
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffects of PNDIT2 end groups on aggregation, thin film structure, alignment and electron transport in field-effect transistors†
Rukiya
Matsidik
ab,
Alessandro
Luzio
c,
Sophie
Hameury
ab,
Hartmut
Komber
d,
Christopher R.
McNeill
e,
Mario
Caironi
c and
Michael
Sommer
*abf
aUniversität Freiburg, Makromolekulare Chemie, Stefan-Meier-Str. 31, 79104 Freiburg, Germany. E-mail: michael.sommer@makro.uni-freiburg.de
bFreiburger Materialforschungszentrum, Universität Freiburg, Stefan-Meier-Str. 21, 79104 Freiburg, Germany
cCenter for Nano Science and Technology @PoliMi, Istituto Italiano di Tecnologia, Via Pascoli 70/3, 20133 Milano, Italy
dLeibniz Institut für Polymerforschung Dresden e.V., Hohe Straße 6, 01069 Dresden, Germany
eDepartment of Materials Science and Engineering, Monash University, Clayton, Victoria 3800, Australia
fFreiburger Institut für Interactive Materialien und Bioinspirierte Technologien, Georges-Köhler Allee 105, 79110 Freiburg, Germany
First published on 18th October 2016
Abstract
To develop greener protocols toward the sustainable production of conjugated polymers, we combine the advantages of atom-economic direct arylation polycondensation (DAP) with those of the green solvent 2-methyltetrahydrofuran (MeTHF). The n-type copolymer PNDIT2 is synthesized from unsubstituted bithiophene (T2) and 2,6-dibromonapthalene diimide (NDIBr2) under simple DAP conditions in MeTHF. Extensive optimization is required to suppress nucleophilic substitution of NDIBr end groups, which severely limits molar mass. Different carboxylic acids, bases, palladium precursors and ligands are successfully screened to enable quantitative yield and satisfyingly high molar masses up to Mn,SEC ∼ 20 kDa. In contrast to PNDIT2 made via DAP in toluene with tolyl-chain termini, nucleophilic substitution of NDIBr chain ends in MeTHF finally leads to NDI-OH termination. The influence of different chain termini on the optical, thermal, structural and electronic properties of PNDIT2 is investigated. For samples with identical molecular weight, OH-termination leads to slightly reduced aggregation in solution and bulk crystallinity, a decreased degree of alignment in directionally deposited films, and a consequently reduced, but not compromised, electron mobility with promising values still close to 0.9 cm2 V−1 s−1.
1. Introduction
Direct arylation polycondensation (DAP) with its unique aspects such as minimized waste, cheap and straightforward synthesis pathways compared to conventional organometallic cross-coupling reactions, and the possibility to make purer materials, is becoming a highly useful tool for the synthesis of π-conjugated polymers.1–4 Eliminating the need for building block functionalization, which adheres to all other conventional organometallic cross-coupling reactions, additionally enables usage of a wider range of substrates in DAP.5,6 In conventional cross-coupling reactions, metalation requires cryogenic or catalytic conditions. This often raises costs and thus renders some substrates poor candidates. Additionally, highly toxic tin derivatives are used in the case of Stille polycondensation.7 As a result, many successful examples of DAP applied to construct conjugated polymers that exhibit comparable or even enhanced properties compared to conventional methods have been reported.8–14 Nevertheless, while high-mobility p-type conjugated polymers have been reported in manifold variation, n-type or electron accepting conjugated polymers with suitable n-channel properties are still rare. Notable recent examples include copolymers based on naphthalene diimide (NDI) and diketopyrrolopyrrole (DPP).14–17 Our recent accomplishment of establishing a very simple and highly efficient DAP protocol for NDI-based copolymers in aromatic solvents successfully addressed the hitherto unsolved issue associated with the copolymerization of the NDI unit and suitable C–H monomers.13,18 Indeed, commonly used Stille polycondensation protocols entail environmental and safety issues, and the few-reported DAP protocols for the synthesis of NDI-based copolymers were either inefficient, led to low molecular weight (MW) or did not allow for structural versatility.16,19–21 Another intriguing feature of the recently established DAP protocol involves MW control through the in situ C–H activation of aromatic solvents, which clearly increases the degree of synthetic control and thus brings DAP to the forefront of synthetic methods also for this class of materials.13,18 However, although aromatic solvents are best suited to overcome limited solubilities of conjugated polymers, to control MW and are less toxic than the often used dimethylacetamide (DMAc) in DAP,7 they are neither ideal considering the principles of green chemistry.22 Tetrahydofuran (THF) is another frequently used solvent in DAP.1,23 Unfortunately, synthesizing NDI copolymers via DAP in THF led to low MW due to nucleophilic substitution of NDI-Br chain ends as observed previously.13,24 2-Methyltetrahydrofuran (MeTHF) is a potential alternative to THF because of its higher boiling point, its lower polarity and most importantly, its availability from biomass.25 Absolute numbers for the environmental friendliness of a solvent have been published elsewhere.26 MeTHF has not yet been applied in DAP where THF is often the solvent of choice. Here, we use MeTHF in the synthesis of PNDIT2 via DAP and optimize DAP conditions with respect to the base, the carboxylic acid, the source of the palladium catalyst and the ligand to minimize nucleophilic substitution and consequently maximize both yield and MW. As a result, quantitative yield and relatively high MWs are achieved. However, in contrast to PNDIT2 made via DAP in toluene which gives tolyl-chain termini, nucleophilic substitution of NDIBr chain ends in MeTHF leads to hydroxyl-terminated PNDIT2-OH (Scheme 1). The optical, thermal and electronic properties of PNDIT2 made in MeTHF were thus investigated and compared to PNDIT2 made in toluene (tol) having the same molecular weight as a reference. PNDIT2-OH exhibits slightly reduced electron mobilities in field-effect transistor devices compared to tolyl-terminated PNDIT2, but still promising values of μe ∼ 0.9 cm2 V−1 s−1, indicating that MeTHF can be a potential green solvent for DAP.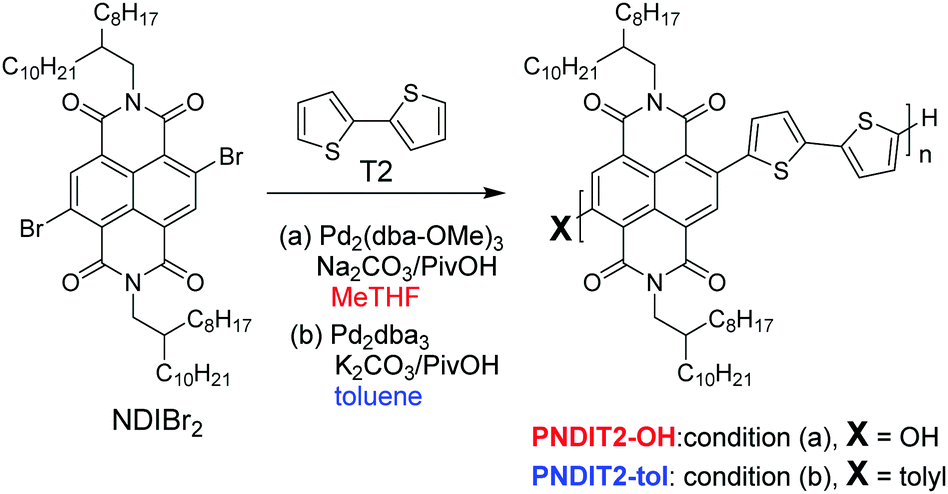 | ||
| Scheme 1 Optimized reaction conditions of PNDIT2 via DAP in MeTHF (condition a) in comparison with in toluene (condition b) leading different chain termination. | ||
2. Results and discussion
Previously, we observed that when using THF as solvent for the synthesis of PNDIT2 via DAP, nucleophilic substitution of the NDI-Br chain ends by pivaloate led to early termination, to low yields <25% and very low number average molecular weights of Mn,SEC ∼ 4.5 kDa.13 As a result, almost quantitative hydroxylation of the NDI chain end was found after saponification of NDI-pivaloate chain ends. Whether saponification occurs during polycondensation due to the presence of base, or during work-up is unclear. Also possible is that OH-termination occurs due to nucleophilic substitution of NDI-Br by hydroxide anions. Importantly, the polarity of the solvents seems to play a major role.13,24 While this side reaction is prevalent in polar solvents such as DMAc24 and THF preventing build-up of long chains, its extent in aromatic solvents is much reduced and only seen for low monomer concentrations when polycondensation is slow.13With the initial aim to find a greener solvent for the synthesis of PNDIT2 and eventually for other conjugated polymers, we replaced THF with MeTHF. The slightly less polar nature of MeTHF could be additionally beneficial in reducing nucleophilic substitution. It is worth noting that all entries were handled with greatest care to ensure a constant stoichiometry and hence comparable results. Using MeTHF at higher monomer concentration increased the reaction yield remarkably to 95% and Mn,SEC to 14.8 kDa (entry 1, Table 1). The same entries in THF and toluene yielded Mn,SEC of 14.5 kDa and 26.0 kDa, respectively, and yields of 79% and 99%, respectively. Compared to samples made in toluene in which nucleophilic substitution is much less prevalent, these values were still unsatisfyingly low and therefore, the reaction conditions in MeTHF were further optimized. We envisioned that, as the carboxylate is responsible for nucleophilic substitution, a change of the structure of the carboxylic acid could give room for improvement. Indeed, carboxylic acid is known to have profound effects in DAP.27–29 Thus, we used pivalic acid (PivOH), cyclopentanecarboxylic acid (CPA) and hexanoic acid (HA) under otherwise identical conditions (entries 1–3). Obviously, no improvement was observed using this strategy. Additionally, we investigated further carboxylic acids with different side chains (entries 4–8). Steric hindrances caused by bulky side chains might be an alternative strategy to reduce nucleophilic attack of the carboxylic acid at NDI chain end. For both linear and cyclic carboxylic acids an increase in the number of carbon atoms improved the reaction yield and molecular weight. However, the best result with Mn,SEC of 15 kDa with 95% yield was achieved when either PivOH or 1-adamantanecarboxylic acid (1-AdaA) were used, with the former enabling a faster reaction than 1-AdaA (entries 1 and 8). Therefore, for all further optimizations, PivOH was used. Next, we varied the base; the use of Na2CO3 instead of K2CO3 further improved Mn and the yield to 17 kDa and 99%, respectively (entries 9 and 10). However, weaker bases like Li2CO3 and CaCO3 did not promote polymerization leading to oligomers only (entries 11 and 12). Next, the amount of catalyst used was altered. An increase of tris(dibenzylideneacetone)dipalladium (Pd2dba3) to 5 mol% led to slight improvements in Mn,SEC with maintained quantitative yield (entries 14 and 15). At this point lowering monomer concentration (entry 13) or switching to microwave heating (entry 16) did not increase MW. Therefore, we fixed the conditions to pivalic acid (1 equiv.) as additive, Na2CO3 (3 equiv.) as base and 2 mol% of catalyst in 0.3 M of monomer concentration with thermal heating, and investigated the reactivity of different types of Pd sources as remaining parameter (Scheme 2). We started with the most simple and commercially available Pd catalyst loaded on activated carbon (Pd/C, 10 wt% loading). The reaction with Pd/C was rather slow and yielded PNDIT2 with Mn,SEC of 13 kDa in 82% yield (entry 17). Pd(PPh3)4 did not lead to any polymer formation (entry 18). We envision that the reason of the lack of reactivity of Pd(PPh3)4 might be the presence of the phosphine ligand. As proven in our previous study, adding phosphines to Pd2dba3 also resulted in nucleophilic substitution of NDI leading to low MW.13 However, quite surprisingly, Buchwald's catalyst Pd2G3 performed better than Pd2dba3 (entry 19) but additionally adding dba as free ligand did not show further improvement (entry 20). Combining Pd2G3 with XPhos (entry 21) was not successful, confirming the detrimental effect of a phosphine ligand to our system.13 The same effect was observed when 1,4-diazabutadiene (DAB) was used as ligand (entry 22). As the combination of Pd2G3 with additional ligands was not promising and Pd2G3 is more cost intensive than Pd2dba3, we settled on Pd2dba3 and investigated potential effects of substituted dba ligands. In order to tune the reactivity of the Pd2dba3 complex, we varied the electronic structure of dba ligands by adding electron rich or poor substituents to the para-position of the phenyl rings.30 Except for the most electron withdrawing group trifluoromethyl (Pd2(dba-CF3)3, entry 27) which slightly underperformed compared to unsubstituted Pd2dba3, all other Pd2dba3 derivatives including Pd2(dba-Me)3 (entry 23), Pd2(dba-tBu)3, (entry 24), Pd2(dba-OMe)3, (entry 25), and Pd2(dba-F)3, (entry 26) equally improved MW compared to unsubstituted Pd2dba3. Polymerization with Pd2(dba-OMe)3 reached the highest MW of Mn,SEC = 20 kDa. It is known that although Pd2dba3 is relatively air-stable, storage for an extended period of time leads to degradation and loss in catalytic activity due to the formation of Pd0 nanoparticles.31 While all these substituted Pd2dba3 complexes were freshly prepared and used, we repeated entry 14 with freshly synthesized Pd2dba3 in order to confirm whether the improvements achieved with substituted Pd2dba3 were caused by substituents or a reduced performance of commercial and aged Pd2dba3. Although freshly made Pd2dba3 gave a slightly better result than commercial Pd2dba3, it still underperformed compared to substituted Pd2dba3 complexes (entry 28).
| Entry | Additive | Base | Catalyst/mol% | Ligand/mol% | T/h | Yield/% | M n,SEC /kDa | M w,SEC /kDa |
|---|---|---|---|---|---|---|---|---|
| a SEC in CHCl3 (0.03 g L−1) at room temperature. b Microwave heating. PivOH: pivalic acid; CPA: cyclopropanoic acid; HA: hexanoic acid; CBA: cyclobutanecarboxylic acid; CHA: cyclohexanecarboxylic acid; OA: octanoic acid; DA: decanoic acid; 1-AdaA: 1-adamantanecarboxylic acid. Structures of catalysts and ligands are given in Scheme 2. | ||||||||
| 1 | PivOH | K2CO3 | Pd2dba3/1 | — | 15 | 95 | 14.8 | 24.8 |
| 2 | CPA | K2CO3 | Pd2dba3/1 | — | 72 | 82 | 10.0 | 14.0 |
| 3 | HA | K2CO3 | Pd2dba3/1 | — | 72 | 86 | 12.0 | 22.0 |
| 4 | CBA | K2CO3 | Pd2dba3/1 | — | 72 | 90 | 14.0 | 21.4 |
| 5 | CHA | K2CO3 | Pd2dba3/1 | — | 24 | 89 | 14.0 | 25.0 |
| 6 | OA | K2CO3 | Pd2dba3/1 | — | 24 | 82 | 13.4 | 21.4 |
| 7 | DA | K2CO3 | Pd2dba3/1 | — | 72 | 92 | 13.3 | 21.3 |
| 8 | 1-AdaA | K2CO3 | Pd2dba3/1 | — | 60 | 95 | 14.0 | 25.0 |
| 9 | PivOH | K2CO3 | Pd2dba3/3 | — | 15 | 97 | 16.2 | 32.3 |
| 10 | PivOH | Na2CO3 | Pd2dba3/3 | — | 22 | 99 | 17.3 | 35.3 |
| 11 | PivOH | Li2CO3 | Pd2dba3/3 | — | 86 | — | — | — |
| 12 | PivOH | CaCO3 | Pd2dba3/3 | — | 72 | — | — | — |
| 13 | PivOH | Na2CO3 | Pd2dba3/3 | — | 20 | 95 | 16.0 | 29.0 |
| 14 | PivOH | Na2CO3 | Pd2dba3/2 | — | 30 | 99 | 16.5 | 33.7 |
| 15 | PivOH | Na2CO3 | Pd2dba3/5 | — | 10 | 97 | 17.5 | 33.9 |
| 16 | PivOH | Na2CO3 | Pd2dba3/3 | — | 2b | 58 | 8.4 | 10.2 |
| 17 | PivOH | Na2CO3 | Pd/C/2 | — | 72 | 82 | 13.0 | 19.0 |
| 18 | PivOH | Na2CO3 | Pd(PPh3)4/2 | — | 72 | — | — | — |
| 19 | PivOH | Na2CO3 | Pd2G3/2 | — | 20 | 98 | 18.1 | 41.6 |
| 20 | PivOH | Na2CO3 | Pd2G3/2 | dba/4 | 20 | 98 | 17.8 | 38.1 |
| 21 | PivOH | Na2CO3 | Pd2G3/2 | XPhos/4 | 20 | — | — | — |
| 22 | PivOH | Na2CO3 | Pd2G3/2 | DAB/4 | 20 | — | — | — |
| 23 | PivOH | Na2CO3 | Pd2(dba-Me)3/2 | — | 20 | 98 | 19.3 | 43.3 |
| 24 | PivOH | Na2CO3 | Pd2(dba-tBu)3/2 | — | 20 | 99 | 19.0 | 42.0 |
| 25 | PivOH | Na2CO3 | Pd2(dba-OMe)3/2 | — | 20 | 98 | 19.9 | 45.3 |
| 26 | PivOH | Na2CO3 | Pd2(dba-F)3/2 | — | 20 | 98 | 19.0 | 42.3 |
| 27 | PivOH | Na2CO3 | Pd2(dba-CF3)3/2 | — | 20 | 98 | 15.0 | 30.0 |
| 28 | PivOH | Na2CO3 | Pd2dba3-fresh/2 | — | 15 | 99 | 18.0 | 34.0 |
 | ||
| Scheme 2 Catalysts and ligands used for the synthesis of PNDIT2 in MeTHF. | ||
Having optimized the reaction conditions in MeTHF, we then investigated the optical and thermal properties of four samples made in MeTHF with varying MW and compared them to main chain defect-free PNDIT2 synthesized in toluene as a reference13 (Table 2). Samples made in MeTHF are referred to as PNDIT2-OH-1–4, while samples made in toluene are referred to as PNDIT2-tol-1–4.
| Sample name | M n,SEC/kDa | Đ | T m/ΔHm/°C/J g−1 | T c/ΔHm/°C/J g−1 |
|---|---|---|---|---|
| PNDIT2-OH-1 | 12.0 | 2.0 | 295.0/2.37 | 279.5/3.21 |
| PNDIT2-OH-2 | 14.8 | 1.7 | 294.5/3.13 | 281.0/3.37 |
| PNDIT2-OH-3 | 17.3 | 2.0 | 297.1/3.94 | 283.3/4.01 |
| PNDIT2-OH-4 | 19.9 | 2.3 | 301.0/5.04 | 287.7/5.43 |
| PNDIT2-tol-1 | 12.0 | 1.7 | 292.0/2.09 | 279.6/2.25 |
| PNDIT2-tol-2 | 14.5 | 1.7 | 300.8/3.28 | 285.0/3.64 |
| PNDIT2-tol-3 | 16.3 | 1.8 | 300.8/4.91 | 286.4/7.20 |
| PNDIT2-tol-4 | 19.3 | 2.1 | 303.1/5.17 | 289.1/6.31 |
UV-vis absorption spectroscopy of samples in the non-aggregating solvent 1-chloronaphthalene (CN) at room temperature revealed that an increase in MW led to gradual increase of the peak wavelength of the charge-transfer (CT) band (λmax) for both samples made in MeTHF and in toluene (Fig. S1, ESI†). This is expected, as the MWs are below the conjugation length in solution, which is around Mn,SEC = 20 kDa.13Fig. 1 shows the UV-vis spectra of four MeTHF samples with molecular weights between 12–20 kDa (PNDIT2-OH-1 (black solid line), PNDIT2-OH-2 (red solid line), PNDIT2-OH-3 (blue solid line) and PNDIT2-OH-4 (magenta solid line)), compared to PNDIT2 made in toluene (dash lines).
 | ||
| Fig. 1 UV-vis absorption spectra of PNDIT2 with increasing molecular weight in 1-chloronaphthalene (CN) and in toluene at room temperature. Solid lines: PNDIT2 made in MeTHF, dashed lines: reference polymers made in toluene. Black: PNDIT2-OH-1 or PNDIT2-tol-1, red: PNDIT2-OH-2 or PNDIT2-tol-2, blue: PNDIT2-OH-3 or PNDIT2-tol-3, and magenta: PNDIT2-OH-4 or PNDIT2-tol-4. The inset shows the CT band absorption intensity versus molecular weight. | ||
On comparing PNDIT2-OH and PNDIT2-tol samples having the same molecular weight, no difference with respect to λmax in CN was spotted, indicating that the two materials do not carry main chain defects. However, UV-vis absorption in toluene, in which PNDIT2 aggregates, is different. Here, most samples made in MeTHF showed a reduced amount of aggregates compared to samples made in tol with the same MW as seen by the reduced intensity at ∼710 nm (Fig. 1, see also inset). Nevertheless, all samples regardless of reaction solvent adopt a same overall shape in absorption spectra indicating they all share same packing mode. The thermal properties were investigated next and the results are collected in Fig. 2, Table 2 and Fig. S2, S3 (ESI†). The melting points Tm and crystallization temperatures Tc and their corresponding melting enthalpies ΔHm obtained from differential scanning calorimetry (DSC) increased as a function of MW for all PNDIT2 samples regardless of the reaction solvent. However, polymers synthesized in MeTHF exhibited slightly lower values of Tm and Tc than the polymers made in toluene with comparable molecular weight. A decrease of these values can be associated with either a reduced crystallisable segment length of the polymer chain arising from structural defects, or from altered end groups.
 | ||
| Fig. 2 Melting (Tm) and crystallization temperature (Tc), and melting enthalpy (ΔHm) of PNDIT2 samples made in MeTHF and reference polymers made in tol with increased MW. | ||
UV-vis absorption spectroscopy in CN already indicated that main chain defects are unlikely to be present, leaving end groups as a potential reason. To further confirm this assumption and additionally investigate the nature of polymer end groups, NMR spectroscopy was performed at high temperature. Based on previous studies,13,18 all minor signals were assigned and the results were compared to reference samples made in toluene. Fig. 3 shows the aromatic region (for full spectra see Fig. S4, ESI†) of PNDIT2-OH-4 in comparison to a sample made in toluene having the same molecular weight (PNDIT2-tol-4). Signals arising from unselective C–H activation leading to kinked structure are not observed for both samples.13 This is in line with UV-vis spectroscopy in CN, which showed no difference suggesting the same conjugation length in solution for the two polymers. It can be expected that any main chain defect structure would lower the conjugation length and hence cause a blue-shifted low energy band, which obviously is not the case. However, polymer end groups were different. Besides T2-H end groups which are characteristic for PNDIT2 synthesized by DAP,13,18 samples made in tol exhibited mainly tolyl-termination at the NDI side, while samples made in MeTHF were almost fully hydroxylated at the NDI side. While these end group patterns are exemplarily shown for PNDIT2-OH-4 and PNDIT2-tol-4 in Fig. 3, they were found to be typical for other samples as well. Thus, the difference between these two samples is the nature of the end groups, while other parameters such as main chain defects, dispersity and molecular weight are identical. Hence, one explanation for the reduced aggregation content in solution and the lower melting and crystallization temperatures and enthalpies of MeTHF samples is the presence of polar hydroxyl end groups at the NDI side of the chain. However, if polar solvents such as MeTHF or THF13 are chosen for synthesis, this type of end group is unavoidable (at least for the monomer couple investigated here). Interestingly, the intensities of T2-H end groups were always higher than NDI-tolyl/OH end group intensity, pointing to chains with symmetric T2-H termination. PNDIT2-OH-4 and PNDIT2-tol-4 exhibit OH/T2 and tol/T2 end group intensity ratios of 0.22 and 0.62, respectively. This behaviour is initially unexpected given precisely controlled stoichiometries, but it can be explained by a ring walking behaviour of the Pd catalyst on the NDI unit, which first leads to PNDIT2 prepolymers that are mainly terminated by T2-H at either side.32 Nevertheless, despite their partial occurrence, NDI-OH and NDI-tolyl chain termini for PNDIT2 made in MeTHF and tol, respectively, represent the only measurable difference in molecular structure.
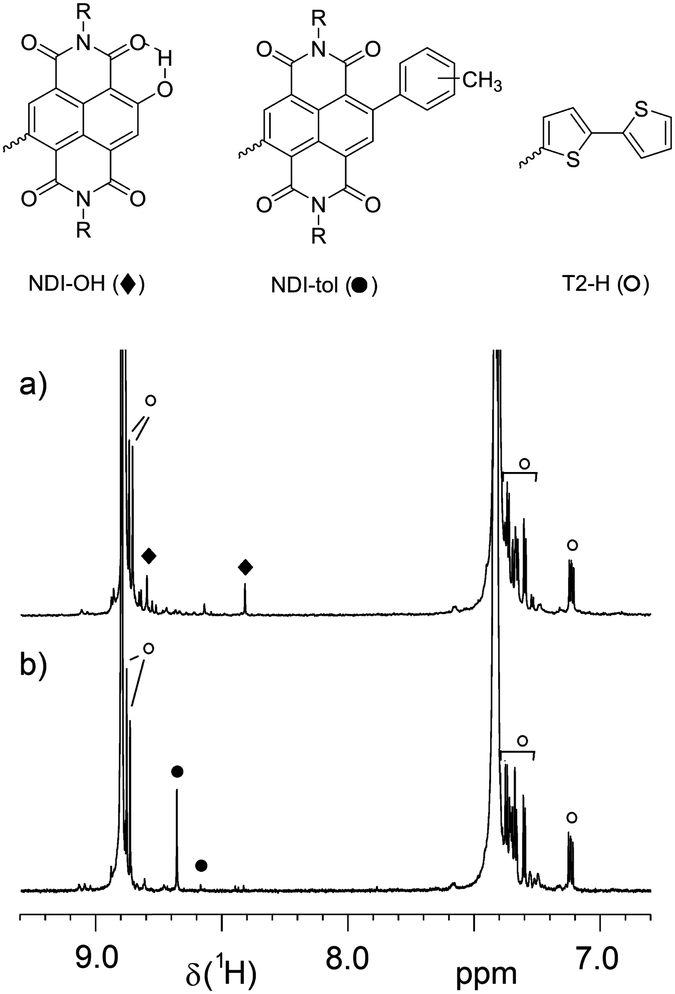 | ||
| Fig. 3 1H NMR spectra (region) of PNDIT2 (a) made in MeTHF (PNDIT2-OH-4) and (b) reference PNDIT2 made in tol (PNDIT2 tol-4) with focus on end group signals. Spectra were taken at 120 °C in C2D2Cl4. | ||
Fig. 4 shows atom force microscopy (AFM) images of PNDIT2-OH-4 and PNDIT2-tol-4. Films were prepared by spin coating from 1,2-dichlorobenzene (o-DCB), resulting in film thicknesses of 25–30 nm. The typical fibrillar microstructure is observed on top of both films, where domains, hundreds of nanometers wide, with coherent fribrillar orientation can be detected, as expected by films deposited from o-DCB solutions.33 These topographical features are strictly connected with an aggregation mechanism initiated by the pre-aggreagated species already present in the solution;33 it is worth mentioning that the dimensionality of such domains, in the case of PNDIT2 films, has been recently found to be critical in defining charge transport properties of devices with planar configuration, like field effect transistors.33–35 Slightly thinner fibrils are seen in films of PNDIT2-OH-4, consistently with the lower values for Tm and ΔHm (cf.Fig. 2 and Table 2) and a lower root mean square roughness (Rq) measured for PNDIT2-OH-4 (Rq ∼ 0.75 nm) films compared to PNDIT2-tol-4 (∼0.86 nm) ones. On the basis of recent reports evidencing the beneficial influence of microstructural macroscopic alignment on charge transport, achievable with a suitable ink formulation,13,36,37 we made films using a highly pre-aggregating solvent (toluene) to enhance the anisotropic interconnectivity of the film and an off-centre spin coating deposition method to control the alignment directionality of the polymer chains (Fig. 5d).13,36,37 A similar topography, with the same Rq ∼ 0.5 nm, consisting of fibril-like domains that are well aligned along the direction radial to the sample rotation, is observed with films from both PNDIT2-OH-4 and PNDIT2-tol-4 (AFM investigation is reported in Fig. S5, ESI†).
 | ||
| Fig. 4 AFM topography (a and c) and phase contrast (b and d) images of PNDIT2 films synthesized in toluene (a and b; PNDIT2 tol-4) and in MeTHF (c and d; PNDIT2-OH-4) spin-coated from o-DCB. | ||
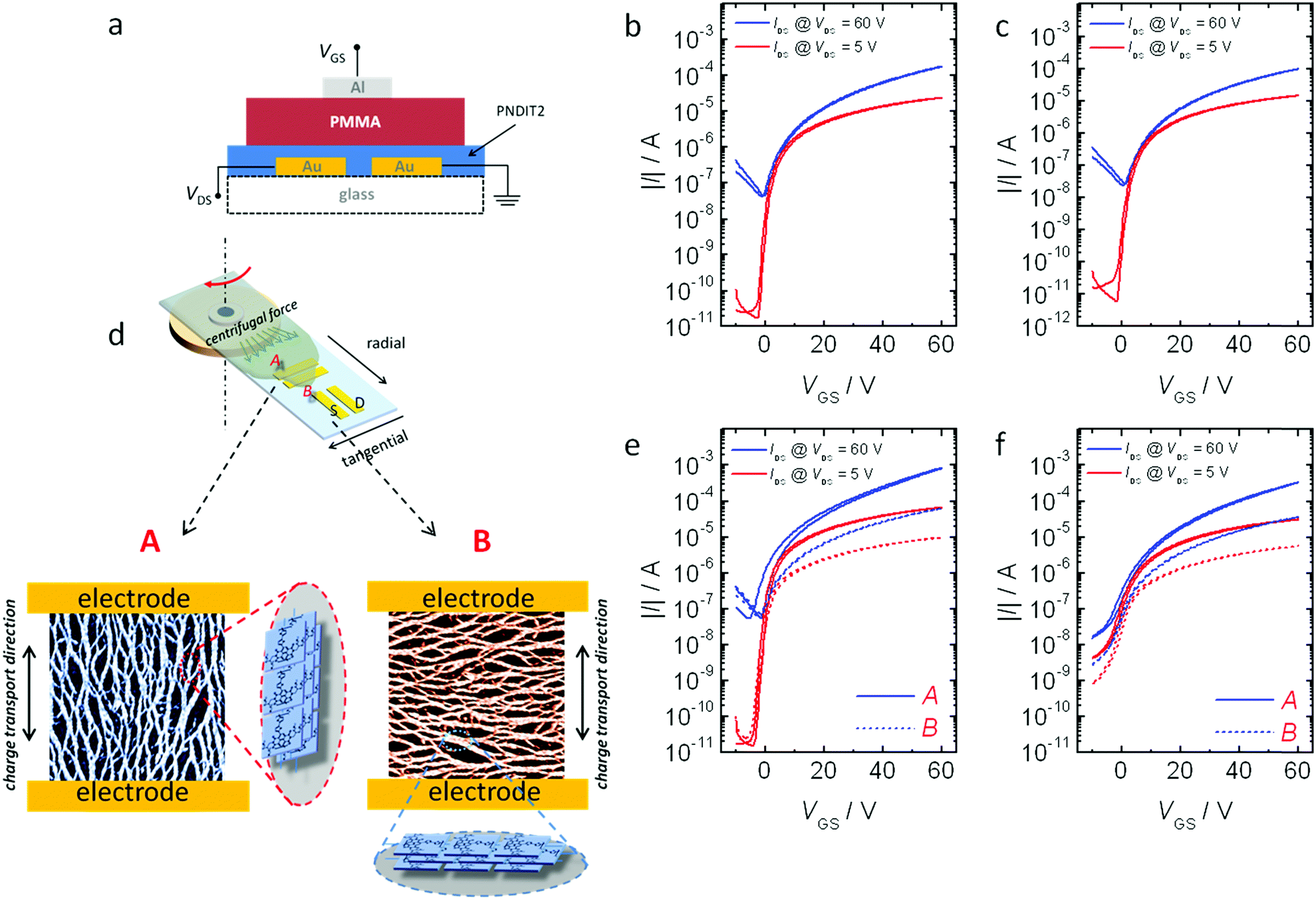 | ||
| Fig. 5 (a) Schematic of bottom contacts, top gate FETs. (b and c) Transfer characteristics of FETs deposited with standard spin coating from o-DCB solution of PNDIT2-tol-4 (b) and of PNDIT2-OH-4 (c); (d) sketch of off-centre spin coating deposition: A and B source and drain patterns are indicated in the sketch, allowing for the check of the transport properties parallel (A) and perpendicular (B) to the backbone/aggregate orientation (representatively indicated by the sketch) resulting from centrifugal flow; (e and f) transfer characteristics of FETs with PNDIT2-tol-4 (e) and PNDIT2-OH-4 (f), deposited using off-centre spin coating from pre-aggregating solvent (toluene). | ||
We investigated charge transport in the reported PNDIT2 thin films in top-gate, bottom-contact field effect transistors (FET) (Fig. 5a). Fig. 5b and c show FETs transfer characteristics of films deposited with standard spin coating from o-DCB. Both samples exhibited the typical unbalanced ambipolar behavior characterized by high electron and low hole mobilities. For the MeTHF sample, an average electron mobility in the saturation regime (μe,sat) of 0.18 cm2 V−1 s−1 was extracted (linear and saturation mobility values of all the FETs of this work are reported in Table 3); these values are in very good agreement with topographical features evidenced by AFM and with many reports on the FET performances of PNDIT2 processed from o-DCB,15,33 demonstrating that the good transport properties of PNDIT2 are generally not compromised by the presence of NDI-OH end groups. However, higher mobilities were extracted for PNDIT2 synthesised in toluene (μe,sat = 0.32 cm2 V−1 s−1), resulting in a reduction of ∼40% in mobility when comparing PNDIT2-tol-4 to PNDIT2-OH-4.
| Sample | Deposition method | μ lin @ VGS = 60 V cm2 V−1 s−1 | μ sat @ VGS = 60 V cm2 V−1 s−1 | SS (V dec−1) |
|---|---|---|---|---|
| PNDIT2-tol-4 | Centred | 0.20 ± 0.06 | 0.32 ± 0.09 | 1.06 ± 0.06 |
| Off-centre A | 0.47 ± 0.03 | 2.40 ± 0.30 | 1.86 ± 0.86 | |
| Off-centre B | 0.07 ± 0.01 | 0.07 ± 0.01 | ||
| PNDIT2-OH-4 | Centred | 0.13 ± 0.03 | 0.18 ± 0.05 | 1.36 ± 0.06 |
| Off-centre A | 0.28 ± 0.03 | 0.89 ± 0.07 | 2.52 ± 0.58 | |
| Off-centre B | 0.04 ± 0.01 | 0.04 ± 0.01 | ||
Fig. 5e and f show the transfer characteristics of FETs with unidirectionally aligned films, deposited with off-centre spin coating from toluene solutions, both parallel (electrodes configuration A in Fig. 5d) and perpendicular (electrodes configuration B in Fig. 5d) to the fibrillar alignment. Strong transport anisotropy is observed in films from both batches, ascribed to the unidirectional chain alignment along the radial direction, introducing a markedly superior interconnectivity of the film and requiring a less number of more energetically demanding inter-chain hopping events through the A channel configuration with respect to the B one.33,36,37 More specifically, at VGS = 60 V, an A/B saturation current ratio of 13 is observed for PNDIT2-tol-4, and a smaller current ratio of 9.3 for PNDIT2-OH-4. From channel configuration B, perpendicular to the fibrils direction, mobility values of 0.07 cm2 V−1 s−1 and 0.04 cm2 V−1 s−1 were extracted for PNDIT2-tol-4 and PNDIT2-OH-4 respectively, confirming the ∼40% drop of mobility already observed using standard spin coating deposition and o-DCB solutions. For channel configuration A, strong charge density dependent effective mobilities are extracted (Fig. S6, ESI†).33,36 Average μe,sat values of 2.4 cm2 V−1 s−1 and 0.89 cm2 V−1 s−1 were extracted from PNDIT2-tol-4 and PNDIT2-OH-4 at VGS = 60 V. Thus, we observe that, in the A channel configuration, where charge flows along the fibrils and main chain alignment direction, the reduction in mobility in PNDIT2-OH-4 with respect to PNDIT2-tol-4 is stronger, accounting to a 50% at low VGS and up to 60% at high VGS (Fig. S6, ESI†). Interestingly, upon equal processing, PNDIT2-OH-4 regularly displays slightly higher subthreshold slope (SS) with respect to PNDIT2-tol-4, which has been generally correlated to a higher density of bulk and superficial trap sites experienced by the charges during the channel accumulation process; however, a contribution from a difference in contact resistance cannot be excluded. To this end, electron trapping by OH end groups might pose a possible mechanism.38 Tolyl end capping has further been reported to be advantageous for crystallinity and transport properties.39
To further explain the FET data, grazing-incidence wide-angle X-ray scattering (GIWAXS) measurements were performed on thin films spin-coated from o-DCB. Fig. 6a and b present 1-dimensional profiles along the in-plane (IP) and out-of-plane (OOP) scattering directions (for two-dimensional images see Fig. S7, ESI†). Both samples show a predominant bulk face-on orientation of crystallites with prominent alkyl stacking (h00) peaks found in-plane and prominent π–π stacking (020) peaks found out-of-plane. Table 4 summarises key crytallographic parameters obtained by peak fitting of the 1-dimensional line profiles. The d-spacing of alkyl, backbone and π–π stacking peaks are the same within experimental uncertainty, indicating that polymer end groups do not significantly affect the crystal packing of PNDIT2 chains. Interestingly the toluene sample has slightly larger coherence lengths reflecting larger crystallites, which is consistent with DSC data on bulk crystallinity and aggregation behavior in solution.
 | ||
| Fig. 6 (a and b) GIWAXS analysis of films prepared via centred spin-coating. (c and d) NEXAFS analysis of the degree of backbone alignment in films prepared via off-centre spin-coating. (a and c) Show data for PNDIT2-tol-4 while (b and d) show data for PNDIT2-OH-4. The insets to (c and d) plot the carbon 1s to Π* resonance intensity as a function of azimuthal angle, φ, with the solid line representing a fit to a cos2φ dependence. | ||
| Reaction solvent | Peak | d-Spacing (Å) | Coherence length (Å) | Area (a.u.) |
|---|---|---|---|---|
| Toluene | IP (100) | 25.0 ± 0.1 | 230 ± 2 | 83 ± 1 |
| IP (001) | 13.9 ± 0.1 | 175 ± 4 | 9.7 ± 0.5 | |
| OOP (020) | 4.0 ± 0.1 | 24 ± 1 | 54 ± 1 | |
| MeTHF | IP (100) | 24.9 ± 0.1 | 210 ± 3 | 131 ± 2 |
| IP (001) | 13.8 ± 0.1 | 168 ± 6 | 5.7 ± 0.5 | |
| OOP (020) | 3.9 ± 0.1 | 21 ± 1 | 55 ± 1 | |
DSC data on bulk crystallinity and aggregation behavior in solution. A significant difference between the scattering profiles of the two samples is the relative prominence of the (001) backbone stacking peak. For PNDIT2-tol-4 this peak is much more prominent relative to the neighboring (200) peak indicating an overall higher degree of backbone ordering, which is more important for charge transport than ordering along the alkyl stacking direction. Note that while the absolute peak area for the IP (100) peak in PNDIT2-OH-4 is larger than that of PNDIT2-tol-4, the absolute scattering intensities in GIWAXS depend upon film thickness, beam footprint and sample alignment with respect to the beam. Thus this should not be taken as evidence of a higher degree of crystallinity for the MeTHF sample, with the DSC data being more reliable in evidencing a higher degree of crystallinity for the toluene-made sample.
To investigate the degree of polymer backbone alignment in samples prepared via off-centre spin-coating, near-edge X-ray absorption fine-structure (NEXAFS) spectroscopy was performed. NEXAFS spectra were acquired at different azimuthal rotations of each sample with respect to a normally incidence polarised X-ray beam, Fig. 6c and d. For aligned samples, variation in the strength of the carbon 1s to Π* resonance intensity (peaks located at ∼285 eV) provides information about the degree of backbone alignment.40 The insets to Fig. 6c and d plot the carbon 1s to Π* resonance intensity as a function of azimuthal angle, φ, with the intensity varying as cos2φ as expected for uniaxially aligned samples. Computing the degree of anisotropy as R = Imax/Imin yields a value of R = 6.6 for the toluene sample and R = 5.3 for the MeTHF sample, with the toluene sample having a higher proportion of chains aligned in the coating direction.
GIWAXS and NEXAFS investigations reveal an inferior backbone alignment in PNDIT2-OH thin films as a result of the –OH end group. This evidence is consistent with solution UV-vis spectroscopy and DSC data that find reduced aggregation content in solution and reduced bulk crystallinity. The reduced degree of structural anisotropy measured by NEXAFS spectroscopy for PNDIT2-OH-4 rationalizes the lower mobility anisotropy of aligned MeTHF films. While we cannot instead be fully conclusive regarding the general reduction in mobility observed in PNDIT2-OH samples with respect to PNDIT2-tol ones, the overall reduced structural order in films made from PNDIT2-OH, particularly along the polymer backbone direction, may provide a partial explanation.
3. Conclusions
In conclusion, we have shown that high electron mobility conjugated copolymers such as PNDIT2 can be made by direct arylation polycondensation in green solvents such as MeTHF for the first time. Extensive optimization of the reaction conditions was successfully performed, thereby largely suppressing nucleophilic substitution of NDIBr chain ends and finally enabling quantitative polymer yield and satisfyingly high molecular weights up to 20 KDa. However, PNDIT2 made in MeTHF was found to exhibit hydroxylated NDI chain termini. Further detailed characterizations showed similar properties of hydroxylated PNDIT2 made in MeTHF compared to tolyl-terminated PNDIT2 controls, demonstrating that the green solvent MeTHF is a suitable candidate to produce conjugated polymers with high performance using atom-economic direct arylation polycondensation. However, OH-termination of PNDIT2 made in MeTHF apparently led to slightly lower aggregation in solution, bulk crystallinity, chain alignment and finally slightly reduced electron mobilities in n-type field effect transistor devices. This behaviour is ascribed to the influence of the polar OH end group, which may partially hinder aggregation and ordering of chains, yet not compromising charge transport and allowing a good n-type field-effect mobility of ∼0.9 cm2 V−1 s−1 in uniaxially aligned films.4. Experimental
General remarks
All the chemicals were obtained from Sigma Aldrich and used without further treatment unless otherwise stated. Anhydrous 2-methyltetrahydrofuran was purchased by Sigma Aldrich and used as received. Monomer NDIBr2,13 Pd2(dba-R)3 complexes30 and DAB ligand41 were synthesized according to previously reported methods. 2,2′-Bithiophene (T2) was purchased from Alfa Aesar (98%) and further purified by filtering through a silica gel plug with iso-hexanes.1H (500.13 MHz) NMR spectra were recorded on a Bruker Avance III spectrometer using a 5 mm gradient probe. 1H NMR spectra of polymers were obtained from C2D2Cl4 solutions at 120 °C. GPC measurements were carried out on four SDV gel 5 μm columns, with pore sizes ranging from 103 to 106 Å (PSS), connected in series with a Knauer K-2301 RI detector, and calibrated with polystyrene standards. CHCl3 was used as eluent at room temperature at a flow rate of 1.0 mL min−1 with sample concentrations of 0.03 mg mL−1. UV-vis measurements were carried out on a Shimadzu UV-1800 spectrophotometer. DSC measurements were acquired on a NETZSCH DSC 204 F1 Phoenix under a nitrogen atmosphere at a heating and cooling rate of 10 °C min−1.
FET fabrication and characterization
FETs with channel width of 2 mm and channel length of 20 μm were fabricated according in top-gate, bottom-contact geometry. Bottom Au contacts were defined by a lift off photolithographic process on glass (Corning Glass 1737F purchased from Apex Optical Services) with a 0.7 nm thick Cr adhesion layer. The thickness of the Au contacts was 15 nm. Solutions of PNDIT2 in 1,2-dichlorobenzene (9 g L−1) were deposited by regular spin-coating at 1000 rpm for 60 s in air. The semiconductor was then annealed for 30 min at 150 °C on a hot plate in a nitrogen atmosphere. PMMA (Sigma-Aldrich) with Mw = 120 kDa was spun from n-butyl acetate (with a concentration of 80 g L−1), resulting in 550 nm thick dielectric layer (ε = 3.6). Al electrodes were thermally evaporated as gate contacts. The electrical characteristics of transistors were measured in a nitrogen glove box on a Wentworth Laboratories probe station with an Agilent B1500A semiconductor device analyzer. Linear and saturation charge carrier mobility values were extracted according to the gradual channel approximation.42GIWAXS and NEXAFS measurements
GIWAXS measurements were performed at the SAXS/WAXS beamline at the Australian Synchrotron.43 11 keV electrons were used with scattering patterns recorded on a Dectris Pilatus 1M detector. The incident angle of the X-ray beam was close to the critical angle of the polymer film but below the critical angle of the substrate, minimizing the background signal from substrate scatter. The X-ray exposure time was 3 s such that no film damage was identified. The sample-to-detector distance was calibrated using a silver behenate sample. The results were analyzed by an altered version of the NIKA 2D44 based in IgorPro.NEXAFS spectroscopy was performed at the Soft X-ray beamline at the Australian Synchrotron.45 Samples were prepared on bare, highly-doped silicon substrates with X-ray absorption measured by measuring the drain current flowing to the sample (total electron yield mode). Spectra were normalised to the incident X-ray flux via the stable monitor method, with further details found elsewhere.46 Data were analysed using QANT.47
General procedure for the synthesis of PNDIT2 in MeTHF
(Entry 25) NDIBr2 (98.51 mg, 0.10 mmol), T2 (16.63 mg, 0.10 mmol), Na2CO3 (31.80 mg, 0.30 mmol), and pivalic acid (10.21 mg, 0.10 mmol) were carefully weighed into a dry vial containing a stir bar. Then, 0.3 mL degassed MeTHF was added under N2 atmosphere and the whole was stirred for 1 min at RT. Then Pd2(dba-OMe)3 (2.19 mg, 2 × 10−3 mmol) was carefully added under nitrogen. The vial was sealed and placed into a preheated oil bath and stirred for 20 h at 90 °C. After cooling to RT, the material was dissolved and precipitated into 200 mL methanol. Then it was filtered and purified via Soxhlet extraction with acetone, ethyl acetate, and iso-hexanes. Finally the material was collected with CHCl3 and filtered to give 98 mg PNDIT2 in 98% yield. Mn,SEC = 20 kDa.Acknowledgements
The DFG (IRTG Soft Matter Science 1645, Project SO-1213/8-1) and the Innovationsfond Forschung of the Universität Freiburg are gratefully acknowledged for funding. We thank M. Hagios for SEC measurements and A. Warmbold for DSC measurements. M. C. and A. L. acknowledge support by the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program ‘HEROIC’, grant agreement 638059. C. R. M. also acknowledges support from the Australian Research Council (DP130102616). This research was undertaken in part on the SAXS/WAXS and Soft X-ray beamlines at the Australian Synchrotron, Victoria, Australia. Dr Eliot Gann is thanked for assistance with GIWAXS measurements.Notes and references
- L. G. Mercier and M. Leclerc, Acc. Chem. Res., 2013, 46, 1597–1605 CrossRef CAS PubMed.
- K. Okamoto, J. Zhang, J. B. Housekeeper, S. R. Marder and C. K. Luscombe, Macromolecules, 2013, 46, 8059–8078 CrossRef CAS.
- S. Kowalski, S. Allard, K. Zilberberg, T. Riedl and U. Scherf, Prog. Polym. Sci., 2013, 38, 1805–1814 CrossRef CAS.
- A. Marrocchi, A. Facchetti, D. Lanari, C. Petrucci and L. Vaccaro, Energy Environ. Sci., 2016, 9, 763–786 CAS.
- J. Zhang, W. Chen, A. J. Rojas, E. V. Jucov, T. V. Timofeeva, T. C. Parker, S. Barlow and S. R. Marder, J. Am. Chem. Soc., 2013, 135, 16376–16379 CrossRef CAS PubMed.
- J. Zhang, T. C. Parker, W. Chen, L. Williams, V. N. Khrustalev, E. V. Jucov, S. Barlow, T. V. Timofeeva and S. R. Marder, J. Org. Chem., 2016, 81, 360–370 CrossRef CAS PubMed.
- N. J. Snoeij, A. A. van Iersel, A. H. Penninks and W. Seinen, Toxicol. Appl. Pharmacol., 1985, 81, 274–286 CrossRef CAS PubMed.
- Q. Wang, R. Takita, Y. Kikuzaki and F. Ozawa, J. Am. Chem. Soc., 2010, 132, 11420–11421 CrossRef CAS PubMed.
- P. Berrouard, A. Najari, A. Pron, D. Gendron, P.-O. Morin, J.-R. Pouliot, J. Veilleux and M. Leclerc, Angew. Chem., Int. Ed., 2012, 51, 2068–2071 CrossRef CAS PubMed.
- C. J. Kudla, D. Dolfen, K. J. Schottler, J.-M. Koenen, D. Breusov, S. Allard and U. Scherf, Macromolecules, 2010, 43, 7864–7867 CrossRef CAS.
- W. Lu, J. Kuwabara, T. Iijima, H. Higashimura, H. Hayashi and T. Kanbara, Macromolecules, 2012, 45, 4128–4133 CrossRef CAS.
- Y. Gao, X. Zhang, H. Tian, J. Zhang, D. Yan, Y. Geng and F. Wang, Adv. Mater., 2015, 27, 6753–6759 CrossRef CAS PubMed.
- R. Matsidik, H. Komber, A. Luzio, M. Caironi and M. Sommer, J. Am. Chem. Soc., 2015, 137, 6705–6711 CrossRef CAS PubMed.
- S. Broll, F. Nübling, A. Luzio, D. Lentzas, H. Komber, M. Caironi and M. Sommer, Macromolecules, 2015, 48, 7481–7488 CrossRef CAS.
- H. Yan, Z. Chen, Y. Zheng, C. Newman, J. R. Quinn, F. Dötz, M. Kastler and A. Facchetti, Nature, 2009, 679–686 CrossRef CAS PubMed.
- M. Sommer, J. Mater. Chem. C, 2014, 2, 3088–3098 RSC.
- J. H. Park, E. H. Jung, J. W. Jung and W. H. Jo, Adv. Mater., 2013, 25, 2583–2588 CrossRef CAS PubMed.
- R. Matsidik, H. Komber and M. Sommer, ACS Macro Lett., 2015, 4, 1346–1350 CrossRef CAS.
- Y. Nohara, J. Kuwabara, T. Yasuda, L. Han and T. Kanbara, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 1401–1407 CrossRef CAS.
- S.-W. Chang, H. Waters, J. Kettle, Z.-R. Kuo, C.-H. Li, C.-Y. Yu and M. Horie, Macromol. Rapid Commun., 2012, 33, 1927–1932 CrossRef CAS PubMed.
- K. Nakabayashi and H. Mori, Chem. Lett., 2013, 42, 717–718 CrossRef CAS.
- P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, New York, 1998 Search PubMed.
- A. Facchetti, L. Vaccaro and A. Marrocchi, Angew. Chem., Int. Ed., 2012, 51, 3520–3523 CrossRef CAS PubMed.
- R. Matsidik, J. Martin, S. Schmidt, J. Obermayer, F. Lombeck, F. Nübling, H. Komber, D. Fazzi and M. Sommer, J. Org. Chem., 2015, 80, 980–987 CrossRef CAS PubMed.
- V. Pace, P. Hoyos, L. Castoldi, P. Domínguez de María and A. R. Alcántara, ChemSusChem, 2012, 5, 1369–1379 CrossRef CAS PubMed.
- D. Prat, A. Wells, J. Hayler, H. Sneddon, C. R. McElroy, S. Abou-Shehada and P. J. Dunn, Green Chem., 2015, 18, 288–296 RSC.
- S. J. Choi, J. Kuwabara and T. Kanbara, ACS Sustainable Chem. Eng., 2013, 1, 878–882 CrossRef CAS.
- M. Wakioka, Y. Kitano and F. Ozawa, Macromolecules, 2013, 46, 370–374 CrossRef CAS.
- A. E. Rudenko and B. C. Thompson, Macromolecules, 2015, 48, 569–575 CrossRef CAS.
- A. R. Kapdi, A. C. Whitwood, D. C. Williamson, J. M. Lynam, M. J. Burns, T. J. Williams, A. J. Reay, J. Holmes and I. J. S. Fairlamb, J. Am. Chem. Soc., 2013, 135, 8388–8399 CrossRef CAS PubMed.
- S. S. Zalesskiy and V. P. Ananikov, Organometallics, 2012, 31, 2302–2309 CrossRef CAS.
- E. Goto, S. Ando, M. Ueda and T. Higashihara, ACS Macro Lett., 2015, 4, 1004–1007 CrossRef CAS.
- A. Luzio, L. Criante, V. D’Innocenzo and M. Caironi, Sci. Rep., 2013, 3, 3425 Search PubMed.
- N. Martino, D. Fazzi, C. Sciascia, A. Luzio, M. R. Antognazza and M. Caironi, ACS Nano, 2014, 8, 5968–5978 CrossRef CAS PubMed.
- D. Fazzi and M. Caironi, Phys. Chem. Chem. Phys., 2015, 17, 8573–8590 RSC.
- S. G. Bucella, A. Luzio, E. Gann, L. Thomsen, C. R. McNeill, G. Pace, A. Perinot, Z. Chen, A. Facchetti and M. Caironi, Nat. Commun., 2015, 6, 8394 CrossRef CAS PubMed.
- N.-K. Kim, S.-Y. Jang, G. Pace, M. Caironi, W.-T. Park, D. Khim, J. Kim, D.-Y. Kim and Y.-Y. Noh, Chem. Mater., 2015, 27(24), 8345–8353 CrossRef CAS.
- L.-L. Chua, J. Zaumseil, J.-F. Chang, E. C.-W. Ou, P. K.-H. Ho, H. Sirringhaus and R. H. Friend, Nature, 2005, 434, 194–199 CrossRef CAS PubMed.
- U. Koldemir, S. R. Puniredd, M. Wagner, S. Tongay, T. D. McCarley, G. D. Kamenov, K. Müllen, W. Pisula and J. R. Reynolds, Macromolecules, 2015, 48, 6369–6377 CrossRef CAS.
- M. M. Nahid, E. Gann, L. Thomsen and C. R. McNeill, Eur. Polym. J., 2016, 81, 532–554 CrossRef CAS.
- A. J. Hickman and M. S. Sanford, ACS Catal., 2011, 1, 170–174 CrossRef CAS.
- S. M. Sze and K. K. Ng, Physics of Semiconductor Devices, John Wiley & Sons, 3rd edn, 2006 Search PubMed.
- N. M. Kirby, S. T. Mudie, A. M. Hawley, D. J. Cookson, H. D. T. Mertens, N. Cowieson and V. Samardzic-Boban, J. Appl. Crystallogr., 2013, 46, 1670–1680 CrossRef CAS.
- J. Ilavsky, J. Appl. Crystallogr., 2012, 45, 324–328 CrossRef CAS.
- B. C. C. Cowie, A. Tadich and L. Thomsen, in AIP Conference Proceedings, Australia, 2010, vol. 1234, pp. 307–310.
- M. M. Nahid, E. Gann, L. Thomsen and C. R. McNeill, Eur. Polym. J., 2016, 81, 532–554 CrossRef CAS.
- E. Gann, C. R. McNeill, A. Tadich, B. C. C. Cowie and L. Thomsen, J. Synchrotron Radiat., 2016, 23, 374–380 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6tc03804b |
| This journal is © The Royal Society of Chemistry 2016 |