α-phases, but they still have a negative energy of formation (4.5 meV per atom for β-Zn4Sb3, 6 meV per atom for β-Zn8Sb7). Surprisingly the α-phase is unstable with respect to the decomposition into ZnSb and Zn.21,53 The predicted structure of Zn3Sb2 has a little lower formation enthalpy (8 meV per atom). Without the SOC the formation energies are a little lower. But this does not change the stability situation for the Zn–Sb binary system.
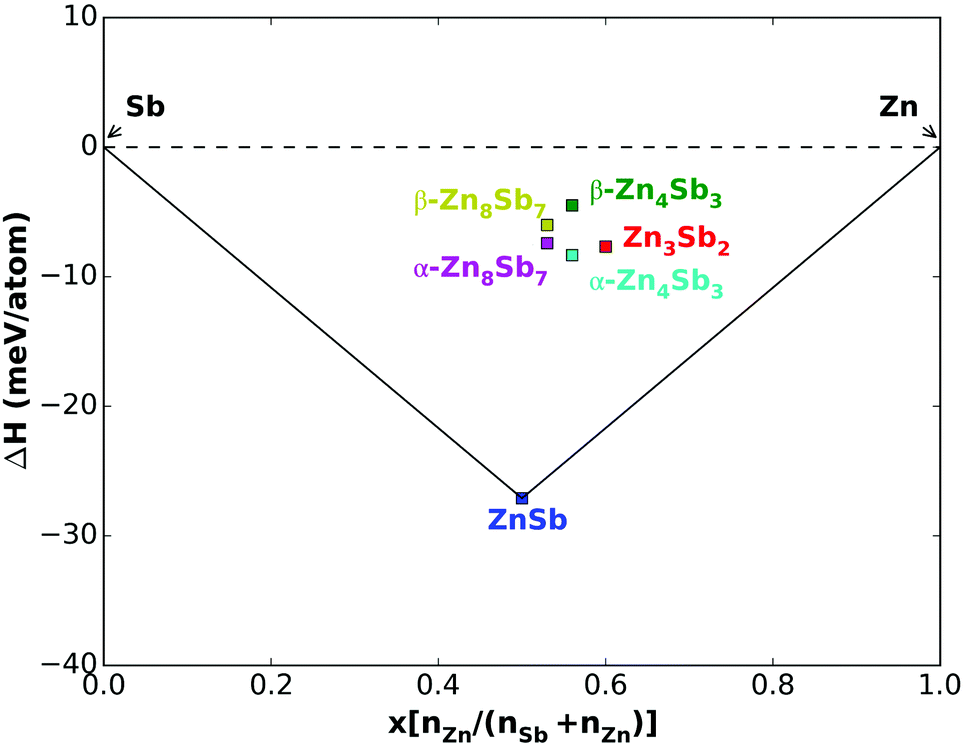 | ||
| Fig. 3 Calculated enthalpy of formation as a function of Zn concentration. | ||
4 Electronic and transport properties
The calculated band structures and total and projected density of states (DOS) obtained with MBJ potential are shown in Fig. 4–9a. The Fermi levels are set to the top of the valence band. The results show that the MBJ method performs consistently better than PBE-GGA and produces band gaps that are in excellent agreement with the experimental values. This is in accord with prior results for other non-d-electron semiconductors.54,55 All the band structures shown are indirect band gap except β-Zn8Sb7. The PDOS show that the valence bands are formed from Zn p, Sb s, and Sb p hybridized states. The conduction band is characterized mainly by 4s-electrons of zinc which are hybridized with 5p-electrons of antimony. The total density of states show a similar shape and magnitude of the valence band edges of Zn4Sb3 and Zn8Sb7, which suggests a similar hole carrier effective mass and similar electronic properties. It is remarkable that in spite of the different Zn–Sb ratios and crystal structures, all these compounds show similar electronic structures. This is an example of the robustness of the Zintl concept for the Zn–Sb system, specifically the balance between the Zn:Sb and the Sb3−:½(Sb2)4− ratios.
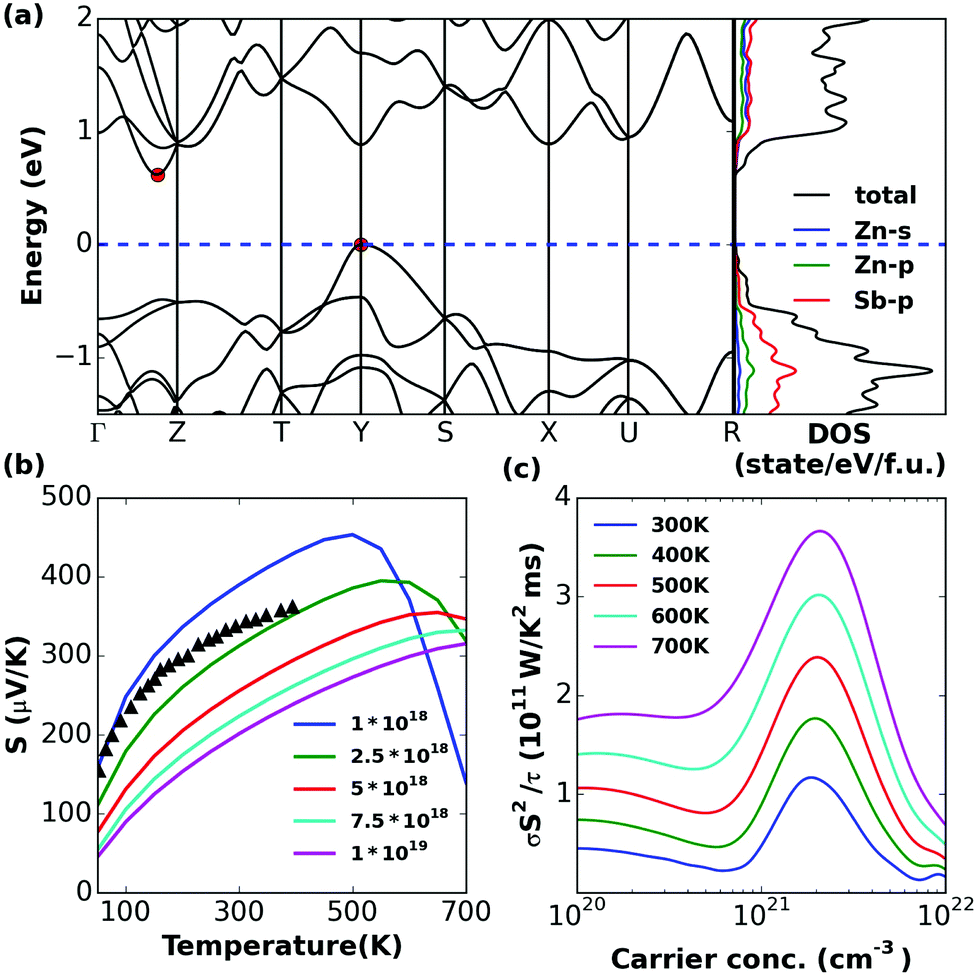 | ||
| Fig. 4 Calculated (a) band structure and (partial) density of states, (b) thermopower (S) versus temperature at different carrier concentrations (with different colorful lines), and (c) power factor divided by scattering time (σS2/τ) versus carrier concentration at different temperatures (with different colorful lines) for ZnSb. In (a) the band edges are marked by red circles and in (b) experimental values (ref. 56) are shown with triangles for comparison. | ||
 | ||
| Fig. 5 Calculated (a) band structure and (partial) density of states, (b) S and (c) σS2/τ of α-Zn4Sb3. Experimental values reported in ref. 57 are shown with triangles for comparison. | ||
 | ||
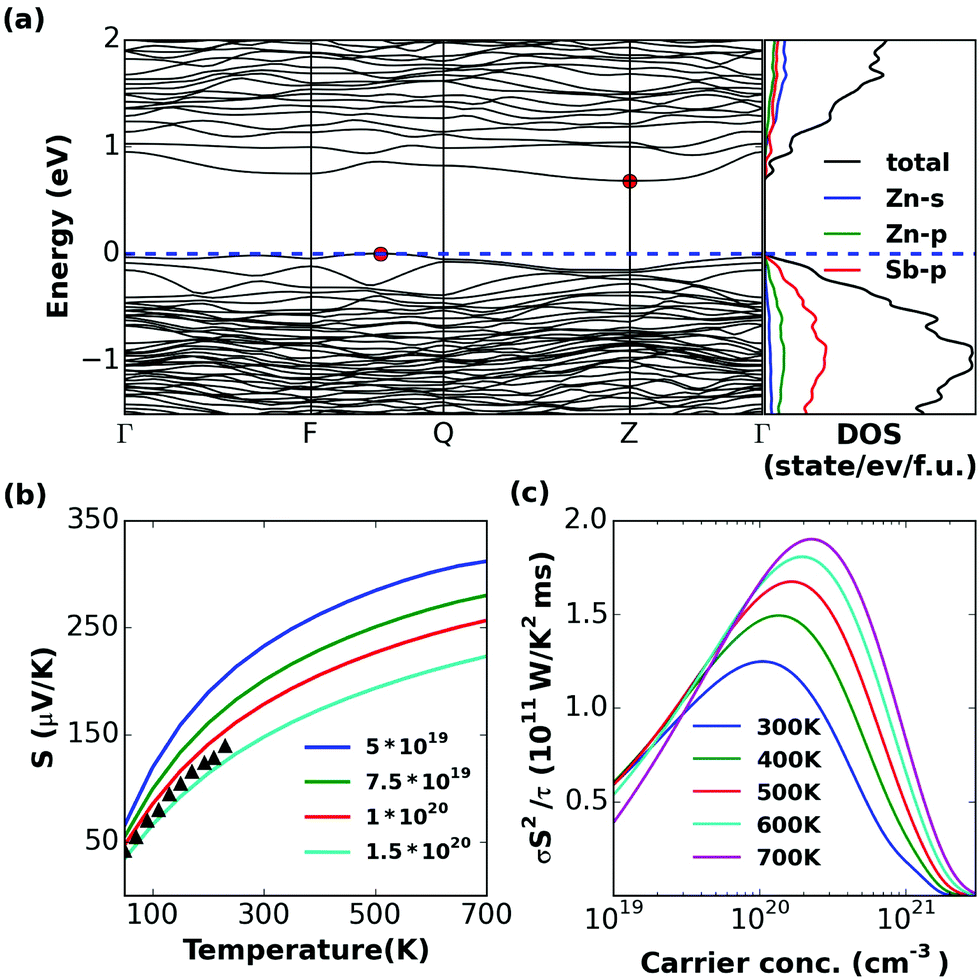
| Fig. 6 Calculated (a) band structure and (partial) density of states, (b) S (c) σS2/τ of β-Zn4Sb3. Experimental values reported in ref. 16 are plotted with triangles. | ||
 | ||
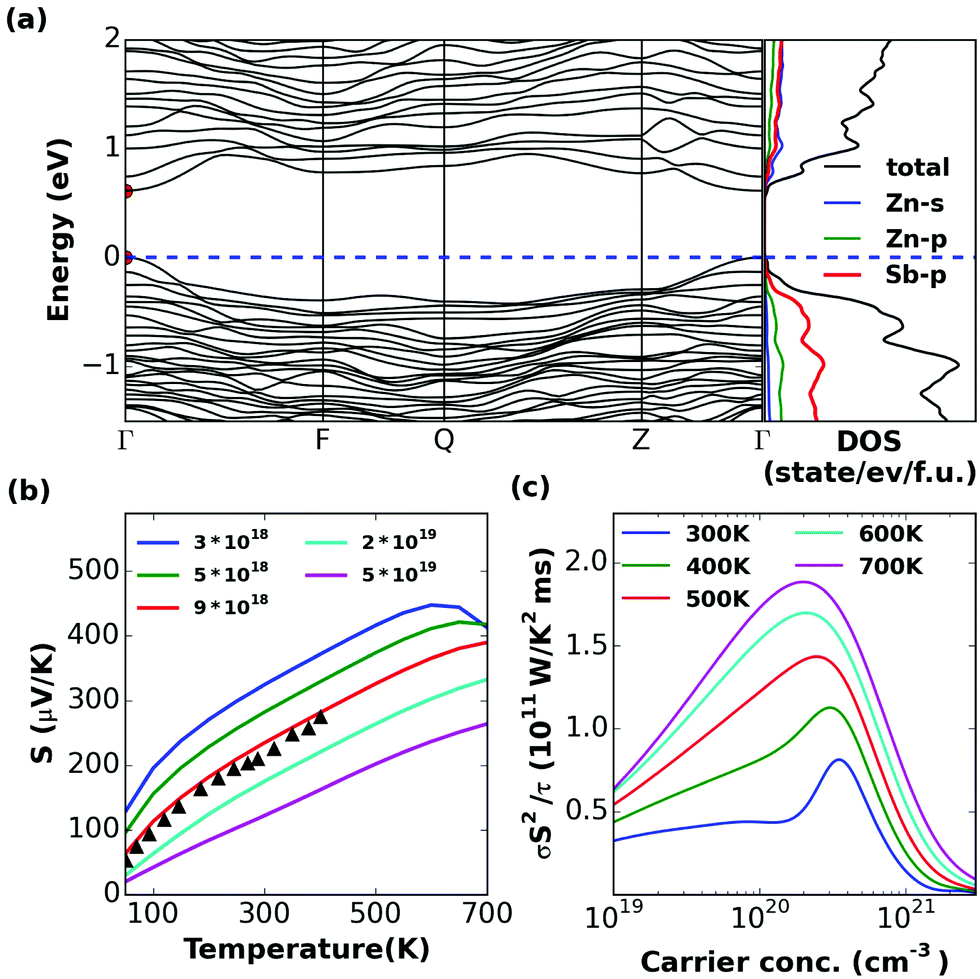
| Fig. 7 Calculated (a) band structure and (partial) density of states, (b) S and (c) σS2/τ of α-Zn8Sb7. | ||
 | ||
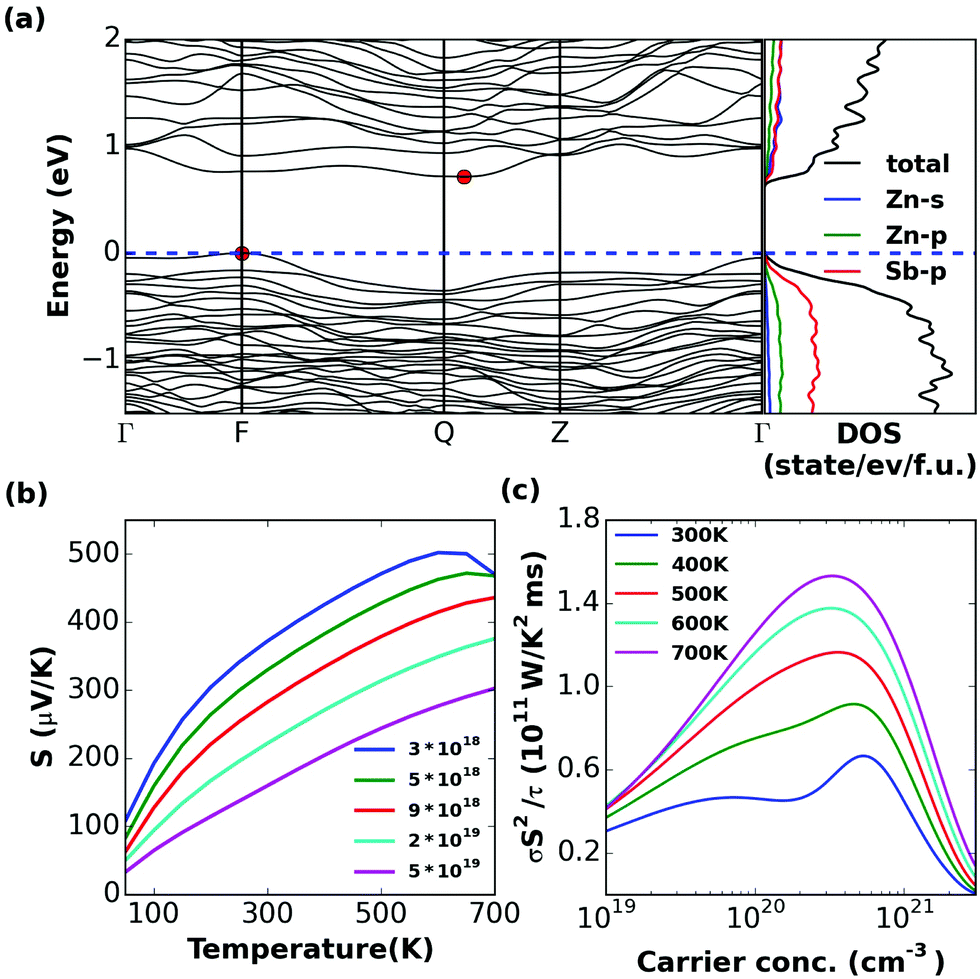
| Fig. 8 Calculated (a) band structure and (partial) density of state, (b) S (c) σS2/τ of β-Zn8Sb7. Experimental values reported in ref. 20 are shown with triangles for comparison. | ||
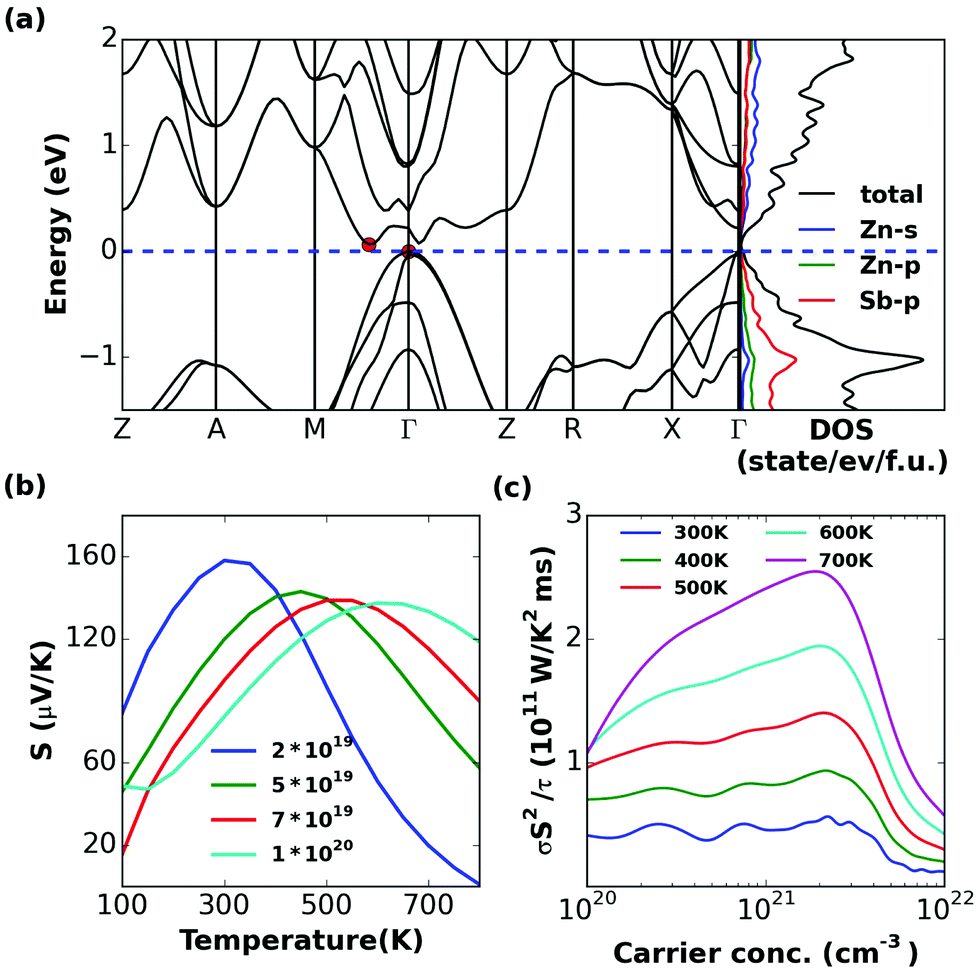 | ||
| Fig. 9 Calculated (a) band structure and (partial) density of states, (b) S and (c) σS2/τ of Zn3Sb2. | ||
Here we focus on the p-type, since experimentally all these compounds form as the p-type. We performed calculations of the thermopower as a function of temperature and the p-type doping level with Boltzmann theory in the CSTA with rigid bands. We also show S2σ/τ as a function of carrier concentration at different temperatures. This is the power factor divided by the scattering time, τ, which is an undetermined parameter that can depend on both doping and temperature. Generally τ decreases with both temperature and carrier concentration, though the carrier concentration dependence is often weaker than the τ dependence. The validity of these approaches can be established by studying the agreement between theoretical results and experimental data shown in Fig. 4–6 and 8 and is discussed further in the text.
ZnSb
The valence bands are nearly parabolic at the band edge. We show the thermopower of ZnSb at carrier concentrations (1018–1019 cm−3), which are in general accord with experimental data56 although the experimental carrier concentration needs to be established in order to determine how close the accord is (Fig. 4b). It should be noted that in ZnSb it has been proven that it is difficult to achieve hole concentrations above 1019 cm−3. The peak σS2/τ lies in the range of 1 × 1020 to 4 × 1020 cm−3 (Fig. 4c). It is highly likely that ZT for ZnSb can be significantly increased if the carrier concentration can be increased.Zn4Sb3
At the edge of the valence bands, we find heavier bands for the α phase than the β phase, resulting in higher DOS near the VBM (Fig. 5 and 6a), which should lead to a little larger thermopower in p-type materials. They display similar band gap Eg = 0.68 and 0.77 eV, respectively. The latter agrees well with the experimental value Eg = 0.8 eV39 for the β phase. The thermopower value of the α phase is indeed a little higher than the β phase in our calculation (Fig. 5 and 6b). Experimental measurements of S for α-Zn4Sb3 are only available for temperatures below 250 K.57 One can see that our interstitial structures can get thermopowers in accord with the experiment again with the caveat of the carrier concentration.16 The doping level at 300–700 K corresponding to the peak σS2/τ lies in the range of 1 × 1020 to 4 × 1020 cm−3 for the α phase and 2 × 1020 to 9 × 1020 cm−3 for the β phase (Fig. 5 and 6c), again implying that the peak ZT is likely to be at high carrier concentration.Zn8Sb7
The band structure of α-Zn8Sb7 shows that it is an indirect band gap semiconductor, with Eg = 0.71 eV. Disordered β-Zn8Sb7 has a direct band gap, with Eg = 0.61 eV. They have very similar transport properties. The doping level at 300–700 K corresponding to the peak σS2/τ lies in the range of 1 × 1020 to 4 × 1020 cm−3 for both α and β phases (Fig. 7 and 8c). It was found that the thermopower values of the disordered β-phase and the ordered α-phase of Zn4Sb3 and Zn8Sb7 were very similar. Hence, the key to the outstanding thermoelectric properties of the β-phase lies in the exceptionally low thermal conductivity, which is as low as that for glass-like materials combined with its favorable electronic properties.Zn3Sb2
A narrow band gap of 0.06 eV separates the valence states to the conduction states. The electronic structure of Zn3Sb2 exhibits the charge carrier pockets with light bands near the VBM and CBM, which leads to low thermopower. As predicted the light band at the edge of bands indeed leads to a low thermopower for Zn3Sb2 (Fig. 9b). The very small gap suggests that this phase will not be a good thermoelectric at least at room temperature and above. This is also seen in the thermopower, which shows strong bipolar effects.5 Conclusions
The β-phases of Zn8Sb7 and Zn4Sb3 have complex crystal structures. These structures challenge crystallography because of the presence of site disorder for the Zn atoms. In this work, we have proposed structural models of β-phases, which are close to experimental studies. All binary Zn/Sb compounds are narrow-gap semiconductors. We have analysed the phase stability, electronic structure, optical property of ZnSb and transport properties by means of first principles density functional theory and Boltzmann transport calculations. Additionally, we have searched for the Zn3Sb2 structure using the PSO method. This yields a structure different from the hexagonal structure of Mg3Sb2, reflecting the different chemistries of Zn and Mg. In terms of the formation energy, this phase is comparable to Zn8Sb7 and Zn4Sb3. In thermodynamic stability analysis, they are all metastable phases.A similar shape and magnitude of the total density of states in the valence band edge of Zn4Sb3 and Zn8Sb7 lead to similar thermopower values. The thermopower data and transport properties from our calculations show that Zn3Sb2 should not be a good candidate for thermoelectric materials at room temperature and above. The existing experimental results show the best performance for β-Zn4Sb3. This material has a reasonable band gap, shows multiple valence bands sufficiently near the band edge to influence the thermoelectric behavior and importantly has a low thermal conductivity characteristic of the β-phases. However, our results also show that there is still room for improvement in ZT of the β phases by optimization of carrier concentration. A comparison of the compounds in terms of the balance between σ/τ and S at 400 K and a carrier concentration of 1019 cm−3 as well as the doping dependence of S is given in Fig. 10.
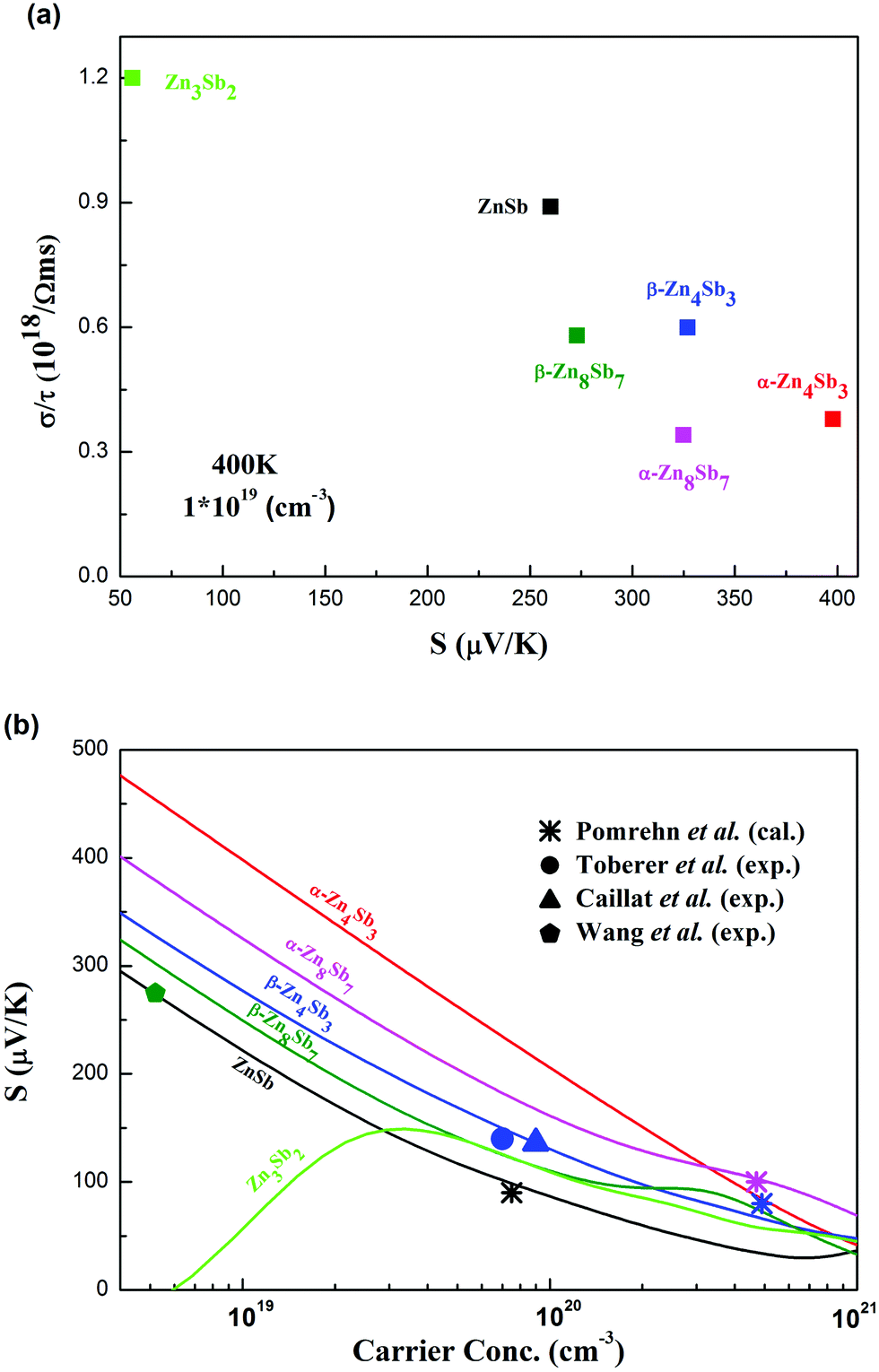 | ||
| Fig. 10 (a) Electrical conductivity divided by scattering time (σ/τ) versus thermopower (S) calculated at a temperature of 400 K and a carrier concentration of 1019 cm−3, and (b) thermopower versus carrier concentration calculated at a temperature of 400 K for different phases of the Zn–Sb materials. In (b) the available experimental and calculated data from the previous work by Pomrehn et al. (ref. 29), Toberer et al. (ref. 57), Caillat et al. (ref. 16), and Wang et al. (ref. 20) are shown for comparison; for the scatters, consistent colors with those of lines are used for the same phases. | ||
Finally it is interesting to note that although some gaps are small, all the compounds are semiconducting. This is a reflection of the Zintl concept in those phases, especially the balance between the Zn:Sb and Sb3−:½(Sb2)4− ratios reflecting stabilization or destabilization of (Sb2)4− polyanions according to the amount of charge available from the Zn2+cations. We note that several of these phases are underdoped for thermoelectric performance. The flexibility of these compounds in terms of carrier compensation via the Sb3−:½(Sb2)4−ratio poses a challenge for achieving the optimum doping level. The solution to this problem may lead to significantly better ZT in the Zn–Sb phases.
Acknowledgements
The authors acknowledge funding support from the Recruitment Program of Global Youth Experts in China and special funds for talent development in Jilin Province. Part of calculations was performed in the high performance computing center of Jilin University. Work of D. J. S is supported by S3TEC an Energy Frontier Research Center funded by the Department of Energy, Office of Science, Basic Energy Sciences under award DE-SC0001299/DE-FG02-09ER46577.References
- H. J. Goldsmid and R. W. Douglas, Br. J. Appl. Phys., 1954, 5, 386–390 CrossRef.
- C. Wood, Rep. Prog. Phys., 1988, 51, 459–540 CrossRef CAS.
- L. E. Bell, Science, 2008, 321, 1457–1461 CrossRef CAS PubMed.
- G. Chen, M. S. Dresselhaus, G. Dresselhaus, J.-P. Fleurial and T. Caillat, Int. Mater. Rev., 2003, 48, 45–66 CrossRef CAS.
- J. Yang, L. Xi, L. Wu, X. Shi, L. Chen, J. Yang, W. Zhang, C. Uher and D. J. Singh, NPJ Comput. Mater., 2016, 2, 15015 CrossRef.
- L. Muchler, F. Casper, B. Yan, S. Chadov and C. Felser, Phys. Status Solidi RRL, 2012, 7, 91–100 CrossRef.
- H. Shi, D. Parker, M. H. Du and D. J. Singh, Phys. Rev. Appl., 2015, 3, 014004 CrossRef.
- Y. Xu, Z. Gan and S. C. Zhang, Phys. Rev. Lett., 2014, 112, 226801 CrossRef PubMed.
- G. Xing, J. Sun, K. P. Ong, X. Fan, W. Zheng and D. J. Singh, APL Mater., 2016, 4, 053201 CrossRef.
- M. Sasaki, H. Negishi and M. Inoui, J. Appl. Phys., 1986, 59, 796–802 CrossRef CAS.
- S. Walia, R. Weber, S. Sriram, M. Bhaskaran, K. Latham, S. Zhuiykov and K. Kalantar-Zadeh, Energy Environ. Sci., 2011, 4, 3558–3564 CAS.
- S. Walia, R. Weber, S. Balendhran, D. Yao, J. T. Abrahamson, S. Zhuiykov, M. Bhaskaran, S. Sriram, M. S. Strano and K. Kalantar-Zadeh, Chem. Commun., 2012, 48, 7462–7464 RSC.
- S. Walia, S. Balendhran, P. Yi, D. Yao, S. Zhuiykov, M. Pannirselvam, R. Weber, M. S. Strano, M. Bhaskaran, S. Sriram and K. Kalantar-Zadeh, J. Phys. Chem. C, 2013, 117, 9137–9142 CAS.
- S. Walia, R. Weber, K. Latham, P. Petersen, J. T. Abrahamson, M. S. Strano and K. Kalantar-Zadeh, Adv. Funct. Mater., 2011, 21, 2072–2079 CrossRef CAS.
- S. Walia, S. Balenhran, H. Nili, S. Zhuiykov, G. Rosengarten, Q. H. Wang, M. Bhaskaran, S. Sriram, M. S. Strano and K. Kalantar-Zadeh, Prog. Mater. Sci., 2013, 50, 1443–1489 CrossRef.
- T. Caillat, J.-P. Fleurial and A. Borshchevsky, J. Phys. Chem. Solids, 1997, 58, 1119–1125 CrossRef CAS.
- K. E. Almin, Acta Chem. Scand., 1948, 2, 400–407 CrossRef CAS.
- Y. Mozharivskyj, Y. Janssen, J. L. Harringa, A. Kracher, A. O. Tsokol and G. J. Miller, Chem. Mater., 2006, 18, 822–831 CrossRef CAS.
- J. Nylén, M. Andersson, S. Lidin and U. Häussermann, J. Am. Chem. Soc., 2004, 126, 16306–16307 CrossRef PubMed.
- J. Wang and K. Kovnir, J. Am. Chem. Soc., 2015, 137, 12474–12477 CrossRef CAS PubMed.
- C. S. Birkel, E. Mugnaioli, T. Gorelik, U. Kolb, M. Panthöfer and W. Tremel, J. Am. Chem. Soc., 2010, 132, 9881–9889 CrossRef CAS PubMed.
- F. Adjadj, E.-d. Belbacha, M. Bouharkat and A. Kerboub, J. Alloys Compd., 2006, 419, 267–270 CrossRef CAS.
- G. J. Snyder, M. Christensen, E. Nishibori, T. Caillat and B. B. Iversen, Nat. Mater., 2004, 3, 458–463 CrossRef CAS PubMed.
- P. H. M. Böttger, S. Diplas, E. Flage-Larsen, Ø. Prytz and T. G. Finstad, J. Phys.: Condens. Matter, 2011, 23, 265502 CrossRef PubMed.
- F. Cargnoni, E. Nishibori, P. Rabiller, L. Bertini, G. J. Snyder, M. Christensen, C. Gatti and B. B. Iversen, Chem. – Eur. J., 2004, 10, 3861–3870 CrossRef CAS PubMed.
- S.-G. Kim, I. I. Mazin and D. J. Singh, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 6199–6203 CrossRef CAS.
- S. Wang, H. Li, D. Qi, W. Xie and X. Tang, Acta Mater., 2011, 59, 4805–4817 CrossRef CAS.
- U. Häussermann and A. S. Mikhaylushkin, Dalton Trans., 2010, 39, 1036–1045 RSC.
- G. S. Pomrehn, E. S. Toberer, G. J. Snyder and A. van de Walle, J. Am. Chem. Soc., 2011, 133, 11255–11261 CrossRef CAS PubMed.
- A. N. Qiu, L. T. Zhang and J. S. Wu, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 035203 CrossRef.
- K. Niedziolka, R. Pothin, F. Rouessac, R. M. Ayral and P. Jund, J. Phys.: Condens. Matter, 2014, 26, 365401 CrossRef CAS PubMed.
- P. H. M. Bottger, S. Diplas, E. Flage-Larsen, O. Prytz and T. G. Finstad, J. Phys.: Condens. Matter, 2011, 23, 265502 CrossRef PubMed.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- J. P. Perdew and A. Zunger, Phys. Rev. B: Condens. Matter Mater. Phys., 1981, 23, 5048–5079 CrossRef CAS.
- L. Sham and M. Schlüter, Phys. Rev. Lett., 1983, 51, 1888–1891 CrossRef.
- F. Tran and P. Blaha, Phys. Rev. Lett., 2009, 102, 226401 CrossRef PubMed.
- H. Komiya, K. Masumoto and H. Fan, Phys. Rev., 1964, 133, A1679–A1684 CrossRef.
- M. Vaida, N. Duteanu and I. Grozescu, J. Serb. Chem. Soc., 2016, 81, 323–332 CrossRef CAS.
- G. K. Madsen and D. J. Singh, Comput. Phys. Commun., 2006, 175, 67–71 CrossRef CAS.
- B. DeWitt and H. Seltz, J. Am. Chem. Soc., 1939, 61, 3170–3173 CrossRef CAS.
- M. Boström and S. Lidin, J. Alloys Compd., 2004, 376, 49–57 CrossRef.
- Y. Wang, J. Lv, L. Zhu and Y. Ma, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 094116 CrossRef.
- Y. Wang, J. Lv, L. Zhu and Y. Ma, Comput. Phys. Commun., 2012, 183, 2063–2070 CrossRef CAS.
- A. Jain, S. P. Ong, G. Hautier, W. Chen, W. D. Richards, S. Dacek, S. Cholia, D. Gunter, D. Skinner, G. Ceder and K. A. Persson, APL Mater., 2013, 1, 011002 CrossRef.
- W. Schweika, R. P. Hermann, M. Prager, J. Person and V. Keppens, Phys. Rev. Lett., 2007, 99, 125501 CrossRef CAS PubMed.
- G. A. Papoian and R. Hoffmann, Angew. Chem., Int. Ed., 2000, 39, 2408–2448 CrossRef.
- E. Toberer, K. Sasaki, C. Chisholm, S. Haile, W. Goddard and G. Snyder, Phys. Status Solidi RRL, 2007, 1, 253–255 CrossRef CAS.
- S. Wang, X. Tan, G. Tan, X. She, W. Liu, H. Li, H. Liu and X. Tang, J. Mater. Chem., 2012, 22, 13977–13985 RSC.
- D. J. Singh and L. Nordstrom, Planewaves Pseudopotentials and the LAPW Method, Springer, Berlin, 2nd edn, 2006 Search PubMed.
- P. Blaha, K. Schwarz, G. Madsen, D. Kvasnicka and J. Luitz, in WIEN2k, An Augmented Plane Wave+Local Orbitals Program for Calculating Crystal Properties, K. Schwarz, Tech. Univ. Wien, Austria, 2001 Search PubMed.
- D. Singh, Phys. Rev. B: Condens. Matter Mater. Phys., 1991, 43, 6388–6392 CrossRef CAS.
- A. S. Mikhaylushkin, J. Nylén and U. Häussermann, Chem. – Eur. J., 2005, 11, 4912–4920 CrossRef CAS PubMed.
- D. J. Singh, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 205102 CrossRef.
- D. Koller, F. Tran and P. Blaha, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 195134 CrossRef.
- D. Eklöf, A. Fischer, Y. Wu, E.-W. Scheidt, W. Scherer and U. Häussermann, J. Mater. Chem. A, 2013, 1, 1407–1414 Search PubMed.
- T. Souma, G. Nakamoto and M. Kurisu, J. Alloys Compd., 2002, 340, 275–280 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2016 |