
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNear-infrared long persistent luminescence of Er3+ in garnet for the third bio-imaging window†
Jian
Xu
 *,
Daisuke
Murata
,
Jumpei
Ueda
and
Setsuhisa
Tanabe
*
*,
Daisuke
Murata
,
Jumpei
Ueda
and
Setsuhisa
Tanabe
*
Graduate School of Human and Environmental Studies, Kyoto University, Yoshida-nihonmatsu-cho, Sakyo-ku, Kyoto 606-8501, Japan. E-mail: xu.jian.57z@st.kyoto-u.ac.jp; tanabe.setsuhisa.4v@kyoto-u.ac.jp
First published on 2nd November 2016
Abstract
By utilizing efficient persistent energy transfer from Ce3+ to Er3+, we have successfully developed a novel garnet persistent phosphor of Y3Al2Ga3O12 doped with Er3+, Ce3+, Cr3+ ions (YAGG:Er–Ce–Cr) exhibiting long (>10 h) near-infrared (NIR) persistent luminescence (PersL) in the broad range from 1450 nm to 1670 nm due to the typical Er3+:4I13/2 → 4I15/2 transition in garnet. The NIR PersL bands of Er3+ match well with the third bio-imaging window (NIR-III, approximately from 1500 nm to 1800 nm) and the response curve of InGaAs detectors. The photon emission rate (8.33 × 1017 cps Sr−1 m−2) of the YAGG:Er–Ce–Cr persistent phosphor at 10 min after ceasing blue light illumination was over two times higher than that of the widely used ZnGa2O4:Cr3+ deep-red persistent phosphor (3.30 × 1017 cps Sr−1 m−2). We also show the first PersL imaging by a commercial InGaAs camera monitoring Er3+ emission indicating that this material can be a promising candidate for in vivo bio-imaging in the NIR-III window.
1. Introduction
Over 20 years have passed since the new generation green persistent phosphor SrAl2O4:Eu2+–Dy3+ (SAO:Eu–Dy) was developed and successfully commercialized for watch dials, luminous paints, safety guidance signs, etc.1 Recently, the luminescence wavelength of persistent phosphors has extended from visible light to near-infrared (NIR),2–6 which is suitable for in vivo bio-imaging due to the reduced scattering and minimal absorption coefficient when compared with visible light, allowing imaging of deep tissues. Furthermore, persistent phosphors in the form of nano-particles charged by ultraviolet (UV) light (visible light in rare cases) before injecting into small animals can emit NIR persistent luminescence (PersL) for minutes to even hours without further in situ excitation. The exclusion of external illumination totally removes the autofluorescence as background noise, avoids the complicated background subtraction procedures and thus improves the signal-to-noise ratio (SNR) remarkably.7–9 This new bio-imaging technology has soon become quite attractive while the most reported emission region of NIR persistent phosphors is located in the first biological window (NIR-I, 650–950 nm),4–13 partially due to the easy availability of Si-detectors that work well in this region.Since the Rayleigh scattering (varies as λ−4, here λ is the wavelength) decreases with increasing wavelength, two more biological windows called the second (NIR-II, 1000–1350 nm)14 and the third (NIR-III, approximately from 1500 nm to 1800 nm)15–17 ones are defined, which fall on either side of a strong water absorption band centered around 1450 nm. Compared with the NIR-I window, these two windows, especially the NIR-III window, give much lower scattering coefficient leading potentially to an improved resolution quality and deeper penetration depth.18–20 Moreover, thanks to the recent development of affordable InGaAs detectors that possess adequate sensitivity and high quantum efficiency in the wavelength region above 1000 nm and up to 1650 nm, the shift of the luminescence wavelength of the bio-imaging probe from the NIR-I to the NIR-III window is definitely demanded. However, even for NIR-to-NIR fluorescence bio-probes using real-time excited photoluminescence (PL) for in vivo imaging, only a few candidates are available for the NIR-III window including the single-walled carbon nanotubes (SWNTs)16,21 and heavy metal based semiconductor quantum dots (QDs),16,22 both of which suffer from their potential cytotoxicity for living beings.15–17 Therefore, the development of nontoxic and biocompatible luminescent materials emitting in the NIR-III window still remains a big challenge.
Due to the typical 4I13/2 → 4I15/2 transition at around 1.55 μm and several important transitions such as the 4S3/2 → 4I15/2 one in green,23 the Er3+ ion has already played a key role in applications such as eye-safe lasers,24 optical amplifiers in fiber telecommunication25,26 and upconversion fluorescence27,28 for long years. On the other hand, Er3+ is also considered as the most promising lanthanide ion whose emitting wavelength matches well with both the NIR-III window and the response curve of InGaAs detectors.15–17 Pan et al.29 firstly observed the PersL from Er3+ through the so-called persistent energy transfer (ET) from Eu2+ in the well-known SAO:Eu–Dy persistent phosphor. However, compared with super long PersL (>10 h) from Eu2+ in the green region, the PersL intensity and duration of Er3+ emission peaking at 1530 nm were much weaker and shorter (less than 10 min) after ceasing UV excitation. On the other hand, based on our previous work, in which a novel garnet persistent phosphor of Ce3+,Cr3+ co-doped Y3Al5−xGaxO12 (YAGG:Ce–Cr) was developed and Cr3+ acts as an efficient electron trap with an ideal trap depth at x = 3 for PersL of Ce3+ working at room temperature (RT),30–33 we successfully developed a persistent phosphor of Y3Al2Ga3O12:Nd3+,Ce3+,Cr3+ (YAGG:Nd–Ce–Cr) emitting long PersL (>10 h) at 880 nm, 1064 nm, and 1335 nm due to f–f transitions of Nd3+ through efficient persistent ET from Ce3+.34 It is also worth noting that, in addition to the Er3+ emission mainly composed of the C-band (1530–1565 nm) and the L-band (1565–1625 nm) usually observed in many hosts,35–40 it exhibits an extended U-band (1625–1675 nm) in garnet hosts owing to the large Stark splitting of the terminal 4I15/2 level.41,42 In this paper, we give an example of how to realize Er3+ long PersL through an efficient persistent ET process from Ce3+ in a garnet host and show the first PersL imaging for the NIR-III window by an InGaAs camera.
2. Experimental section
YAGG:Er–Ce–Cr, YAGG:Ce–Er and YAGG:Er ceramic phosphors with the compositions of Y2.925Ce0.015Er0.06Al1.999Cr0.001Ga3O12, Y2.925Ce0.015Er0.06Al2Ga3O12 and Y2.94Er0.06Al2Ga3O12, respectively, were fabricated by a conventional solid-state reaction method. Y2O3 (99.99%), Al2O3 (99.99%), Ga2O3 (99.99%), CeO2 (99.99%), Er2O3 (99.99%) and Cr2O3 (99.9%) were used as raw materials. The starting powder was mixed by a ball milling method (Premium Line P-7, Fritsch Co. Ltd) with anhydrous alcohol for several hours. The mixed powder was dried at 80 °C for 36 h, compacted to form a ceramic green body (ϕ 20 mm, 2 mm thickness) under uniaxial pressing of 50 MPa, and finally sintered at 1600 °C for 10 h in air. The YAGG:Ce–Cr (Y2.985Ce0.015Al1.999Cr0.001Ga3O12) and YAGG:Ce (Y2.985Ce0.015Al2Ga3O12) ceramic phosphors prepared by the same experimental procedure were used as reference samples. All the as-prepared samples were confirmed to be single phase (ICPDS: No. 089-6660) by XRD measurements (see Fig. S1, ESI†).The diffuse reflectance spectrum of the YAGG:Er ceramic sample was measured using a spectrophotometer (UV3600, Shimadzu) equipped with an integrating sphere. The lifetime measurements of the YAGG:Ce and YAGG:Ce–Er samples were made using a compact fluorescence lifetime spectrometer (Quantaurus-Tau-C11367, Hamamatsu Photonics & Co. Ltd) using a 405 nm picosecond pulsed LED as an excitation source. Photoluminescence (PL) spectra of the YAGG:Ce, YAGG:Er and YAGG:Ce–Er ceramic samples by pumping with a 405 nm laser diode (LD) (SDL-405-LM-100T, Shanghai Dream Lasers Technology Co. Ltd) and the YAGG:Ce–Cr and YAGG:Er–Ce–Cr samples by pumping with a 442 nm LD (NDHB510APA-E, Nichia Co. Ltd) were recorded in the range of 350–1670 nm. The PL spectra were measured using a monochromator (G250, Nikon) equipped with a Si photodiode (PD) detector (S-025-H, Electro-Optical System Inc.) from 350 to 1100 nm and an InGaAs PD detector (IGA-030-H, Electro-Optical System Inc.) from 1100 to 1670 nm (see the schematic illustration of the measurement setup in Fig. S2, ESI†). All the PL spectra were calibrated by using a standard halogen lamp (SCL-600, Labsphere). The persistent luminescence (PersL) spectrum of the YAGG:Er–Ce–Cr sample was measured using a Si CCD spectrometer (QE65-Pro, Ocean Optics) from 350 to 1075 nm and an InGaAs CCD spectrometer (NIR-Quest512, Ocean Optics) from 1075 to 1700 nm connected with an optical fiber. All the PersL spectra were calibrated using the same halogen lamp. A 300 W Xe lamp (MAX-302, Asahi Spectra) with an UV mirror module (250–400 nm) was used as the excitation source for thermoluminescence (TL) two-dimensional (2D) plot measurements. The ceramic sample was set in a cryostat (Helitran LT3, Advanced Research Systems) to control the temperature and firstly illuminated by UV light at 100 K for 10 min, then heated up to 600 K at a rate of 10 K min−1 for 10 min after ceasing the illumination, and the TL signals were recorded using a PMT detector (R11041, Hamamatsu Photonics & Co. Ltd) covered with 475 nm short-cut and 650 nm long-cut filters to monitor Ce3+ emission and the same InGaAs PD detector covered with a 1000 nm short-cut filter to monitor the NIR emission of Er3+. The same Si CCD and InGaAs CCD spectrometers were operated simultaneously with the TL measurement to monitor the emission spectra at different temperatures. All persistent luminescence decay curves of the YAGG:Er–Ce–Cr sample after being excited for 5 min by the Xe lamp with a 460 nm band-pass filter were measured at 25 °C using the same PMT or InGaAs PD detectors (see the schematic illustration of the measurement setup in Fig. S3, ESI†). In order to monitor the Ce3+ emission, the PMT detector was covered with 475 nm short-cut and 650 nm long-cut filters to filter out all but the Ce3+ luminescence. Then the decay curves were calibrated to absolute luminance (in units of mcd m−2) using a radiance meter (Glacier X, B&W Tek Inc.). In order to monitor the Er3+ luminescence, the InGaAs PD was covered with a 1000 nm short-cut filter to filter out all but the Er3+ luminescence. Then the decay curves were calibrated to the photon emission rate (in units of cps Sr−1 m−2) using the same radiance meter calculated by the absolute radiance (in units of mW Sr−1 m−2) divided by photon energy (photon energy: E = hc/λ, where h is the Planck constant, c is the speed of light in vacuum and λ is the wavelength). The photographs of the YAGG:Ce–Cr and YAGG:Er–Ce–Cr samples after charging by a 455 nm LED lamp (M445L3, THORLABS, Inc.) for 5 min were taken using a digital camera (EOS kiss X5, Canon) for the visible light region, and the settings remained constant: exposure time – 1 s, ISO value – 1600, aperture value (F value) – 5.0. The photographs for the short-wave infrared (SWIR) region (∼900–1700 nm) were taken using a bio-imaging machine (NIS-OPT, Shimadzu) equipped with an InGaAs camera (Xeva-1.7-320, TE3 cooling system down to 223 K) after charging by the same 455 nm LED for 5 min, and the integrating time was set to be 0.04 s.
3. Results and discussion
3.1. Efficient energy transfer from Ce3+ to Er3+ in the YAGG host
Fig. 1(a)–(c) show the PL spectra of the YAGG:Ce, YAGG:Er and YAGG:Ce–Er samples under 405 nm laser excitation. Both the broad band emission from Ce3+:5d1 → 4f peaking at 500 nm and sharp-line emission bands from Er3+:4S3/2 → 4I15/2 peaking at 555 nm, the 4S3/2 → 4I13/2 band peaking at 862 nm, and the 4I13/2 → 4I15/2 band peaking at 1532 nm in the YAGG:Ce–Er sample are clearly observed. Especially for the 4I13/2 → 4I15/2 transition, similar to that in the Y3Al5O12 (YAG) host,40–44 broadly split bands ranging from 1450 to 1670 nm in the YAGG host are also observed. On the other hand, the emission bands due to the Er3+:4I11/2 → 4I15/2 transition peaking at 966 nm and 1013 nm are almost quenched in the YAGG:Ce–Er sample compared with that in the YAGG:Er sample, which can be attributed to the ET from Er3+:4I11/2 → 4I13/2 to Ce3+:2F7/2 ← 2F5/2, the famous “cross-relaxation process” in the Ce3+–Er3+ pair45 as shown in Fig. 1(d). This process can efficiently facilitate the population of the 4I13/2 level and simultaneously improve the emission intensity of the Er3+:4I13/2 → 4I15/2 transition dramatically.45 | ||
| Fig. 1 PL spectra of the (a) YAGG:Ce, (b) YAGG:Er and (c) YAGG:Ce–Er ceramic samples under 405 nm laser excitation; (d) energy level diagrams of Ce3+ and Er3+; (e) fluorescence decay curves of the YAGG:Ce and YAGG:Ce–Er ceramic samples (λex = 405 nm and λem = 500 nm). | ||
Fig. 1(e) shows that the lifetime of the Ce3+:5d1 level declines from 34.1 ns to 12.2 ns after Er3+ co-doping, indicating extra decay pathways due to the non-radiative ET from Ce3+ to Er3+ in the YAGG:Ce–Er sample. The total decay rate (Wtot) of the 5d1 level in the YAGG:Ce sample is given by
| Wtot = A + WMP = τCe−1 | (1) |
| Wtot = A + WMP + WET = τCe,Er−1 | (2) |
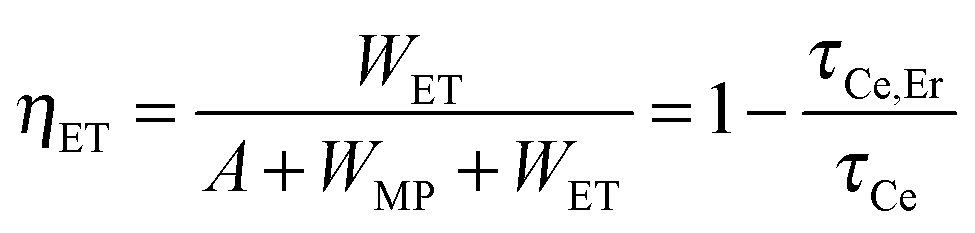 | (3) |
3.2. PL and PersL spectra
Fig. 2(a) shows the PL spectra of the YAGG:Ce–Cr and YAGG:Er–Ce–Cr samples under blue laser (442 nm) excitation. The YAGG:Ce–Cr sample exhibits an intense emission band peaking at 505 nm due to the parity allowed 5d1 → 4f transition of Ce3+.46–48 Besides, a weak emission band peaking at around 690 nm is attributed to the Cr3+:2E(2G) → 4A2(4F) transition (the so-called R-line). Comparing the PL spectrum of the YAGG:Ce–Cr sample with the diffuse reflectance of the Er3+ singly doped YAGG sample (YAGG:Er), the absorption bands (4I15/2 → 4F3/2,5/2,7/2, 2H11/2, 4S3/2) of Er3+ are largely overlapped with the emission band of Ce3+ indicating that the ET process from Ce3+ to Er3+ can efficiently occur. This is confirmed by the decrease of the Ce3+ emission intensity in the visible range and the presence of several sharp emission bands at around 555 nm, 862 nm and 1532 nm owing to the f–f transitions of Er3+:4S3/2 → 4I15/2, 4I13/2, and 4I13/2 → 4I15/2, respectively, in the YAGG:Er–Ce–Cr sample. The quenching of the Er3+:4I11/2 → 4I15/2 bands peaking at 966 nm and 1013 nm is also observed as in the YAGG:Ce–Er sample due to the cross-relaxation process mentioned in Section 3.1.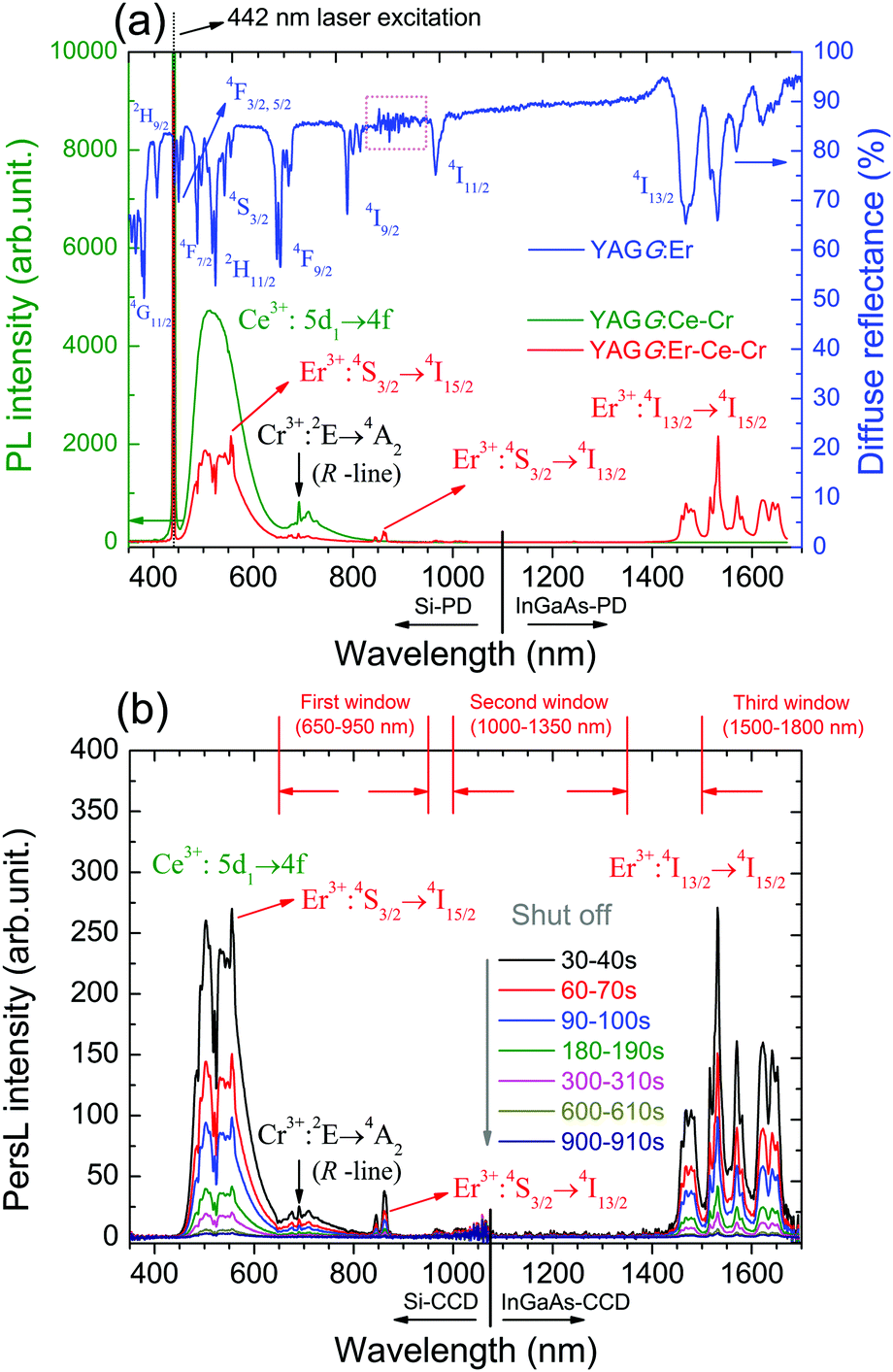 | ||
| Fig. 2 (a) PL spectra of the YAGG:Ce–Cr and YAGG:Er–Ce–Cr ceramic samples (λex = 442 nm) as well as the diffuse reflectance of the YAGG:Er ceramic sample (pink-dotted-line area is the detector change region from PMT to PbS). (b) PersL spectra of the YAGG:Er–Ce–Cr ceramic sample (integrating time: 10 s) after ceasing the blue light illumination. | ||
The PersL spectra of the YAGG:Er–Ce–Cr sample recorded at different times after ceasing blue light illumination are shown in Fig. 2(b). The persistent emission bands not only exhibit a broad band peaking at around 500 nm due to Ce3+:5d1 → 4f transition but also intense sharp bands located in the NIR region (around 862 nm and 1532 nm) due to the f–f transitions of Er3+. Especially the PersL bands in the range of 1450–1670 nm match well with the NIR-III window. The identical shape of the PL and PersL spectra of the YAGG:Er–Ce–Cr sample suggests that the emission centers are the same under and after excitation. Since the efficient ET process from Ce3+ to Er3+ was confirmed in the YAGG:Ce–Er sample shown in Fig. 1(e), we assume that the efficient persistent ET also occurs in the YAGG:Er–Ce–Cr phosphor after ceasing the blue light illumination.
3.3. Persistent luminescence decay curves
The persistent luminescence decay curve monitoring the Ce3+ emission (475–650 nm) of the YAGG:Er–Ce–Cr sample after ceasing the blue light illumination is shown in Fig. 3(a), in which the decay curves of the standard YAGG:Ce–Cr ceramic phosphor and a compacted ceramic pellet made of the commercial SAO:Eu–Dy phosphor (LumiNova-GLL300FFS, Nemoto & Co. Ltd) under the same experimental condition are also plotted as references.30 The luminance values at 10 min after ceasing the excitation are 58 mcd m−2 for YAGG:Er–Ce–Cr, 627 mcd m−2 for YAGG:Ce–Cr, and 211 mcd m−2 for SAO:Eu–Dy [see the photographs of the two samples after ceasing blue LED illumination in Fig. 3(c)]. Persistent luminescence duration to reach a luminance value of 0.32 mcd m−2 in the YAGG:Er–Ce–Cr sample is around 213 min, which is much shorter than that of the YAGG:Ce–Cr sample (about 808 min), due to the quenching effect of the green Ce3+ emission by the non-radiative persistent ET to Er3+ (note that the luminance value 0.32 mcd m−2 is the minimum value commonly used by the safety signage industry, about 100 times the sensitivity of the dark-adapted eye).49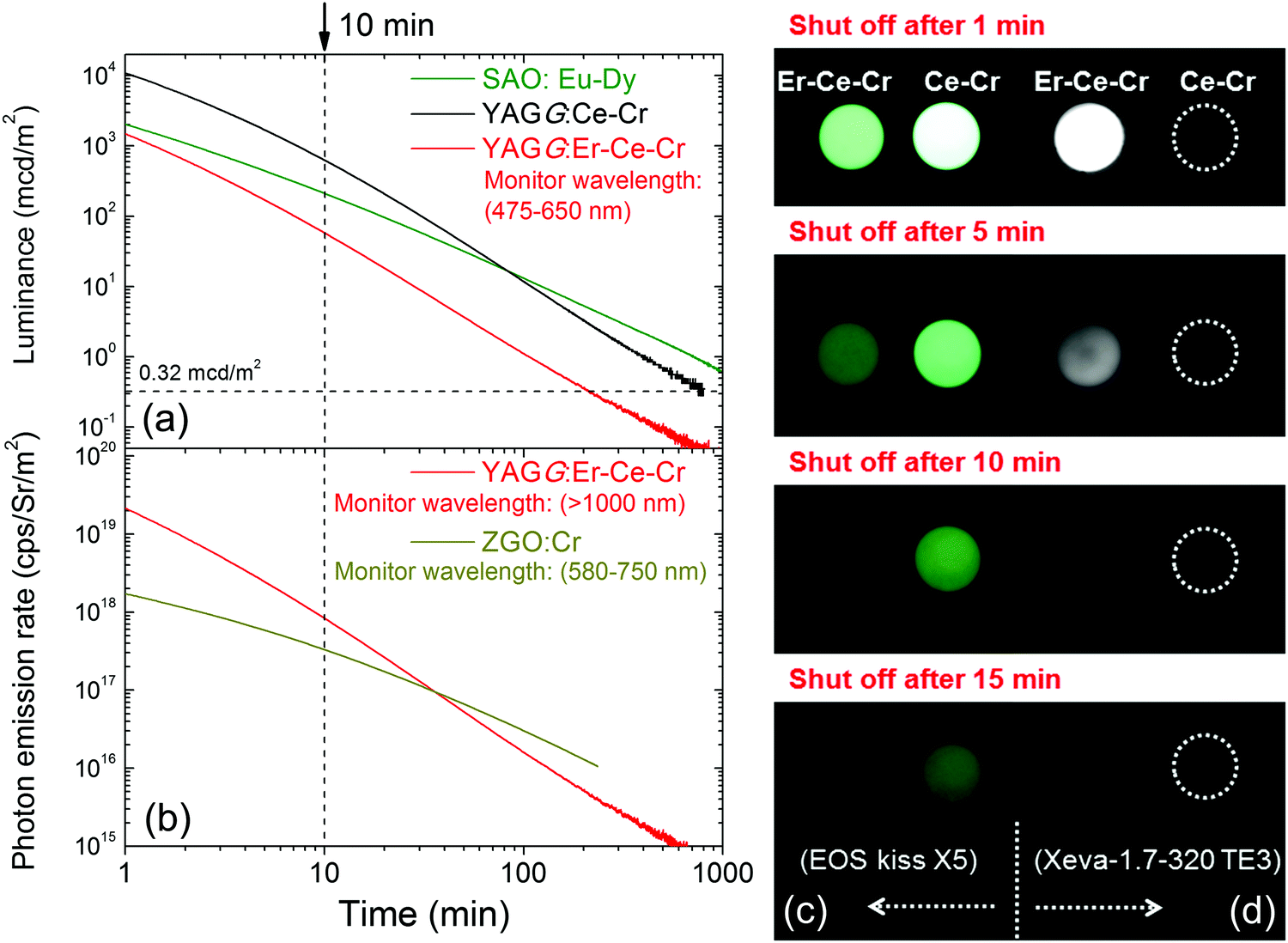 | ||
| Fig. 3 Persistent luminescence decay curves of the YAGG:Er–Ce–Cr ceramic sample: (a) luminance monitoring Ce3+ emission (YAGG:Ce–Cr and SAO:Eu–Dy ceramic samples as references) and (b) photon emission rate monitoring Er3+ emission (ZGO:Cr ceramic sample as a reference); and photo images of the YAGG:Ce–Cr and YAGG:Er–Ce–Cr ceramic samples after blue LED (455 nm, 1 W output) illumination for 5 min: (c) taken by a digital camera (EOX kiss X5) with exposure time: 1 s, ISO value: 1600, aperture value (F value): 5.0 and (d) taken by a SWIR camera (Xeva-1.7-320 TE3) with integrating time: 0.04 s. | ||
The persistent luminescence decay curve monitoring Er3+ NIR emission (>1000 nm) of the YAGG:Er–Ce–Cr sample after ceasing the same blue light illumination is shown in Fig. 3(b), in which the decay curve of the standard ZnGa2O4:Cr3+ (ZGO:Cr, emitting wavelength peaking at 695 nm) sample under the same experimental condition is also plotted as a reference.50 The NIR photon emission rate of the YAGG:Er–Ce–Cr sample for the NIR-III window at 10 min after ceasing the blue light excitation (8.33 × 1017 cps Sr−1 m−2) is over two times higher than that of the widely used deep-red persistent phosphor ZGO:Cr (3.30 × 1017 cps Sr−1 m−2) for the NIR-I window (summarized in Table 1), which indicates that this persistent phosphor exhibits excellent PersL in the NIR region by Er3+ as well as in the visible light region by Ce3+. In Fig. 3(d), we exhibit the first PersL imaging by a commercial InGaAs camera51 for the two garnet ceramic phosphors. Although the YAGG:Ce–Cr sample shows bright green PersL by a digital camera in the visible light region [see Fig. 3(c)], no signal can be captured by the InGaAs camera due to its lack of PersL in the short-wave infrared (SWIR, ∼900–1700 nm) region. Since the InGaAs camera is only sensitive to the luminescence located in the SWIR region, the PersL imaging from Er3+ in the YAGG:Er–Ce–Cr sample was nicely recorded even when using a very short integrating time (0.04 s, the maximum value to avoid the saturation of Er3+ PersL intensity at 1 min after ceasing the blue light excitation). This result clearly proves that Er3+ PersL is intense and long enough to be recorded by an InGaAs camera.
3.4. Thermoluminescence (TL) glow curves and 2D contour plots
Fig. 4(a) and (c) show the TL glow curves of the YAGG:Ce–Cr and YAGG:Er–Ce–Cr samples monitoring only Ce3+ emission, respectively. Although the intensity of the Ce3+ emission in the YAGG:Er–Ce–Cr sample is much weaker than that in the YAGG:Ce–Cr sample due to the persistent ET from Ce3+ to Er3+, both of the glow peaks are located at around 300 K. This suggests that PersL from Ce3+ is originated from the same electron trap, Cr3+, in both samples with the same trap depth or more precisely with the same trap distribution.31,34 Therefore, the thermal activation energy which is required to clean up the electrons captured by the traps is the same.31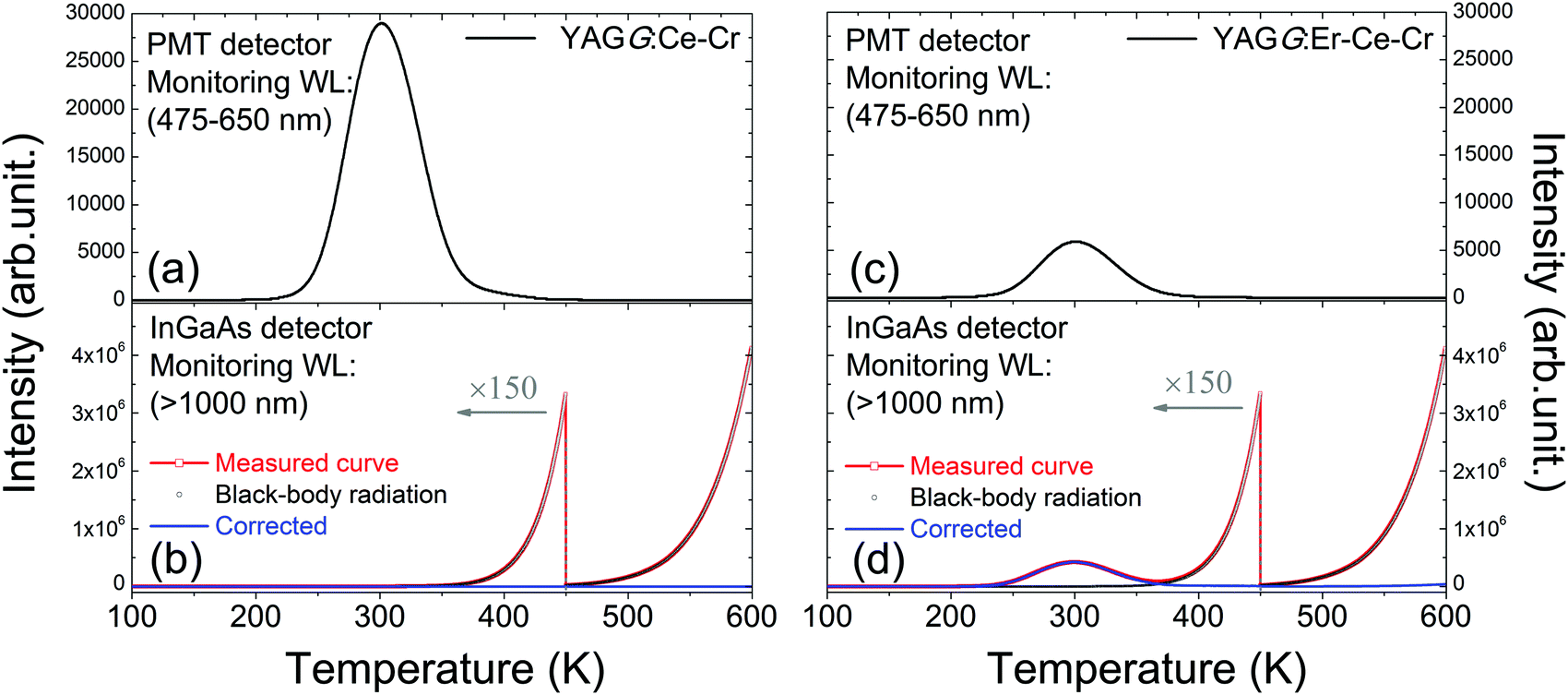 | ||
| Fig. 4 TL glow curves of the YAGG:Ce–Cr ceramic sample monitored by the (a) PMT detector in the range of 475–650 nm, (b) InGaAs detector in the range of over 1000 nm and that of the YAGG:Er–Ce–Cr ceramic sample monitored by the (c) PMT detector in the range of 475–650 nm, and (d) InGaAs detector in the range of over 1000 nm. | ||
For TL measurements monitoring the visible light region, the black-body radiation has nearly no effects on the TL read-out when the heating temperature is lower than 600 K. On the other hand, when monitoring NIR emission, the excessive effect of black-body radiation on the TL read-out must be taken into account.52,53Fig. 4(b) and (d) show the TL glow curves of the YAGG:Ce–Cr and YAGG:Er–Ce–Cr samples monitoring only NIR emission (>1000 nm), respectively. Since there is no PersL contribution from the YAGG:Ce–Cr sample in the wavelength region over 1000 nm, only intense black-body radiation from the sample and partially from the measurement setup is observed. However, for the YAGG:Er–Ce–Cr sample, the additional TL glow curve centered at around 300 K is also observed after subtracting the intense black-body radiation signal (blue-line by subtracting the black-dotted-curve from the red-dotted-line), which is ascribed to the PersL from Er3+ in the NIR region.
Fig. 5 shows the TL two-dimensional (2D) contour plots of the YAGG:Ce–Cr and YAGG:Er–Ce–Cr samples in order to see what kind of emission bands contribute to the TL glow peak at different temperatures. From the contour plots of the YAGG:Ce–Cr sample in Fig. 5(a) and (b), it can be seen that at increased temperatures, the TL spectrum is simply composed of two emission bands from Ce3+ and Cr3+, and no contributed emission except strong black-body radiation in the NIR region (1300–1700 nm, above ∼500 K) is observed. On the other hand, in the YAGG:Er–Ce–Cr sample as shown in Fig. 5(c) and (d), besides the emission bands from Ce3+ and Cr3+, the NIR emission band of Er3+ appears in the same temperature range, which agrees well with its PersL spectrum in Fig. 2(b). The intense TL glow peaks of both YAGG:Ce–Cr and YAGG:Er–Ce–Cr samples lie in almost the same temperature range (around 300 K), close to RT and body temperature of Mammalia (around 310 K). Since the TL peak temperature is correlated to the energy gap between the bottom of the conduction band and the electron trap,49 the identical glow temperature of the two samples indicates the same electron trapping and de-trapping process54 in both, where Cr3+ works as an efficient electron trap with ideal trap depth for long PersL in the host of Y3Al2Ga3O12.30–33
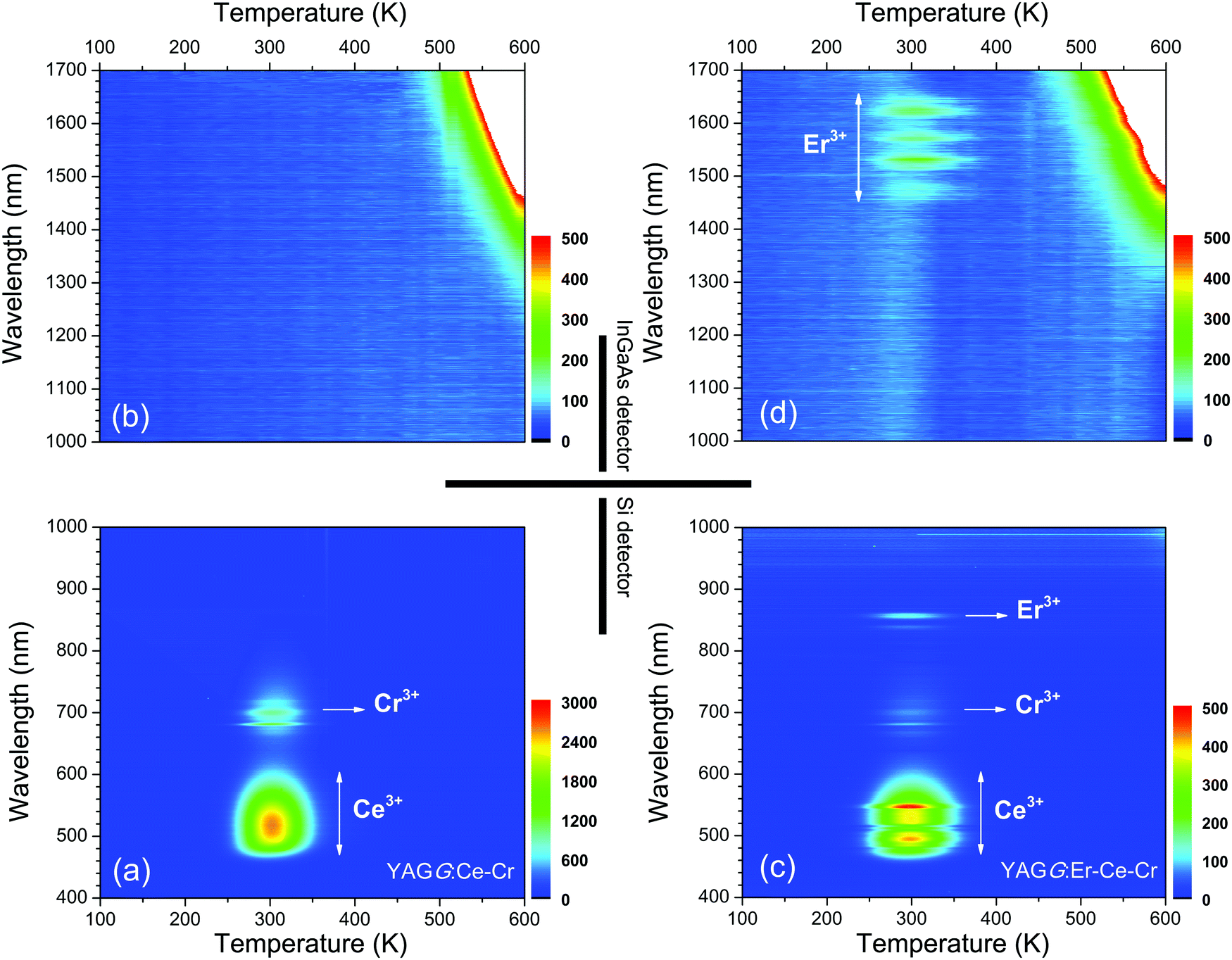 | ||
| Fig. 5 Wavelength–temperature (λ–T) contour plots of the (a), (b) YAGG:Ce–Cr and (c), (d) YAGG:Er–Ce–Cr ceramic samples. | ||
3.5. Persistent luminescence mechanism
The persistent luminescence mechanism of the YAGG:Er–Ce–Cr persistent phosphor is briefly explained by constructing the vacuum referred binding energy (VRBE) diagram31,55,56 composed of Ce3+, Er3+, Cr2+, conduction band (C.B.), and valence band (V.B.) energy levels in the Y3Al2Ga3O12 host (see Fig. 6). When the YAGG:Er–Ce–Cr sample is charged by blue light, the electron located at the ground state (2F5/2) of Ce3+ is promoted to the excited state of the lowest 5d energy level (5d1). Since the energy gap between the bottom of C.B. and the 5d1 level is small, the excited electron can “jump” to C.B. with thermal assisted activation energy at RT and then be trapped by the electron trapping center (Cr3+).30,31 At that time, Ce3+ is photo-oxidized into Ce4+ or (Ce3+ + h+) and Cr3+ changes into Cr2+ or (Cr3+ + e−) after capturing one electron (process ①).31,56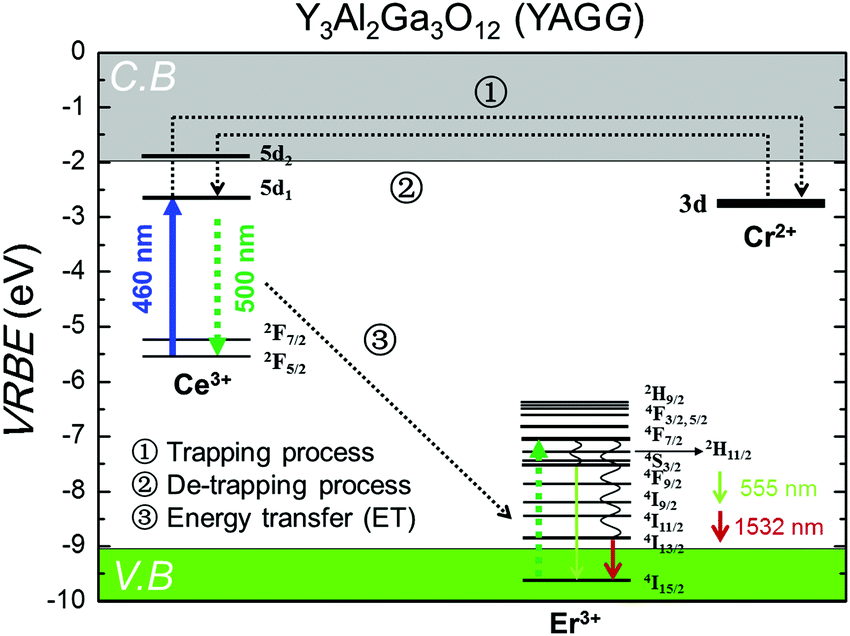 | ||
| Fig. 6 The VRBE diagram including selected energy levels of Ce3+, Er3+, and Cr2+ in the Y3Al2Ga3O12 (YAGG) host. | ||
Then the de-trapping process occurs with the thermal release of the captured electron from the Cr2+ (Cr3+ + e−) trap, and finally the excited state of the Ce ion, (Ce3+)*, appears after capturing the released electron in the re-combination process (process ②). The radiative relaxation gives a broad band emission of Ce3+:5d1 → 2F5/2, 2F7/2, and the resonant ET process takes place nearly at the same time to the Er3+ ion (process ③), which is followed by rapid multi-phonon relaxation down to the 4S3/2 and 4I13/2 excited levels leading to the sharp luminescence bands of Er3+: 4S3/2 → 4I15/2 in the green and 4I13/2 → 4I15/2 in the NIR region.
4. Conclusion
In summary, we have successfully developed a blue-light-chargeable NIR persistent phosphor (Y3Al2Ga3O12:Er3+,Ce3+,Cr3+) with long (>10 h) persistent luminescence due to the typical Er3+:4I13/2 → 4I15/2 transition ranging from 1450 nm to 1670 nm in garnet. The NIR PersL bands from Er3+ through efficient persistent energy transfer from Ce3+ match well with the NIR-III window, and its photon emission rate (8.33 × 1017 cps Sr−1 m−2) at 10 min after ceasing blue light (460 nm) illumination was over two times higher than that of the widely used ZnGa2O4:Cr3+ deep-red persistent phosphor (3.30 × 1017 cps Sr−1 m−2). We also showed the first persistent luminescence imaging with high intensity and long duration by a commercial InGaAs camera. In vivo bio-imaging in the NIR-III window with improved optical resolution quality and deep tissue penetration depth can be expected in the near future by using this material (in the form of nano-particles) as a functionalized bio-probe.Acknowledgements
We would like to acknowledge Dr Morgane Pellerin from Chimie-Paris Tech and Dr Laura Wortmann and Prof. Kohei Soga from the Tokyo University of Science for supporting persistent luminescence imaging by the Xeva-1.7-320 TE3 InGaAs camera.This work was financially supported by a Grant-in-Aid for JSPS Fellows (No. 16J09849).
References
- T. Matsuzawa, Y. Aoki, N. Takeuchi and Y. Murayama, J. Electrochem. Soc., 1996, 143, 2670–2673 CrossRef CAS.
- K. Van Den Eeckhout, D. Poelman and P. F. Smet, Materials, 2013, 6, 2789–2818 CrossRef CAS.
- D. Dutczak, T. Jüstel, C. Ronda and A. Meijerink, Phys. Chem. Chem. Phys., 2015, 17, 15236–15249 RSC.
- S. K. Singh, RSC Adv., 2014, 4, 58674–58698 RSC.
- Y. Zhuang, Y. Katayama, J. Ueda and S. Tanabe, Opt. Mater., 2014, 36, 1907–1912 CrossRef CAS.
- B. Viana, S. K. Sharma, D. Gourier, T. Maldiney, E. Teston, D. Scherman and C. Richard, J. Lumin., 2016, 170, 879–887 CrossRef CAS.
- Q. le Masne de Chermont, C. Chanéac, J. Seguin, F. Pellé, S. Maîtrejean, J. P. Jolivet, D. Gourier, M. Bessodes and D. Scherman, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 9266–9271 CrossRef CAS PubMed.
- F. Liu, W. Yan, Y. J. Chuang, Z. Zhen, J. Xie and Z. Pan, Sci. Rep., 2013, 3, 1554–1562 Search PubMed.
- T. Maldiney, A. Bessière, J. Seguin, E. Teston, S. K. Sharma, B. Viana, A. J. J. Bos, P. Dorenbos, M. Bessodes, D. Gourier, D. Scherman and C. Richard, Nat. Mater., 2014, 13, 418–426 CrossRef CAS PubMed.
- A. Bessière, S. Jacquart, K. Priolkar, A. Lecointre, B. Viana and D. Gourier, Opt. Express, 2011, 19, 10131–10137 CrossRef PubMed.
- Z. W. Pan, Y. Y. Lu and F. Liu, Nat. Mater., 2012, 11, 58–63 CrossRef CAS PubMed.
- Y. Katayama, J. Ueda and S. Tanabe, Opt. Mater. Express, 2014, 4, 613–623 CrossRef CAS.
- J. Xu, J. Ueda, Y. Zhuang, B. Viana and S. Tanabe, Appl. Phys. Express, 2015, 8, 042602 CrossRef.
- A. M. Smith, M. Mancini and S. Nie, Nat. Nanotechnol., 2009, 4, 710–711 CrossRef CAS PubMed.
- E. Hemmer, N. Venkatachalam, H. Hyodo, A. Hattori, Y. Ebina, H. Kishimoto and K. Soga, Nanoscale, 2013, 5, 11339–11361 RSC.
- E. Hemmer, A. Benayas, F. Légaré and F. Vetrone, Nanoscale Horiz., 2016, 1, 168–184 RSC.
- J.-C. G. Bünzli, J. Lumin., 2016, 170, 866–878 CrossRef.
- D. J. Naczynski, M. C. Tan, M. Zevon, B. Wall, J. Kohl, A. Kulesa, S. Chen, C. M. Roth, R. E. Riman and P. V. Moghe, Nat. Commun., 2013, 4, 2199 CAS.
- L. A. Sordillo, Y. Pu, S. Pratavieira, Y. Budansky and R. R. Alfano, J. Biomed. Opt., 2014, 19, 056004 CrossRef PubMed.
- L. A. Sordillo, S. Pratavieira, Y. Pu, K. S. Ramirez, L. Shi, L. Zhang, Y. Budansky and R. R. Alfano, Proc. SPIE, 2014, 8940, 89400V CrossRef.
- K. Welsher, Z. Liu, D. Daranciang and H. Dai, Nano Lett., 2008, 8, 586–590 CrossRef CAS PubMed.
- W. Y. Liu, A. Y. Chang, R. D. Schaller and D. V. Talapin, J. Am. Chem. Soc., 2012, 134, 20258–20261 CrossRef CAS PubMed.
- S. Tanabe, Int. J. Appl. Glass Sci., 2015, 6, 305–328 CrossRef CAS.
- E. Snitzer and R. Woodcock, Appl. Phys. Lett., 1965, 6, 45–46 CrossRef CAS.
- M. Nakazawa, M. Tokuda, K. Washio and Y. Asahara, Opt. Lett., 1984, 9, 312–314 CrossRef CAS PubMed.
- E. Desurvire, J. Simpson and P. C. Becker, Opt. Lett., 1987, 12, 888–890 CrossRef CAS PubMed.
- F. Auzel, C. R. Acad. Sci., 1966, 262, 1016–1019 Search PubMed.
- F. Auzel, C. R. Acad. Sci., 1966, 263, 819–821 Search PubMed.
- N. Yu, F. Liu, X. Li and Z. W. Pan, Appl. Phys. Lett., 2009, 95, 231110 CrossRef.
- J. Ueda, K. Kuroishi and S. Tanabe, Appl. Phys. Lett., 2014, 104, 101904 CrossRef.
- J. Ueda, P. Dorenbos, A. J. J. Bos, K. Kuroishi and S. Tanabe, J. Mater. Chem. C, 2015, 3, 5642–5651 RSC.
- J. Ueda, J. Ceram. Soc. Jpn., 2015, 123, 1059–1064 CrossRef CAS.
- J. Xu, J. Ueda, K. Kuroishi and S. Tanabe, Scr. Mater., 2015, 102, 47–50 CrossRef CAS.
- J. Xu, S. Tanabe, A. D. Sontakke and J. Ueda, Appl. Phys. Lett., 2015, 107, 081903 CrossRef.
- A. A. Kaminskii, Crystalline Lasers: Physical Processes and Operating Schemes, CRC Press, New York, 1996, pp. 187–188 Search PubMed.
- S. Tanabe and T. Hanada, J. Non-Cryst. Solids, 1996, 196, 101–105 CrossRef CAS.
- S. Tanabe, N. Sugimoto, S. Ito and T. Hanada, J. Lumin., 2000, 87, 670–672 CrossRef.
- M. Nishi, S. Tanabe, K. Fujita, K. Hirao and G. Pezzotti, Solid State Commun., 2004, 132, 19–23 CrossRef CAS.
- S. Ono and S. Tanabe, IEEE J. Quantum Electron., 2004, 40, 1704–1708 CrossRef CAS.
- M. Nishi, S. Tanabe, M. Inoue, M. Takahashi, K. Fujita and K. Hirao, Opt. Mater., 2005, 27, 655–662 CrossRef CAS.
- Y. Shiota, S. Tanabe, T. Hanada and Y. Maeda, J. Ceram. Soc. Jpn., 1999, 107, 1201–1205 CrossRef CAS.
- M. Nishi, S. Tanabe, K. Fujita and K. Hirao, J. Alloys Compd., 2006, 408–412, 788–790 CrossRef CAS.
- J. Zhou, Y. Teng, X. Liu, S. Ye, Z. Ma and J. Qiu, Phys. Chem. Chem. Phys., 2010, 12, 13759–13762 RSC.
- Q. Wang, L. Su, H. Li, L. Zheng, X. Xu, H. Tang, X. Guo, D. Jiang and J. Xu, Phys. Status Solidi A, 2011, 208, 2839–2842 CrossRef CAS.
- Z. Meng, T. Yoshimura, K. Fukue, M. Higashihata, Y. Nakata and T. Okada, J. Appl. Phys., 2000, 88, 2187–2190 CrossRef CAS.
- Y. Luo and Z. Xia, J. Phys. Chem. C, 2014, 118, 23297–23305 CAS.
- J. Ueda, P. Dorenbos, A. J. J. Bos, A. Meijerink and S. Tanabe, J. Phys. Chem. C, 2015, 119, 25003–25008 CAS.
- V. Khanin, I. Venevtsev, P. Rodnyi and C. Ronda, Radiat. Meas., 2016, 90, 104–108 CrossRef CAS.
- K. Van den Eeckhout, P. F. Smet and D. Poelman, Materials, 2010, 3, 2536–2566 CrossRef CAS.
- Y. Zhuang, J. Ueda and S. Tanabe, Appl. Phys. Express, 2013, 6, 052602 CrossRef.
- http://www.xenics.com/sites/default/files/leaflets/xb-081_01_xeva-1.7-320_te3_lowres.pdf .
- E. P. Manche, Rev. Sci. Instrum., 1978, 49, 715–717 CrossRef CAS PubMed.
- F. Liu, Y. Liang, Y. Chen and Z. W. Pan, Adv. Opt. Mater., 2016, 4, 562–566 CrossRef CAS.
- P. Dorenbos, J. Electrochem. Soc., 2005, 152, H107–H110 CrossRef CAS.
- P. Dorenbos, J. Lumin., 2013, 134, 310–318 CrossRef CAS.
- J. Xu, J. Ueda and S. Tanabe, J. Mater. Chem. C, 2016, 4, 4380–4386 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6tc04027f |
| This journal is © The Royal Society of Chemistry 2016 |