
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
[{VOCl2(CH2(COOEt)2)}4] as a molecular precursor for thermochromic monoclinic VO2 thin films and nanoparticles†
Ben
Blackburn
,
Michael J.
Powell
,
Caroline E.
Knapp
,
Joseph C.
Bear
*,
Claire J.
Carmalt
and
Ivan P.
Parkin
Department of Chemistry, University College London, 20 Gordon Street, London, WC1H 0AJ, UK. E-mail: joseph.bear.11@ucl.ac.uk
First published on 20th October 2016
Abstract
The synthesis of thermochromic monoclinic vanadium(IV) oxide (VO2 (M)) thin films and vanadium oxide nanocrystals from a molecular precursor, [{VOCl2(CH2(COOEt)2)}4] is described. Thin films were synthesised using aerosol assisted chemical vapour deposition (AACVD) onto glass substrates at high temperatures and were subsequently characterised and tested for thermochromic efficiency. The film's suitability as smart, energy efficient window coatings was investigated by calculating their solar modulation potential. Thin films were also doped with tungsten to lower their metal to semiconductor transition temperature (MST) through the addition of tungsten(VI) phenoxide during the AACVD process, lowering the MST from a typical ∼70 °C for an undoped VO2 (M) thin film to ∼50 °C for a typical W-doped example. The optimum deposition temperature of 550 °C produced films with thermochromic properties (solar modulation of 15.9%) comparable to the highest values reported. In addition, vanadium oxide nanostructures were synthesised using the thermal decomposition of [{VOCl2(CH2(COOEt)2)}4] and their shape controllably tailored by varying surfactant concentration and type. Thin films and nanostructures were characterised using X-ray diffraction (XRD), scanning electron microscopy (SEM), transmission electron microscopy (TEM), X-ray photoelectron spectroscopy (XPS) and energy-dispersive X-ray spectroscopy (EDS). Thermochromic measurements were measured using UV-Vis spectroscopy with a variable temperature stage.
Introduction
The synthesis of monoclinic vanadium dioxide thin films and nanocrystals for energy saving, infrared radiation reflecting windows is currently an area of intense research due in no small part to the ever-increasing pressure placed on world energy feedstocks.1–4 Therefore the need to reduce energy consumption or switch to sustainable energy sources have never been greater. Buildings contribute approximately 1/3 of greenhouse gas emissions, with the majority of these being due to heating and cooling loads to maintain a narrow range of temperatures.5 The employment of smart, energy saving materials such as thermochromic VO2 window coatings is one such initiative to moderate this vast energy consumption.The monoclinic phase vanadium(IV) oxide (VO2 (M)) undergoes a structural transition to the rutile phase (VO2 (R)) at a metal to semiconductor transition temperature (MST) of ca. 68 °C, a phenomenon known as thermochromism.6,7 VO2 (M) is of particular interest as its MST is relatively close to that of room temperature, unlike other thermochromic VxOy species, such as V2O3.8 Below the MST, VO2 (M) is a semiconductor that transmits infrared and ultraviolet radiation consistently, whereas above the MST, VO2 (R) exhibits metallic conduction, reflecting in the same region.1,9 This property is crucial in the design of smart window coatings, that are transmissive of near IR wavelengths at low temperatures, but reflective at high temperatures, thus alleviating the use of expensive and energy-intensive air conditioning units in hot countries.10 In order to decrease the MST, it is common to incorporate dopant atoms into the VO2 matrix. Tungsten is by far the most common and most effective dopant in lowering the MST, and has the added advantage of giving the VO2 a more aesthetically acceptable blue tint rather than the dirty yellow of VO2 (M).11 Other dopants such as: niobium,12 fluorine,13 magnesium14 and molybdenum15 have also been shown to reduce the MST.
The tungsten dopant in W–VO2 has tetragonal symmetry, which distorts the monoclinic VO2 towards the tetragonal form. Atoms with large atomic radii such as tungsten introduces distortions into the VO2 lattice around the W centre,16 as well as introducing areas of greater electron density into the antibonding orbitals of the V–V bond, and so destabilising the monoclinic phase.17 These factors are thought to lower the energy barrier to the MST.
The synthesis of VO2 (M) is exacting due to the large number of stable oxide polymorphs of vanadium.8,18 Vanadium oxides will easily form as both phase pure oxides (V2O3, VO2 and V2O5) and mixed oxidation states (V2O7, V3O7etc.) and thus isolating phase pure VO2 (M) requires precise control of the partial pressure of oxygen in the process.8 Thin films of VO2 have been very well studied, with highly effective thermochromic VO2 (M) and doped VO2 (M) films deposited on a variety of substrates.19–21 The paucity of low molecular weight, volatile precursors containing vanadium(IV) has led to the use of air-sensitive VCl422,23 and stable VO(acac)224 precursors. It is also noteworthy that VO2 (M) thin films have been deposited using VOCl3, a vanadium(V) source.23,25,26
Thin films of VO2 (M) have been deposited using both atmospheric pressure chemical vapour deposition (APCVD)27–30 and aerosol assisted chemical vapour deposition (AACVD) methods,11,31,32 both of which are highly effective methods for coating large substrates with thin films of VO2. The ability to tailor the vanadium precursor could lead to improved properties in the deposited films. This could be achieved by the synthesis of a single molecular precursor, which would have benefits such as the ability to precisely control dopant levels to achieve a given reduction in the phase transition temperature. To this end, AACVD is the preferred method for the CVD of single molecular precursors, due to the typical low volatility of such species.33–35
The majority of synthetic methods for the synthesis of nanoparticles and nanostructures have focussed on high temperature methods.36–38 Vanadium oxide nanoparticles have been synthesised using a variety of methods, including: pyrolysis,39 sol–gel,40 hydrothermal methods41 and ball milling42 for use in coatings. Hydrothermal (autoclaving) and sol–gel methods tend to produce large, highly crystalline VO2 (M) nanostructures, from vanadium precursors in the presence of reducing agents such as acids to prevent over-oxidation to V2O5.37,43,44 Autoclave methods have also shown to be effective in the growth of large nanowires, and remain the most widely used bottom-up method for the synthesis of VO2 (M) nanocrystals. It is noteworthy that the manipulation of the gaseous environment to contain partial amounts of oxygen in order to prevent the reduction/oxidation of V(IV) species has met with considerable success in aforementioned high temperature syntheses.45–47
However, to date, the use of single molecular precursors for vanadium oxide nanocrystals and thin films has been under-explored. Our work describes the synthesis of a single molecular precursor; dichloro(oxo) vanadium(IV) diethyl malonate [{VOCl2(CH2(COOEt)2)}4] [1] for VO2 (Section 1), fabrication of doped and undoped VO2 (M) thin films using AACVD (Section 2), and anisotropic vanadium oxide nanomaterials whose growth is controlled by surfactants (Section 3). Compound [1] has two key advantages over typical vanadium compounds used for the formation of VO2 (M) thin films; superior solubility in preferred solvents for AACVD such as hexane, dichloromethane, chloroform, toluene etc. whereas compounds such as vanadyl acetylacetonate are sparingly soluble, and higher reactivity with oxygen due to the presence of volatile halide ligands. The films were doped with tungsten by adding small amounts of tungsten(VI) hexaphenoxide [W(OPh)6] to the precursor solution, and a lowering of the MST observed. Vanadium oxide nanomaterials were synthesised by the thermal decomposition of [1] in the presence of a high boiling point solvent (1-octadecene) and structure directing surfactants (oleic acid and oleylamine). The structure of dichloro(oxo) vanadium(IV) diethyl malonate [{VOCl2(CH2(COOEt)2)}4] [1] was determined by X-ray crystallography, elemental analysis, 1H and 13C {1H} NMR spectroscopy. Thin films and nanostructures were characterised using X-ray diffraction (XRD), scanning electron microscopy (SEM), transmission electron microscopy (TEM), X-ray photoelectron spectroscopy (XPS) and energy-dispersive X-ray spectroscopy (EDS). Thermochromic measurements were measured using UV-Vis spectroscopy with a variable temperature stage.
Experimental
Materials
Vanadium(V) oxychloride (99%), diethyl malonate (ReagentPlus®, 99%), oleic acid (techn. grade, 90%), oleylamine (techn. grade, 70%), 1-octadecene (techn. grade, 90%) (dried over sodium) and 1,2-tetradecanediol (techn. grade, 90%) were purchased from Sigma Aldrich Ltd and used as received. Laboratory solvents were purchased from Sigma Aldrich Ltd and of analytical grade. Solvents were dried over activated alumina via the Grubbs method using anhydrous engineering equipment, ensuring that the concentration of water in the solvents was below 5–10 ppm.48 Deuterated chloroform (CDCl3) was obtained from GOSS Scientific and was degassed and dried over 3 Å molecular sieves.Instrumentation
1H and 13C{1H} NMR spectroscopy was carried out on a Bruker A-600 MHz spectrometer, operating at 295 K and 600.13 MHz (1H). Signals are reported relative to SiMe4 (δ = 0.00 ppm) and the following abbreviations are used s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet), b (broad). Scanning electron microscope images were recorded on a Jeol JSM-6301F SEM at an acceleration voltage of 15 kV. Transmission electron microscopy (TEM) images and were obtained using a high resolution TEM Jeol 2100 with a LaB6 source operating at an acceleration voltage of 200 kV. Images were recorded on a Gatan Orius Charge-coupled device (CCD). Samples were prepared by drop-casting a nanoparticle dispersion in n-hexane onto a 400 mesh gold grid with a thin holey carbon film (Agar Scientific). Energy dispersive X-ray spectra (EDS) were recorded on an Oxford Instruments XMax EDS detector running AZTEC software. X-Ray diffraction (XRD) patterns were recorded using a Bruker D8 Discover X-ray diffractometer using monochromatic Cu Kα1 and Cu Kα2 radiation of wavelengths 1.54056 and 1.54439 Å respectively, emitted in an intensity ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 with a voltage of 40 kV and a current of 40 mA. The incident beam angle was 1° and data was collected between 10° and 66° 2θ with a step size of 0.05° at 2 s per step. XRD studies on the nanostructures were carried out using a Stoe (Mo) StadiP diffractometer with a Mo X-ray source (Mo tube 50 kV 30 mA), monochromated (Pre-sample Ge (111) monochromator selects Kα1 only) and a Dectris Mython 1K silicon strip detector covering 18° 2θ. Samples were run in transmission mode, with the sample under rotation in the X-ray beam. All diffraction patterns obtained were compared with database (ICSD) standards. X-ray photoelectron spectroscopy was conducted on a Thermo Scientific K-alpha spectrometer with monochromated Al Kα radiation, a dual beam charge compensation system and constant pass energy of 50 eV (spot size 400 μm). Survey scans were collected in the binding energy range 0–1200 eV. High-resolution peaks were used for the principal peaks of V (2p), O (1s), and C (1s). Data was fitted using CASA XPS software, with doublet separations of 7.5 eV for V (2p) and 2.1 eV for W (4f).49 Thermochromic measurements were recorded using UV/Vis spectroscopy with a variable temperature stage on a Perkin Elmer Lambda 950 UV/Vis/NIR Spectrophotometer in diffuse reflectance mode. Heating of samples in the UV/Vis spectrometer was achieved by an aluminium temperature cell controlled by RS cartridge heaters, Eurotherm temperature controllers and k-type thermocouples. A Labsphere reflectance standard was used as a reference for the UV/Vis measurements.
:1 with a voltage of 40 kV and a current of 40 mA. The incident beam angle was 1° and data was collected between 10° and 66° 2θ with a step size of 0.05° at 2 s per step. XRD studies on the nanostructures were carried out using a Stoe (Mo) StadiP diffractometer with a Mo X-ray source (Mo tube 50 kV 30 mA), monochromated (Pre-sample Ge (111) monochromator selects Kα1 only) and a Dectris Mython 1K silicon strip detector covering 18° 2θ. Samples were run in transmission mode, with the sample under rotation in the X-ray beam. All diffraction patterns obtained were compared with database (ICSD) standards. X-ray photoelectron spectroscopy was conducted on a Thermo Scientific K-alpha spectrometer with monochromated Al Kα radiation, a dual beam charge compensation system and constant pass energy of 50 eV (spot size 400 μm). Survey scans were collected in the binding energy range 0–1200 eV. High-resolution peaks were used for the principal peaks of V (2p), O (1s), and C (1s). Data was fitted using CASA XPS software, with doublet separations of 7.5 eV for V (2p) and 2.1 eV for W (4f).49 Thermochromic measurements were recorded using UV/Vis spectroscopy with a variable temperature stage on a Perkin Elmer Lambda 950 UV/Vis/NIR Spectrophotometer in diffuse reflectance mode. Heating of samples in the UV/Vis spectrometer was achieved by an aluminium temperature cell controlled by RS cartridge heaters, Eurotherm temperature controllers and k-type thermocouples. A Labsphere reflectance standard was used as a reference for the UV/Vis measurements.
Synthesis of [{VOCl2(CH2(COOEt)2)}4][1]
Synthesis of [{VOCl2(CH2(COOEt)2)}4] was carried out under nitrogen obtained from BOC Ltd. in anhydrous solvents using standard Schlenk techniques. VOCl3 (2 ml, 21.1 mmol) was added to 30 ml of anhydrous n-hexane under an atmosphere of nitrogen using a cannula. Diethyl malonate (0.5 ml, 3.3 mmol) was added dropwise and the mixture stirred under nitrogen for 2 h. An excess of VOCl3 was used to ensure complete malonate complexation. Unreacted VOCl3 was removed along with the n-hexane via filtration, leaving behind a dark red/black solid precipitate. The precipitate was filtered and washed 3 times with ca. 20 ml hexane and dried in vacuo to give the target compound 1, in good yield (0.96 g, 83%). 1H and 13C NMR were carried out on this product under nitrogen. A 5 cm3 concentrated solution of [1] in dichloromethane was made up and layered with 15 ml of n-hexane. Small crystals formed over approximately a week. 1H NMR (CDCl3): δ 4.18 (q, 4H, –CH2), δ 3.35 (s, 2H, –CH2), δ 1.34 (t, 6H, –CH3). 13C {1H} NMR (CDCl3): δ 14.2 (CH3), 55.9 (CH2), 63.6 (CH2CH3), 163 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O). All peaks were highly broadened due to the paramagnetic nature of the vanadium species. Elemental analysis calculated for V4Cl8C28O20H48: C, 28.21; H, 4.06. Found: C, 27.33; H, 3.87. Single crystal XRD of [1] gave a crystal structure matching that of the tetrameric complex first synthesised by Sobota et al. by addition of diethyl malonate to VCl4 and subsequent exposure to atmospheric oxygen (CCDC: 1307348).50
O). All peaks were highly broadened due to the paramagnetic nature of the vanadium species. Elemental analysis calculated for V4Cl8C28O20H48: C, 28.21; H, 4.06. Found: C, 27.33; H, 3.87. Single crystal XRD of [1] gave a crystal structure matching that of the tetrameric complex first synthesised by Sobota et al. by addition of diethyl malonate to VCl4 and subsequent exposure to atmospheric oxygen (CCDC: 1307348).50
Deposition of thin films
All films were deposited using AACVD. Compound [1] (0.30 g, 0.25 mmol) was dissolved in anhydrous toluene (20 ml) and transferred to a glass bubbler under nitrogen using a cannula. The resulting solution was atomized for 5 minutes to ensure complete solvation of the precursor. An aerosol was generated using a piezoelectric humidifier (Ultrasonic Liquids Atomizer LIQUIFOG®). The depositions were carried out using a carrier gas of 2% O2 in N2 at a flow rate of 3 L min−1. The glass substrate was standard float glass of 4 mm thickness with a 50 nm SiO2 barrier layer, supplied by Pilkington/NSG. A top plate was suspended 0.5 cm above the substrate to ensure a laminar flow. A stream of air was introduced into the baffle via a second tube, as a means of introducing oxygen to oxidise a precursor.51 Depositions were carried out at 540 °C, 550 °C and 560 °C, each taking approximately 30 minutes.Tungsten doping
Tungsten doping was affected by the addition of tungsten(VI) hexaphenoxide [W(OPh)6] into the precursor solution prior to an AACVD deposition. W(OPh)6 was synthesised using a literature protocol by Cross et al.521H NMR (600 MHz, CDCl3) δ 7.21 (12H, m), 6.88 (16H, H), 13C{1H} NMR (600 MHz, CDCl3): δ 161.94 (O–C(CH)2), δ 128.87(CH, ortho), δ 123.52(CH, para), δ 120.51 (CH, meta). Elemental analysis calculated for C36H30O6W (%): C, 58.2; H, 4.05. Found: C, 56.5; H, 3.88.Synthesis of nanoparticles
Nanoparticle samples were prepared using the thermal decomposition of [1] in the presence of a high boiling point solvent and alkyl surfactants. Briefly, compound [1] (0.5 g, 0.41 mmol) was weighed out into a nitrogen-purged three-necked 250 ml flask with a condensor. At this stage, 1-octadecene (dried over sodium) (20 ml), oleylamine (Table 1), oleic acid (Table 1) and 1,4-tetradecanediol were added. The flask was subjected to 3 vacuum/back fill cycles with nitrogen and heated to the required synthesis temperature (Table 1) at a heating rate of 3.3 °C min−1 under dynamic nitrogen. Once the desired temperature was reached, the temperature was maintained for 1 hour before cooling to room temperature naturally. The reaction remained under nitrogen throughout the cooling period. The nanoparticles were precipitated with ethanol (ca. 100 ml) and centrifuged at 3000 × g. The precipitate was subjected to three further ethanol washes (3 × 100 ml)/centrifugation cycles before storage in a vacuum desiccator. Due to the presence of the alkyl surfactants, the nanomaterials exhibited a high level of dispersibility in organic solvents such as chloroform and n-hexane.Annealing
Nanoparticle samples were annealed in a tube furnace under a constant argon flow of 0.6 L min−1 for 10 hours at 550 °C at a heating rate of 10 °C min−1 and allowed to cool to room temperature naturally under argon flow before analysis.Results and discussion
1. Precursor synthesis
[{VOCl2(CH2(COOEt)2)}4] [1], was synthesised under an atmosphere of nitrogen as per literature procedure.53 It is widely reported that the Achilles heel of vanadium(V) precursors for the formation of VO2 is the 5+ oxidation state (these precursors preferring the formation of V2O5) and as such vanadium(IV) precursors were sought for this work. Recently our group reported a synthetic study on the reactivity of vanadium(V) oxytrichloride with a range of ligands in order to isolate and fully characterise a library of novel vanadium precursors.53 Interestingly, striking ligand dependant structural and chemical variations were exhibited, altering the functionality of these compounds. This recent work described the reaction of vanadium(V) oxytrichloride with the smaller β-diketonate: acetylacetonate (acac) which resulted in the formation of HCl and a vanadium(V) monomeric species. Additionally, reaction with the larger diethyl succinate ligand was reported to proceed with elimination of Cl2 and reduction of the vanadium centre, which yielded the more reactive (although polymeric) vanadium(IV) species. It was shown that the use of a ligand slightly smaller than the succinate: diethyl malonate, prevented the unwanted polymerisation, whilst still reducing the vanadium(V) oxytrichloride to vanadium(IV) which was desirable, forming the tetrameric precursor: [{VOCl2(CH2(COOEt)2)}4].Since the reaction of vanadium(V) oxytrichloride with diethyl malonate does not chemically alter the ligand, and instead, it coordinates via both carbonyl oxygen atoms to the vanadium centre, we propose that this coordination then facilitates a bond between V and a Cl atom to break, thus reducing the metal centre. Four of these units then come together, the vanadium(IV) centre being stabilised through the coordinating VO from another identical unit, resulting in the tetramer shown below (Scheme 1). This mechanism is confirmed through spectroscopic analysis: of particular interest the 1H NMR which indicates no loss of proton from the diethylmalonate backbone as well as the longer C–O bond lengths reported in the single crystal X-ray structure, 1.492(7) and 1.498(7) Å (compared with 1.398(4) Å and 1.383(4) in the [VOCl2(acac)] compound).53
 | ||
| Scheme 1 Synthesis of [{VOCl2(CH2(COOEt)2)}4], designated compound [1]. | ||
2. Film deposition
All films were deposited using AACVD. This differs from traditional CVD insofar as AACVD involves delivery of the precursor to the substrate in the liquid phase at room temperature rather than the deposition of films from high temperature vapours, requiring volatile precursors. In AACVD, the precursor is dissolved in a suitable solvent, from which an aerosol is generated. The precursor-containing aerosol droplets are transported into the reaction vessel via a carrier gas, at which point the high temperature inside the reactor causes the solvent to evaporate bringing the precursor in contact with the substrate. The precursor then undergoes decomposition, nucleation and crystal growth to form a thin film. As the transport of the precursor to the substrate is a liquid phase process, the volatility of the precursor becomes unimportant, the suitability of the precursor instead being dependant on its solubility in a suitable solvent system. This enables a range of new precursor molecules to be employed. Additionally, the cost and complexity of the delivery process is reduced considerably, thus making AACVD suitable for industrial scale-up.In all depositions, toluene was used as the solvent. The precursor for both the VO2 film and the tungsten dopant were both dissolved in the same solution and transported to the reactor using a carrier gas consisting of 98% nitrogen and 2% oxygen. It is noteworthy that in order to attain VO2 (M), a separate oxygen source was required in order to counteract the reducing nature of an almost total azotic atmosphere. Due to the air sensitive nature of [1], a dual stream of air and nitrogen would be highly detrimental as the precursor would oxidise before entering the reaction chamber. Therefore, a separate gas stream has to be introduced into the chamber itself. This is shown in the schematic in Scheme S1 (ESI†).
Thin films of VO2 were deposited via AACVD of [1] in toluene over a temperature range of 540–560 °C using a carrier gas of 2% O2 in N2 at a flow rate of 3 L min−1 in order to encourage oxidation of the precursor. The deposited films were of good coverage and pale yellow in colour, with some darker, metallic patches formed at higher temperatures. It is noteworthy that AACVD of films at 500 °C did not deposit, whilst raising the temperature to 600 °C yielded poorly crystalline films (see Fig. S4 and S5, ESI†).
Tungsten was doped into the films over a range of concentrations from (0.4–0.8%) by adding small amounts of tungsten(VI) hexaphenoxide [W(OPh)6] to the precursor solution. [W(OPh)6] was chosen as the tungsten source as it is better suited to lower temperature applications due to its ease of handling in air and good solubility in the chosen AACVD solvent (toluene) when compared to other tungsten sources such and WF6 and WCl6. The molecular mass of [W(OPh)6] as a percentage of [1] present in the precursor solution ranged from 5% to 1%. The W doped films each had greater optical transparency than the undoped films deposited at the same temperature, with colours ranging from pale yellow to azure, becoming more blue with increased concentration. All films were fairly adherent, resisting the “Scotch Tape” test.54,55
SEM images were obtained for films of VO2 deposited at 550 °C, both with and without the incorporation of tungsten into VO2 film. The undoped film consisted of highly disordered rod-like parties of width approximately 50 nm and length 300–500 nm (Fig. 1a). Incorporation of tungsten into the film appears to greatly alter the morphology, with the film consisting of densely packed, rounded, vertical protrusions with a width of around 100 nm, typical of doped VO2 thin films obtained by CVD (Fig. 1b).11,26,29
 | ||
| Fig. 1 SEM images of the VO2 film deposited from [1] in toluene at (a) 550 °C using a 2% O2 in N2 carrier gas, and (b) the film W doped VO2 film deposited from [1] with 5 at% W(OPh)6 in toluene at 550 °C using N2 carrier gas. (c) Shows XRD patterns for a VO2 (M) film deposited at 550 °C (c) (top) and a W-doped VO2 (M) film also deposited at 550 °C (c) (bottom). | ||
XRD patterns were referenced against those of VO2 (M) (ICSD: 34033), VO2 (A) (ICSD: 51213), VO2 (B) (ICSD: 199) V2O5 (ICSD: 15798) and V2O3 (ICSD: 1473). Films deposited in the temperature range 540–560 °C were indexed as VO2 (M) as seen in Fig. 1c. There were, however, additional peaks in the XRD patterns, which indicated low levels of an impurity phase, such as a Magneli.56 There was no evidence of either V2O3 or V2O5 in the XRD patterns. Tungsten incorporation induces a slight shift in the XRD pattern (27.8° (011) for undoped VO2versus 27.9° (011) for W-doped) and a loss of crystallinity. This, coupled with the lowering of the MST is highly indicative of successful doping. Film thickness, measured by side-on SEM (see Fig. S7, ESI†), gave increasing thicknesses ranging between 1.8 ± 0.2 μm for the VO2 grown at 540 °C, 2.1 ± 0.6 μm for 550 °C and 4.1 ± 1.6 μm for 560 °C. Tungsten doping at the same temperatures gave thicker films with W–VO2 grown at 550 °C giving a thickness of 2.8 ± 0.5 μm.
Typical XPS data from VO2 (M) and W-doped VO2 (M) thin films are shown in Fig. 2. The V2p spectra are split into two regions which were then fitted with two separate environments as it was clear that more than one oxidation state was present. The visible shoulders of the main V2p3/2 peak indicate the presence of V(IV) in the films. XPS is highly surface sensitive, and as such a higher proportion of V2O5 was present at the surface than in the film, hence a large V(V) peak. Surface vanadium species will always be oxidised and this is commonly observed for XPS spectra of thin films of VO2.57,58 The fitted peak positions of 516.5 eV and 516.7 eV for V2p3/2 in the undoped and doped films respectively are indicative of V(IV) in VO2.49 The second feature at higher binding energy in both spectra (518.1 eV and 518.2 eV respectively) was assigned to V2O5.49
 | ||
| Fig. 2 Detailed XPS scans of: (a) V2p3/2 of a VO2 (M) thin film formed from the AACVD of [{VOCl2(CH2(COOEt)2)}4] [1] at 550 °C (showing V(IV) at lower binding energy (thin solid line) vs. V(V) (dotted line)), (b) V2p3/2 of a W-doped VO2 (M) thin film at 550 °C and (c) corresponding W 4f scan. | ||
The W 4f environment showed the presence of only one oxidation state. This gave a value of 35.28 eV (W 4f7/2) and 37.38 eV (W 4f5/2) which is consistent with W6+. This has been previously seen for W-doped VO2 in both thin film and nanoparticle synthesis.1,29,59
Variable temperature transmission UV/Vis spectroscopy was carried out on the films deposited from [1] in order to determine whether the films underwent the structural transition associated with VO2 (M). A spectrum was taken of the film at room temperature. Following this the films were rapidly heated to 90 °C and the measurement repeated. The films were then cooled to room temperature and the initial measurement repeated to ensure that the sample has remained in the same position throughout the heating (Fig. 3).
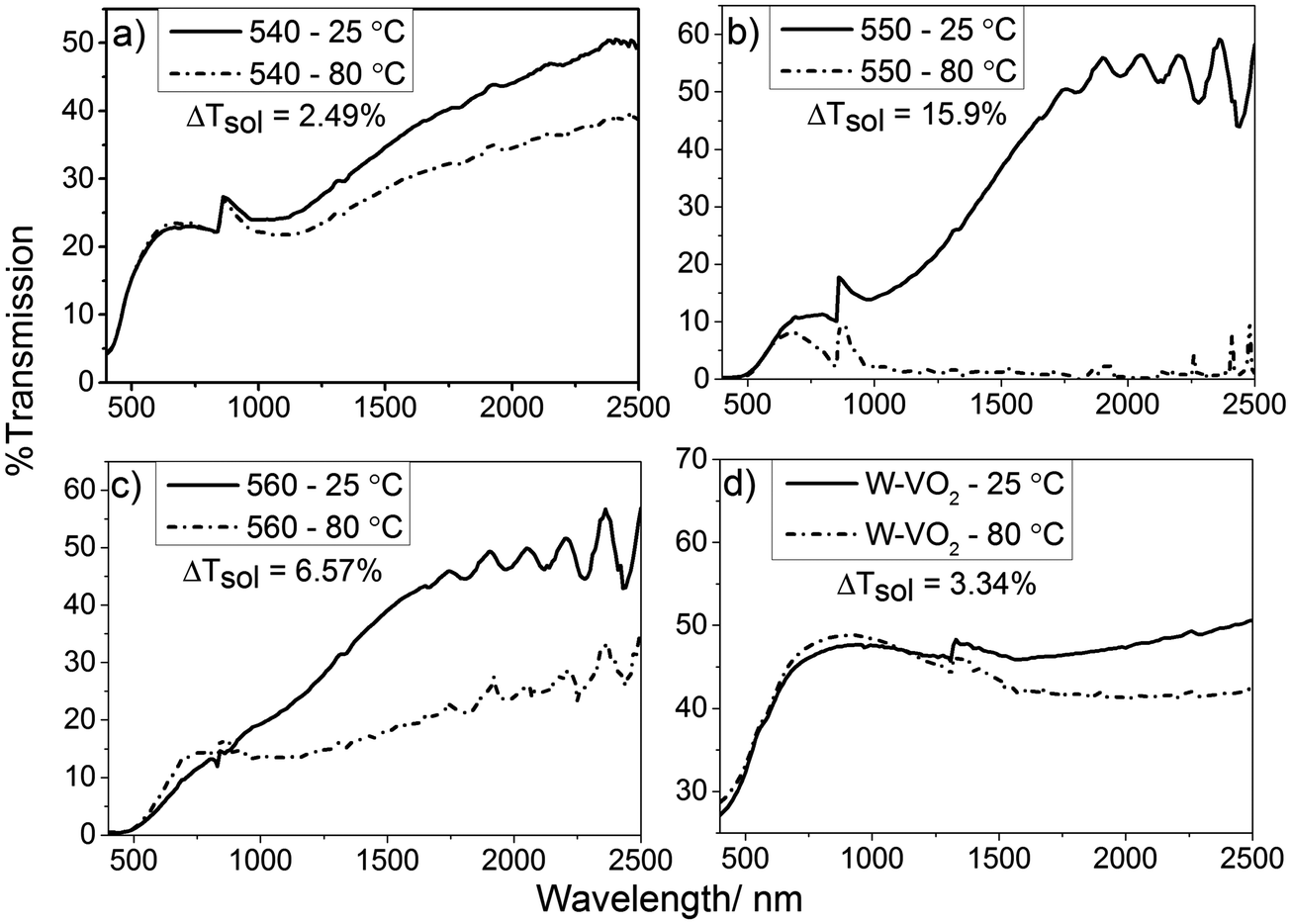 | ||
| Fig. 3 Variable temperature UV/Vis/NIR spectra showing the change in optical properties of thin films of undoped and W-doped VO2 thin films deposited by AACVD of synthesised vanadium precursor solutions. Solar modulation values (ΔTsol) for each film have been included in the figure. (a) VO2 (M) film deposited at 540 °C, (b) 550 °C, (c) 560 °C and (d) W-doped VO2 (M) thin film deposited at 550 °C. | ||
Films deposited at 550 °C showed the highest MST by a considerable margin, with the transmittance at 2500 nm falling from 60% to around 2% when the sample was heated from room temperature to 90 °C. Films deposited at 560 °C demonstrated similar characteristics at room temperature to those deposited at 550 °C. Testing the samples for thermochromic activity revealed that the monoclinic phase of the VO2 is far less pure, roughly halving the transmission to 30%. The VO2 film deposited at 540 °C was by far the least effective, seeing a reduction in transmission from 50% to 40% at 2500 nm. These measurements reveal that the optimum temperature for the formation of monoclinic VO2 from [1] was 550 °C. All undoped films displayed a phase transition temperature of ca. 68 °C, which is typical of undoped VO2 (M).60 Tungsten doped films exhibited a reduction in MST, to ca. 50 °C. This was determined from hysteresis data obtained via UV/Vis spectroscopy, giving a narrow temperature window for the transition of ca. 5 °C (see Fig. S6, ESI†).
Solar modulation calculations (ΔTsol) were performed as described by Taylor et al.61 Solar modulation values take into account the effect that gases, such as CO2 and H2O, have on the intensity of solar radiation at ground level. This gives a value which can be used to give a better representation of the absolute change in the optical properties of the material. The majority of the energy in the solar spectrum, at ground level, is accounted for by the UV/Visible region (380–780 nm); this leaves a maximum change of ca. 20% for the near IR region (780–2500 nm). VO2 (M) shows negligible changes in visible wavelength absorption/transmission when it passes through the MST. When comparing the solar modulation values for the VO2 films, it can be seen that the sample deposited at 550 °C shows a solar modulation of 15.9% which is close to this maximum allowed solar modulation for single layer VO2 (M) and is comparable to the best results seen for VO2 thin films by other research groups.60
3. Nanostructure synthesis and characterisation
Nanostructures were also synthesised from [{VOCl2(CH2(COOEt)2)}4] [1] using thermal decomposition in the presence of a high boiling point solvent and structure-directing surfactants under an inert atmosphere. Thermal decomposition of metal–organic precursors has been shown to be extremely effective at producing nanoparticles with low polydispersity and shape anisotropy which are subsequently dispersible in a wide range of [organic] solvents.62–65 A large number of the metal oxide nanoparticle syntheses are based on the in or ex situ formation of metal–fatty acid complexes and their subsequent decomposition to yield the metal oxide nanoparticles forested by surfactants (fatty acids) rendering them readily dispersible in organic solvents.66–70Examples using this methodology for VO2 nanocrystals are few and far between, however it is noteworthy that Paik et al. utilised a similar methodology to synthesise VOx nanocrystals from VOCl3 in the presence of oleylamine and octadecanol, resulting in colloidally stable, monodisperse mixed-phase VOx nanocrystals.71 The VOx nanocrystals obtained were flash-annealed at 500 °C for 5 minutes in air at 1 mTorr in order to give thermochromic VO2 (M), as the formation of phase-pure VO2 (M) at low (<450 °C) temperature and atmospheric pressure is thermodynamically unfavourable.18 The authors make reference to the fact that no reaction occurs from VOCl3 without 1-octadecanol as vanadium–oxygen bonds are formed from the non-hydrolitic reaction between a primary alcohol and a metal halide (sic 1-octadecanol and VOCl3).71–73 Although the synthesis described herein is conspicuous by the absence of a primary alcohol, the composition of the precursor with co-ordinated oxygen species is the de facto product of the non-hydrolitic reaction described above. This, coupled with the high temperature of the synthesis, allows the nucleation and growth of VOx nanoparticles to proceed.
The investigation on the decomposition of [1] in the presence of surfactants in a high boiling point solvent was designed to investigate the effect of differing surfactant ratios, temperature and the inclusion of a fatty alcohol, 1,4-tetradecanediol. The sample preparation conditions are summarised in Table 1. All reactions were undertaken in 20 ml of dry 1-octadecene (dried over sodium) and after 3 vacuum/re-fill cycles of nitrogen. The reaction was heated from room temperature to the target temperature at 3.3 °C min−1 under a dynamic flow of nitrogen, during which the reaction mixture darkened in colour from green to a final black. The addition of oleylamine also induced a colour change of the precursor to blue, presumably due to complexation with the vanadium centre, most likely a d → d transition due to the formation of an octahedral complex. On precipitation with ethanol and centrifugation all samples (bar sample 7) gave a black precipitate which could be readily dispersed in organic solvents.
From TEM analysis of the different samples, it was clear that different surfactant blends affected the morphology of the final product. Oleylamine in particular promoted the growth of spine-like structures as well as nanoparticles which were not seen in its absence, whereas oleic acid purely promoted particle growth. Mixtures of oleic acid and oleylamine were found to be the most effective at generating readily dispersible products, with the optimum blend of equimolar amounts of oleic acid and oleylamine [used by Sun and Zeng in their seminal paper on iron oxide nanoparticle synthesis]74 in samples 1, 6, 7 and 8. The “6,6” blend produced a mixture of some small particles but predominantly large, spine-like structures of 50–100 nm in length. These features were preserved on annealing in nitrogen for 8 hours, but were not as pronounced in sample 6, in which the synthesis temperature is lower (280 °C), leading us to deduce that the formation of the vanadium oxide spines is temperature as well as oleylamine dependent. However, the features emerged with annealing treatment at 550 °C under nitrogen for 10 hours (Fig. 4d and e). Sample 7, in which the synthesis temperature was reduced to 240 °C was evidently lower than the nucleation temperature for this system as no nanoparticles were produced. The addition of an oxygen source, 1,4-tetradecanediol in sample 9 with the “6,6” blend of oleic acid and oleylamine had no effect on the phase and seemed to the 1,4-tetradecanediol counteracted the structure-directing effects of oleylamine to form almost exclusively particulate material (Fig. S2d, ESI†).
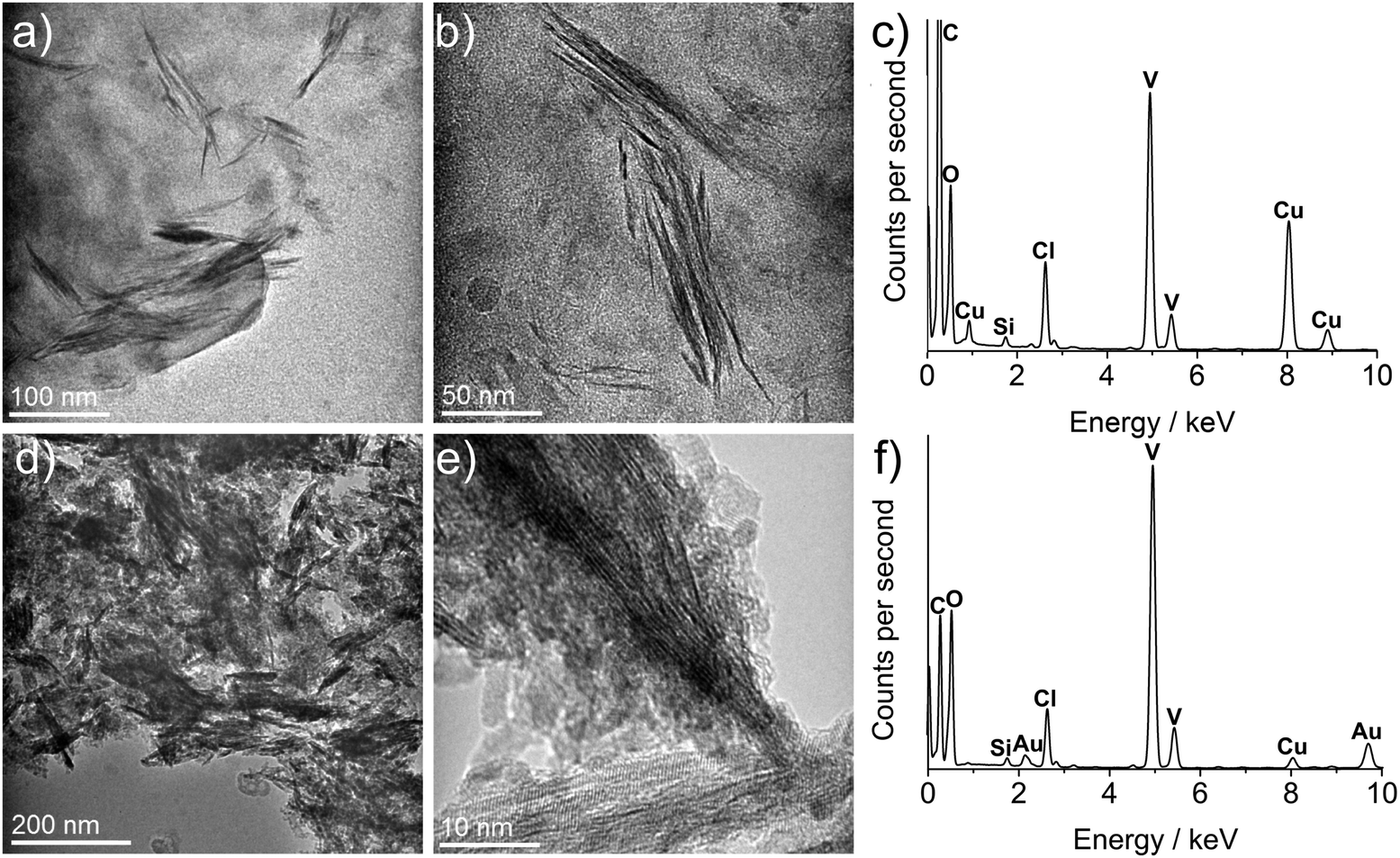 | ||
| Fig. 4 TEM images and EDS spectra of sample 1 pre- and post annealing. (a and b) are TEM images of pre-annealed sample 1, showing the directing effects of oleylamine and the formation of a spine-like morphology. (c) is the corresponding EDS spectrum of the pre-annealed sample 1. (d and e) are annealed TEM images from sample 1 with corresponding EDS spectrum, (f). | ||
Detailed TEM analysis of the as-synthesised VOx nanostructures (especially lattice plane images) were exacting due to the presence of high carbon contamination, predominantly caused by surfactant layers and decomposition products from the precursor itself. The latter was in evidence through the presence of chlorine by EDS as residue from initial VOCl3 (Fig. 4c and f).
Samples 1 and 6 were both annealed at 550 °C under argon in order to increase crystallinity. Analysis of lattice planes in the annealed samples (Fig. 4e) gave a d-spacing of 0.326 nm corresponding to the 〈111〉 plane of vanadium oxide (V4O9, ICSD 15041). Analysis of lattice planes in Fig. S3 (ESI†) gave a d-spacing of 0.327 nm, again corresponding to the 〈111〉 plane of V4O9, confirming that heat treatment had an identical effect on both samples despite initial differences with synthesis temperatures.
EDS spectra showed the persistence of chlorine in the system as a consequence of the chlorine in the precursor. It also showed the reduction in carbon from the heavily carbon contaminated pre-annealed samples versus post annealed samples, attributed to the alkyl ligands. The at% ratios of V:O for samples 1 and 6 pre-anneal was 26.20:73.80 and 1.91:98.09 respectively, with heat treatment dramatically effecting sample 6 with the ratio changing to 26.22:73.78. The ratio for sample 1 however, was relatively unchanged (26.40:73.60). These non-stoichiometric amounts, along with HRTEM and XRD analysis, leads us to conclude that a mixture of vanadium oxide phases were formed in the nanoparticle synthesis. Ratios of V:O for all samples are listed in the Table S1 (ESI†).
Powder X-ray diffraction (PXRD) was performed on the as synthesised and the post-annealed nanoparticle samples as shown in Fig. 5. The as-synthesised nanoparticles, Fig. 5a and b, have only a few diffraction peaks- it was not possible to confirm the phase of the material from these. The broad reflections between 5–12° 2θ also suggest that there was a large amorphous component to the samples.
 | ||
| Fig. 5 PXRD diffraction patterns for as synthesised powders, (a) and (b) and following annealing treatment, (c) and (d). λ = 0.7093 Å. Sample 1 and 6 were synthesised from the decomposition of [{VOCl2(CH2(COOEt)2)}4] in the presence of oleic acid (6 mmol) and oleylamine (6 mmol) at 320 °C (Sample 1) and 280 °C (Sample 6). | ||
The post-annealed samples, Fig. 5c and d, have a greater number of diffraction peaks in the data. The intensities of these diffraction peaks are also greater, with a reduction in the number of broad diffraction peaks-suggesting higher crystallinity for the annealed samples. Phase analysis of these samples suggested a mix of Magneli phases, with V3O7 – 22.2° 〈020〉, V4O9 – 11.1° 〈102〉, 16.5° 〈113〉 and V5O9 – 5.2° 〈001〉, 24.1° 〈211〉 and 28.8° 〈203〉 lattice planes all being identified in the data. This is not surprising, as vanadium is known to readily form oxides with multiple oxidation states of vanadium present.75,76
XPS was used as a comparison between annealed and non-annealed samples. XPS spectra for the V2p region in Sample 1 pre (a) and post (b) anneal are shown in Fig. 6. Both show similar profiles to the thin films in Fig. 2, with V(IV) peaks reported at 516.5 eV and 516.3 eV for non-annealed and annealed samples respectively (V2p3/2) and assigned as VO2. The V(V) peaks, at 517.7 eV and 517.8 eV respectively were again assigned as V2O5.49
 | ||
| Fig. 6 Detailed XPS scans of: (a) V2p of sample 1, VO2 nanomaterials before annealing and (b) after annealing showing V(IV) at lower binding energy (thin solid line) vs. V(V) (dotted line). | ||
Conclusions
The synthesis of [{VOCl2(CH2(COOEt)2)}4] [1] was demonstrated, and its versatility as an AACVD precursor for vanadium oxide thin films and nanocrystals investigated. [1] was shown to be a highly effective precursor for the formation of thermochromic VO2 (M) thin films on glass by AACVD. The AACVD process has numerous advantages over conventional CVD methods including: the wide range of precursors including those with low volatility, high control over dopant concentration and is applicable to industrial scale-up.6 VO2 (M) films produced at 550 °C (ΔTsol = 15.9%) were shown to have superior solar modulation when compared to films produced at 540 °C and 560 °C and compare favourably with literature values. The thermochromic switching temperature was reduced through the addition of [W(OPh)6] to the precursor solution, reducing the MST to ∼50 °C. This demonstrates the viability of the AACVD method when using a designed molecular precursor to form thermochromic materials with tunable properties.In terms of nanomaterial synthesis, [1] was decomposed in the presence of alkyl surfactants at elevated temperatures in an inert atmosphere. It was shown that different surfactants affected the shape of the final product, with oleylamine promoting “spine-like” growth, and oleic acid particulate material. The control over shape and size of the resultant nanomaterials by manipulation of surfactant type and concentration in the decomposition of [1] bodes well for the future for designed molecular vanadium oxide nanomaterial precursors. [1] represents the first example in a class of activated vanadium complexes for the synthesis of a variety of VO2 thin films and shape tunable VxOy nanomaterials. The design of these molecules may lead to a single precursor for the low temperature, one-pot synthesis of colourless thermochromic VO2 (M) nanocrystals – a key step towards the routine use of smart windows, potentially saving millions of kWh per annum worldwide.
Acknowledgements
JCB and CEK acknowledge the Ramsay Memorial Trust for funding by means of Ramsay Memorial Fellowships. BB acknowledges Huntsman Pigments and the Energy and Physical Research Council (EPSRC) for an EngD studentship (Grant No. EP/G036675). IPP, CJC and MJP acknowledge EPSRC Grant No. EP/L017709 for funding.References
- M. E. A. Warwick and R. Binions, J. Mater. Chem. A, 2014, 2, 3275 CAS.
- C. G. Granqvist, J. Vac. Sci. Technol., B, 2014, 32, 60801 Search PubMed.
- M. Kamalisarvestani, R. Saidur, S. Mekhilef and F. S. Javadi, Renewable Sustainable Energy Rev., 2013, 26, 353–364 CrossRef CAS.
- C. G. Granqvist, Sol. Energy Mater. Sol. Cells, 2007, 91, 1529–1598 CrossRef CAS.
- P. Huovila, Buildings and Climate Change: Status, Challenges, and Opportunities, UNEP/Earthprint, 2007 Search PubMed.
- P. Marchand, I. A. Hassan, I. P. Parkin and C. J. Carmalt, Dalton Trans., 2013, 42, 9406–9422 RSC.
- S. M. Babulanam, T. S. Eriksson, G. A. Niklasson and C. G. Granqvist, Sol. Energy Mater., 1987, 16, 347–363 CrossRef CAS.
- J. B. Goodenough, Annu. Rev. Mater. Sci., 1971, 1, 101–138 CrossRef CAS.
- M. Saeli, C. Piccirillo, I. P. Parkin, R. Binions and I. Ridley, Energy Build., 2010, 42, 1666–1673 CrossRef.
- C. Batista, R. M. Ribeiro and V. Teixeira, Nanoscale Res. Lett., 2011, 6, 1–7 CrossRef PubMed.
- C. Piccirillo, R. Binions and I. P. Parkin, Thin Solid Films, 2008, 516, 1992–1997 CrossRef CAS.
- G. V. Jorgenson and J. C. Lee, Sol. Energy Mater., 1986, 14, 205–214 CrossRef CAS.
- W. Burkhardt, T. Christmann, S. Franke, W. Kriegseis, D. Meister, B. K. Meyer, W. Niessner, D. Schalch and A. Scharmann, Thin Solid Films, 2002, 402, 226–231 CrossRef CAS.
- S. Hu, S.-Y. Li, R. Ahuja, C. G. Granqvist, K. Hermansson, G. A. Niklasson and R. H. Scheicher, Appl. Phys. Lett., 2012, 101, 201902 CrossRef.
- T. J. Hanlon, J. A. Coath and M. A. Richardson, Thin Solid Films, 2003, 436, 269–272 CrossRef CAS.
- X. Tan, T. Yao, R. Long, Z. Sun, Y. Feng, H. Cheng, X. Yuan, W. Zhang, Q. Liu, C. Wu, Y. Xie and S. Wei, Sci. Rep., 2012, 2, 466 Search PubMed.
- R. Long, B. Qu, R. Tan, Y. Sun, X. Tan, W. Ying, B. Pan, Y. Xiong and Y. Xie, Phys. Chem. Chem. Phys., 2012, 14, 7225–7228 RSC.
- C. H. Griffiths and H. K. Eastwood, J. Appl. Phys., 1974, 45, 2201–2206 CrossRef CAS.
- M. Borek, F. Qian, V. Nagabushnam and R. K. Singh, Appl. Phys. Lett., 1993, 63, 3288–3290 CrossRef CAS.
- N. R. Mlyuka, G. A. Niklasson and C. G. Granqvist, Sol. Energy Mater. Sol. Cells, 2009, 93, 1685–1687 CrossRef CAS.
- T. D. Manning, I. P. Parkin, C. Blackman and U. Qureshi, J. Mater. Chem., 2005, 15, 4560–4566 RSC.
- M. N. Field and I. P. Parkin, J. Mater. Chem., 2000, 10, 1863–1866 RSC.
- U. Qureshi, T. D. Manning and I. P. Parkin, J. Mater. Chem., 2004, 14, 1190–1194 RSC.
- M. B. Sahana, M. S. Dharmaprakash and S. A. Shivashankar, J. Mater. Chem., 2002, 12, 333–338 RSC.
- T. D. Manning and I. P. Parkin, Polyhedron, 2004, 23, 3087–3095 CrossRef CAS.
- T. D. Manning and I. P. Parkin, J. Mater. Chem., 2004, 14, 2554–2559 RSC.
- T. D. Manning, I. P. Parkin, R. J. H. Clark, D. Sheel, M. E. Pemble and D. Vernadou, J. Mater. Chem., 2002, 12, 2936–2939 RSC.
- D. Vernardou, P. Paterakis, H. Drosos, E. Spanakis, I. M. Povey, M. E. Pemble, E. Koudoumas and N. Katsarakis, Sol. Energy Mater. Sol. Cells, 2011, 95, 2842–2847 CrossRef CAS.
- C. S. Blackman, C. Piccirillo, R. Binions and I. P. Parkin, Thin Solid Films, 2009, 517, 4565–4570 CrossRef CAS.
- R. Binions, G. Hyett, C. Piccirillo and I. P. Parkin, J. Mater. Chem., 2007, 17, 4652–4660 RSC.
- C. Piccirillo, R. Binions and I. P. Parkin, Eur. J. Inorg. Chem., 2007, 4050–4055 CrossRef CAS.
- M. E. A. Warwick, I. Ridley and R. Binions, J. Nanosci. Nanotechnol., 2011, 11, 8158–8162 CrossRef CAS PubMed.
- S. S. Garje, J. S. Ritch, D. J. Eisler, M. Afzaal, P. O’Brien and T. Chivers, J. Mater. Chem., 2006, 16, 966–969 RSC.
- N. O. Boadi, M. A. Malik, P. O’Brien and J. A. M. Awudza, Dalton Trans., 2012, 41, 10497–10506 RSC.
- J. M. Clark, G. Kociok-Köhn, N. J. Harnett, M. S. Hill, R. Hill, K. C. Molloy, H. Saponia, D. Stanton and A. Sudlow, Dalton Trans., 2011, 40, 6893–6900 RSC.
- M. J. Powell, P. Marchand, C. J. Denis, J. C. Bear, J. A. Darr and I. P. Parkin, Nanoscale, 2015, 7, 18686–18693 RSC.
- T.-D. Nguyen and T.-O. Do, Langmuir, 2009, 25, 5322–5332 CrossRef CAS PubMed.
- J.-H. Son, J. Wei, D. Cobden, G. Cao and Y. Xia, Chem. Mater., 2010, 22, 3043–3050 CrossRef CAS.
- H. Zhang, X. Xiao, X. Lu, G. Chai, Y. Sun, Y. Zhan and G. Xu, J. Alloys Compd., 2015, 636, 106–112 CrossRef CAS.
- J. Nag and R. F. Haglund Jr., J. Phys.: Condens. Matter, 2008, 20, 264016 CrossRef.
- L. Zhong, M. Li, H. Wang, Y. Luo, J. Pan and G. Li, CrystEngComm, 2015, 17, 5614–5619 RSC.
- P. Billik, M. Čaplovičová, J. Maňka, Ľ. Čaplovič, A. Cigáň, A. Koňakovský, R. Bystrický and A. Dvurečenskij, Meas. Sci. Technol., 2011, 11, 29–33 Search PubMed.
- K. C. Kam and A. K. Cheetham, Mater. Res. Bull., 2006, 41, 1015–1021 CrossRef CAS.
- Y. Zhang, J. Zhang, X. Zhang, Y. Deng, Y. Zhong, C. Huang, X. Liu, X. Liu and S. Mo, Ceram. Int., 2013, 39, 8363–8376 CrossRef CAS.
- B.-G. Chae, H.-T. Kim, S.-J. Yun, B.-J. Kim, Y.-W. Lee, D.-H. Youn and K.-Y. Kang, Electrochem. Solid-State Lett., 2006, 9, C12–C14 CrossRef CAS.
- H. Wang, X. Yi, S. Chen and X. Fu, Sens. Actuators, A, 2005, 122, 108–112 CrossRef CAS.
- S. A. Pauli, R. Herger, P. R. Willmott, E. U. Donev, J. Y. Suh and R. F. Haglund Jr, J. Appl. Phys., 2007, 102, 73527 CrossRef.
- A. B. Pangborn, M. A. Giardello, R. H. Grubbs, R. K. Rosen and F. J. Timmers, Organometallics, 1996, 15, 1518–1520 CrossRef CAS.
- NIST X-Ray Photoelectron Spectrosc. XPS Database Version 35.
- P. Sobota, J. Ejfler, S. Szafert, T. Glowiak, I. O. Fritzky and K. Szczegot, J. Chem. Soc., Dalton Trans., 1995, 1727–1732 RSC.
- N. Noor and I. P. Parkin, J. Mater. Chem. C, 2013, 1, 984–996 RSC.
- W. B. Cross, I. P. Parkin, S. A. O’Neill, P. A. Williams, M. F. Mahon and K. C. Molloy, Chem. Mater., 2003, 15, 2786–2796 CrossRef CAS.
- B. J. Blackburn, J. H. Crane, C. E. Knapp, M. J. Powell, P. Marchand, D. Pugh, J. C. Bear, I. P. Parkin and C. J. Carmalt, Mater. Des., 2016, 108, 780–790 CrossRef CAS.
- A. Mills, N. Elliott, I. P. Parkin, S. A. O’Neill and R. J. Clark, J. Photochem. Photobiol., A, 2002, 151, 171–179 CrossRef CAS.
- J. G. Buijnsters, P. Shankar, W. Fleischer, W. J. P. van Enckevort, J. J. Schermer and J. J. ter Meulen, Diamond Relat. Mater., 2002, 11, 536–544 CrossRef CAS.
- U. Schwingenschlögl and V. Eyert, Ann. Phys., 2004, 13, 475–510 CrossRef.
- R. Quesada-Cabrera, M. J. Powell, P. Marchand, C. J. Denis, F. D. Maggio, J. A. Darr and I. P. Parkin, J. Nanosci. Nanotechnol., 2016, 16, 10104–10111 CrossRef.
- S. Lu, L. Hou and F. Gan, J. Mater. Sci., 1993, 28, 2169–2177 CrossRef CAS.
- G. P. Halada and C. R. Clayton, J. Vac. Sci. Technol., A, 1993, 11, 2342–2347 CAS.
- A. Taylor, I. Parkin, N. Noor, C. Tummeltshammer, M. S. Brown and I. Papakonstantinou, Opt. Express, 2013, 21(suppl 5), A750–A764 CrossRef PubMed.
- M. J. Powell, R. Quesada-Cabrera, A. Taylor, D. Teixeira, I. Papakonstantinou, R. G. Palgrave, G. Sankar and I. P. Parkin, Chem. Mater., 2016, 28, 1369–1376 CrossRef CAS.
- C. R. Crick, J. C. Bear, P. Southern and I. P. Parkin, J. Mater. Chem. A, 2013, 1, 4336–4344 CAS.
- J. Park, K. An, Y. Hwang, J.-G. Park, H.-J. Noh, J.-Y. Kim, J.-H. Park, N.-M. Hwang and T. Hyeon, Nat. Mater., 2004, 3, 891–895 CrossRef CAS PubMed.
- M. Lattuada and T. A. Hatton, Langmuir, 2007, 23, 2158–2168 CrossRef CAS PubMed.
- J. C. Bear, N. Hollingsworth, P. D. McNaughter, A. G. Mayes, M. B. Ward, T. Nann, G. Hogarth and I. P. Parkin, Angew. Chem., Int. Ed., 2014, 53, 1598–1601 CrossRef CAS PubMed.
- L. M. Bronstein, J. E. Atkinson, A. G. Malyutin, F. Kidwai, B. D. Stein, D. G. Morgan, J. M. Perry and J. A. Karty, Langmuir, 2011, 27, 3044–3050 CrossRef CAS PubMed.
- L. M. Bronstein, X. Huang, J. Retrum, A. Schmucker, M. Pink, B. D. Stein and B. Dragnea, Chem. Mater., 2007, 19, 3624–3632 CrossRef CAS.
- S. G. Kwon, Y. Piao, J. Park, S. Angappane, Y. Jo, N.-M. Hwang, J.-G. Park and T. Hyeon, J. Am. Chem. Soc., 2007, 129, 12571–12584 CrossRef CAS PubMed.
- S. G. Kwon and T. Hyeon, Small, 2011, 7, 2685–2702 CrossRef CAS PubMed.
- S. G. Kwon and T. Hyeon, Acc. Chem. Res., 2008, 41, 1696–1709 CrossRef CAS PubMed.
- T. Paik, S.-H. Hong, E. A. Gaulding, H. Caglayan, T. R. Gordon, N. Engheta, C. R. Kagan and C. B. Murray, ACS Nano, 2014, 8, 797–806 CrossRef CAS PubMed.
- M. Niederberger, Acc. Chem. Res., 2007, 40, 793–800 CrossRef CAS PubMed.
- M. Niederberger, M. H. Bartl and G. D. Stucky, J. Am. Chem. Soc., 2002, 124, 13642–13643 CrossRef CAS PubMed.
- S. Sun and H. Zeng, J. Am. Chem. Soc., 2002, 124, 8204–8205 CrossRef CAS PubMed.
- C. Wu, F. Feng and Y. Xie, Chem. Soc. Rev., 2013, 42, 5157–5183 RSC.
- M. Demeter, M. Neumann and W. Reichelt, Surf. Sci., 2000, 454–456, 41–44 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6tc03482a |
| This journal is © The Royal Society of Chemistry 2016 |