
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIncreased thermal stabilization of polymer photovoltaic cells with oligomeric PCBM†
Hasina H.
Ramanitra
ab,
Simon A.
Dowland
*c,
Bruna A.
Bregadiolli
d,
Michael
Salvador
e,
Hugo
Santos Silva
abf,
Didier
Bégué
f,
Carlos F. O.
Graeff
d,
Heiko
Peisert
b,
Thomas
Chassé
b,
Sambatra
Rajoelson
c,
Andres
Osvet
e,
Christoph J.
Brabec
eg,
Hans-Joachim
Egelhaaf
g,
Graham E.
Morse
h,
Andreas
Distler
c and
Roger C.
Hiorns
 *i
*i
aUniversité de Pau et des Pays de l'Adour (UPPA), IPREM (EPCP, CNRS-UMR 5254), 2 Avenue Président Angot, 64053 Pau, France
bInstitut für Physikalische und Theoretische Chemie, Eberhard Karls Universität Tübingen, Auf der Morgenstelle 18, 72076 Tübingen, Germany
cBelectric OPV GmbH, Landgrabenstr. 94, 90443 Nürnberg, Germany. E-mail: simon.dowland@gmx.de
dDepartamento de Física – FC – UNESP, Av. Luiz Edmundo Carrijo Coube, 14-01, 17033-360 Bauru, Brazil
eZAE Bayern, Auf AEG, Bau 16, 1. OG Fürther Str. 250, 90429 Nürnberg, Germany
fUPPA, IPREM (ECP, CNRS-UMR 5254), 64053 Pau, France
gInstitute of Materials for Electronics and Energy Technology (i-MEET), Friedrich-Alßexander-University of Erlangen-Nürnberg, Martensstrasse 7, 91058, Erlangen, Germany
hMerck Chemicals Ltd., Chilworth Technical Centre, University Parkway, SO16 7QD, Southampton, UK
iCNRS, IPREM (EPCP, CNRS-UMR 5254), 64053 Pau, France. E-mail: roger.hiorns@univ-pau.fr
First published on 12th August 2016
Abstract
The first oligomerisation of phenyl-C61-butyric acid methyl ester (PCBM) using a facile atom transfer radical addition polymerization (ATRAP) and its exploitation for organic photovoltaic devices is described. Oligo{(phenyl-C61-butyric acid methyl ester)-alt-[1,4-bis(bromomethyl)-2,5-bis(octyloxy)benzene]} (OPCBMMB) shows opto-electronic properties equivalent to those of PCBM but has a higher glass transition temperature. When mixed with various band gap semiconducting polymers, OPCBMMB delivers performances similar to PCBM but with an enhanced stabilization of the bulk heterojunction in photovoltaic devices on plastic substrates under thermal stress, regardless of the degree of crystallinity of the polymer and without changing opto-electronic properties.
Introduction
Phenyl-C61-butyric acid methyl ester (PCBM) is a ubiquitous material in organic photovoltaics due to its high electron affinities and mobilities.1–3 As the electron acceptor of choice in photo-active layers in bulk heterojunction (BHJ) devices, it has dominated research studies for the last decade or so in combination with poly(3-hexylthiophene).4 More recently it has clearly demonstrated its aptness for use with a wide range of low band gap electron donor polymers,5,6 small molecules,7 and has even found employment in perovskite solar cells as an electron extraction layer,8 confirming its exceptional position in device manufacturing and research. Specific to BHJs is its nano-scale morphology,9 with a tendency to organise in amorphous and crystalline domains,10 ideally within the exciton diffusion length, and with a beneficial vertical distribution that facilitates charge percolation.11 However, the morphology of PCBM blends is not stable in BHJs. Fullerenes tend to diffuse through layers to form large μm-scale crystals or to accumulate at active layer-electrode interfaces,12 both occurring notably when heated above the glass transition temperature and potentially leading to reductions in device efficiencies.11,13,14 While photo-induced reactions between PCBM molecules may lead to morphological stability,15,16 they can also give rise to dimers that act as charge traps and reduce short circuit current (Jsc) values.17 PCBM is also difficult to cast as a thin film, resulting in rough surfaces of large aggregates.In some cases, it has been established that the addition of small amounts of nucleating agents may lead to controlled fullerene crystal growth and thus hinder coarsening of the morphology.18 This works well in very amorphous, nucleation-limited polymer donor blends but is less effective in systems that per se have a high degree of fractional crystallinity.19 PCBM oligomers could be an alternative route, as the molecules would no longer be able to easily diffuse through the device layer stack, and should form stable thin films. Ideally, the oligomer should retain the opto-electronic properties of the PCBM molecule. In the past, fullerene (C60) and PCBM were incorporated into macromolecules as pendent moieties.20 While this remains one route, it can let the fullerene sphere continue to form aggregates,21 possibly because of the high degrees of freedom. Nevertheless, pendent fullerenes have been preferred because it is generally easier to control the chemistry; as soon as conditions such as concentration and temperature are increased to force more than one addition, multi-additions can occur at any of 30 available [6,6]-double bonds. Other promising strategies for attaining morphological stability include the use of cross-linkable, conjugated materials and BHJ compatibilizers,22 the latter envisioning control over interfacial tensions, and block copolymers.23
A higher concentration of fullerene and greater control over its aggregative properties can be reached by incorporating it into the main-chain, effectively turning C60 into a monomer. Work has been performed to these ends, for example via Diels–Alder chemistry,24 or by pre-modifying the sphere. However, these techniques can lead to crosslinking or be quite complex to perform.25 In an attempt to resolve these problems it was discovered possible to polymerize C60 in a controlled manner via radical 1,4-additions to a single phenylene ring. This atom transfer radical addition polymerization (ATRAP) gave high yields (ca. 70%) of oligo(fullerene)s and poly(fullerene)s from a one-pot reaction.26,27 However, we also found that the materials were not very stable due to instabilities around the methylene–C60 bond.28,29 This hindered purification, an important point given that even minute quantities of metal catalysts and other impurities can reduce device productivities.30
Here we describe the revelation that it is possible to perform a very simple one-step ATRAP of PCBM to yield soluble, low molecular weight oligomers that retain PCBM's essential opto-electronic properties. It has shown how this new material can be used in BHJs to give improved thermal properties against those of PCBM, and importantly, we demonstrate that this approach works on solar cells fabricated on plastic substrates featuring active areas larger than 25 mm2. This technology can thus prevent thermal degradation associated with elevated temperatures during high-throughput device fabrication with no processing changes. Furthermore, we find by theoretical treatments, that the oligomers are more stable than their C60 equivalents.
Experimental
Materials
All materials were used as received and obtained from Aldrich (France), except PCBM (99.5%) which was obtained from Merck KGaA (Germany). Solvents were used after drying over their respective drying agents under dry nitrogen.Synthesis of OPCBMMB
Oligomerisations were performed in flame-dried, dry-nitrogen flushed glass reactors. Where possible, air and light were excluded from handling of the oligomers and their solutions. This is a representative procedure. PCBM (3 g, 3.28 × 10−3 mol) and 1,4-bis(bromomethyl)-2,5-bis(octyloxy)benzene (1.71 g, 3.28 × 10−3 mol) were dissolved in toluene (150 mL) at room temperature. CuBr (0.158 g, 1 × 10−3 mol) and bipyridine (2.06 g, 13.2 × 10−3 mol) were added and the temperature was slowly raised to 100 °C. After 16 h, and recovery by precipitation in methanol (500 mL), the product was Soxhlet washed with acetone for three days. Drying under reduced pressure yielded a dark brown powder (2.07 g, 49%). 1H NMR (400 MHz, C6D6) δ = 0.9 (s, –OC7H14–CH3), 1.20–1.71 (m, broad, O–CH2–C6H12–CH3), 1.95–2.38 (m, –C3H6–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) OOCH3), 2.75 (s, O–CH2–C6H12–CH3), 3.33 (s, –O–CH3), 3.65–3.87 (broad, O–CH2–C6H12–CH3), 3.88–4.30 (broad, C60–CH2–ph), 4.31–4.65 (broad, –CH2–Br), 7.22–7.68 (m, aromatic) ppm; 13C NMR (101 MHz, C6D6) δ = 14.1 (–OC7H14–CH3), 22.3–33.0 (O–CH2–C6H12–CH3, CH2–Br), 50.8–51.9 (–O–CH3), 69.5 (O–CH2–C6H12–CH3), 114.7–117.8 (aromatic), 132.0 (aromatic-PCBM), 136.8–148.8 (fullerene cage), 172.3 (CO) ppm.
OOCH3), 2.75 (s, O–CH2–C6H12–CH3), 3.33 (s, –O–CH3), 3.65–3.87 (broad, O–CH2–C6H12–CH3), 3.88–4.30 (broad, C60–CH2–ph), 4.31–4.65 (broad, –CH2–Br), 7.22–7.68 (m, aromatic) ppm; 13C NMR (101 MHz, C6D6) δ = 14.1 (–OC7H14–CH3), 22.3–33.0 (O–CH2–C6H12–CH3, CH2–Br), 50.8–51.9 (–O–CH3), 69.5 (O–CH2–C6H12–CH3), 114.7–117.8 (aromatic), 132.0 (aromatic-PCBM), 136.8–148.8 (fullerene cage), 172.3 (CO) ppm.
Film formation
Films of PCBM and OPCBMMB were prepared from solutions in neat o-xylene at a concentration of 2 mg mL−1. Films of P3HT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PCBM and P3HT:OPCBMMB were prepared from solutions in neat o-xylene at various ratios of the semiconductors. The solutions were left overnight at 80 °C prior to deposition by doctor blade deposition on glass substrates. The depositions were at 65 °C with a blade rate of 20 mm s−1. UV-visible absorption measurements on these films were taken with a Perkin Elmer Lambda 950 UV-Vis spectrometer. Photoluminescence measurements were conducted with a Jasco Spectrofluorimeter FP-8500.
:PCBM and P3HT:OPCBMMB were prepared from solutions in neat o-xylene at various ratios of the semiconductors. The solutions were left overnight at 80 °C prior to deposition by doctor blade deposition on glass substrates. The depositions were at 65 °C with a blade rate of 20 mm s−1. UV-visible absorption measurements on these films were taken with a Perkin Elmer Lambda 950 UV-Vis spectrometer. Photoluminescence measurements were conducted with a Jasco Spectrofluorimeter FP-8500.
Device fabrication
Flexible ITO substrates were cleaned by sonication in acetone and isopropyl alcohol. A ZnO nanoparticle suspension (5 wt% in ethanol) was then coated onto the clean substrates and annealed at 140 °C in air. The active layer was coated from a blend solution containing various ratios of P3HT:OPCBMMB in 27 mg ml−1o-xylene (+5% 1-methylnaphthalene) solutions. Si-PCPDTBT and PCDTBT blends were coated from a 16.5 mg ml−1 (1:2) o-xylene (+5% 1-methylnaphthalene) and a 30 mg ml−1 (1:2) 1,2-dichlorobenzene, respectively. A layer of PEDOT:PSS (Heraeus) was coated from a stock solution diluted with 3 parts isopropanol and subsequent annealing in nitrogen at 140 °C for 5 min. All aforementioned depositions were carried out with a doctor blade. The devices were completed by thermal evaporation of silver under high vacuum to produce cells with 27 mm2 active area. The current voltage characteristics were measured under N2 using a Keithley 2400 source meter and a Xenon arc lamp with an intensity of approximately 100 mW cm−2.
Results and discussion
The reaction is shown in Scheme 1. The mechanism is based on the formation of a methylene radical on 1,4-bis(bromomethyl)-2,5-bis(octyloxy)benzene, via the abstraction of a bromine with CuBr, and the attack of this group at one C60 [6,6]-bond.26,27 As the radical migrates within a single phenylene ring, and as 1,2-additions are excluded due to steric effects, the second addition occurs at the opposing 1,4-phenylene carbon.27 The reaction is in effect a polycondensation forming oligo{(phenyl-C61-butyric acid methyl ester)-alt-[1,4-bis(bromomethyl)-2,5-bis(octyloxy)benzene]} (OPCBMMB) and CuBr2. | ||
| Scheme 1 Synthesis of OPCBMMB using ATRAP. | ||
Fig. 1a shows the SEC chromatogram of the representative sample OPCBMMB. The toluene marker elutes at around 43.5 mL, and PCBM at 35 mL. Fullerene does not elute from the SEC in the same manner as polystyrene standards,31 and therefore molecular weights are well underestimated. Prior work has shown that it is possible to use shoulders on the curve to give a better indication by assuming that each one is due to an incremental increase of repeating units.25,27,32 Counting off peaks and shoulders, OPCBMMB was indicated to contain oligomers with between around one and four repeating units. Assuming the same UV absorbance for PCBM as each repeating unit, deconvolution of the GPC showed OPCBMMB to contain 65% oligomers and ca. 35% PCBM. The presence of a significant PCBM fraction in OPCBMMB was further confirmed by X-ray photoelectron spectroscopy (Fig. S1, S2 and Table S1 in the ESI†).
 | ||
| Fig. 1 (a) SEC chromatogram of OPCBMMB (THF, 30 °C, UV 300 nm; toluene flow marker at 43.5 mL); (b) 1H NMR of OPCBMMB. Note peak due to water at 0.4 ppm; (c) 13C NMR of OPCBMMB; and (d) DOSY NMR of OPCBMMB. All spectra in C6D6 at ambient temperature. Note peaks due to grease (0.3 ppm), water (0.4 ppm) and acetone (1.5 ppm) in 1H and DOSY spectra picked up due to long experiment times. | ||
Fig. 1b shows the 1H NMR of OPCBMMB. Peaks associated with the PCBM alkyl-ester side-chain are easily discernable and give distinctive peaks due to their ease of relaxation. Similarly the chain-ends (–CH2Br) relax well at 4.5 ppm. Poorly relaxing protons on the methylenes next to the C60 at around 4.3 ppm indicate that they are constrained. Similarly weak relaxations from the protons at ca. 4.1 ppm in the etherated comonomer side confirm a tightly bound structure. Integration could not be accomplished, due to this high variability in proton relaxations. The 13C NMR of OPCBMMB in Fig. 1c shows the expected peaks consistent with the presence of PCBM and of the comonomers. In particular the chain-end bromomethylenes show a peak at ca. 28 ppm. However, peaks from C60–methylenes are not visible, due to their poor relaxation arising from their constrained positions. Diffusion-ordered spectroscopy (DOSY) permits identification of components in a mixture by using their diffusion coefficient.33 While far from quantitative, it can be used to elucidate the mixture compositions. The diffusion planes indicated in Fig. 1d further confirm the presence of PCBM within OPCBMMB.
Both OPCBMMB and pure PCBM and were characterized by thermogravimetric analysis (TGA). Both show initial losses at 85 °C associated with solvent trapping within a crystalline matrix.34,35 From 260 °C upwards it is noticeable that OPCBMMB loses ca. 2 wt% more than PCBM, most likely due to rupture of octyl side-chains, but otherwise the materials show similar degradation patterns (Fig. S3, ESI†). Surprisingly, the OPCBMMB is considerably more stable than the equivalent oligomer made using C60 which is known to undergo degradation at low temperatures through cleavage of C60–methylene links.29 This change in temperature is most likely due to a redistribution of thermal energy through the structure, as discussed vide infra.
DSC thermograms for OPCBMMB and PCBM were performed and the PCBM curves (Fig. S4, ESI†) were found consistent with the literature data.34 An endothermic peak at 248 °C was attributed to an evaporation of residual solvents in the PCBM crystalline structure. However, OPCBMMB shows a different curve, with a reduction in a peak arising from the melting of PCBM at 272 °C. Remarkably, the crystallization of PCBM in the OPCBMMB sample is arrested with no peak arising in either first or second cycles. OPCBMMB does, however, show a Tg at 136 °C. Although generally difficult to observe, it has been found at ca. 118 °C for pure PCBM.35 These results indicate a close and indeed intimate mixing between PCBM and OPCBMMB where the latter restrains PCBM crystallization.
The thermal stability of methylene bonds on C60 can be low but vary greatly with the size of the addend and the number of additions around the sphere.31 First principle calculations (i.e., B3LYP/6-31G*, MP2/def2-TZVPP and B3LYP/def2-TZVPP theoretical levels)36 predict a very low bond-dissociation energy range (35–50 kcal mol−1) for the fullerene-comonomer link, and ATRAP-poly(fullerene)s made from C60 in the place of PCBM can undergo methylene–C60 bond cleavage at temperatures below the annealing temperature used here (120 °C).29 As mentioned above, however, it is apparent from TGA and DSC measurements that OPCBMMB is more stable than other ATRAP-based oligomers made using C60. To understand this difference, it is necessary to consider the specific heat of the system,37 wherein the greater number of vibrators present on each repeating unit of OPCBMMB allows more heat to be absorbed, thereby delivering greater thermal stability as compared to unsubstituted C60 oligo(fullerene)s. In effect, the macromolecules are stabilized by the existence of a sum of vibrators that absorb energy thereby arresting disassociation of weak bonds and oligomer unzipping. Full details on AM1 calculations on the available vibrational states for these comparable systems are given in the ESI.†
UV-visible spectroscopic characterisations of OPCBMMB and PCBM in solution are shown in Fig. 2a. There is a clear overlap of the spectra of OPCBMMB and PCBM. This is again confirmed in the solid state spectra shown in Fig. S5 of the ESI.† While OPCBMMB contains a large proportion of PCBM which may mask many of the electronic properties of the oligomers, the similarity in these spectra indicate that additional peaks arising from asymmetric additions have not occurred. This suggest that the ATRAP attack has occurred at a 1,4-phenylene position on the C60 sphere which retains the same symmetry as the starting material.38,39 ATRAP cis attacks are inhibited by steric effects to the benefit of the equatorial and the trans positions. Given the known mechanism,26 the perfect match in the absorption spectra is thus an indication of the ATRAP substitution on PCBM occurring at a symmetrical 1,4-phenylene trans position.
 | ||
| Fig. 2 (a) Normalized UV-visible absorption spectra of OPCBMMB (red dashed) and PCBM (blue) (toluene, ambient temperature); and intermittent contact mode AFM height images (5 × 5 μm) of films of: (b) PCBM (RMS = 2.5 nm); and (c) OPCBMMB (RMS = 0.4 nm). Films prepared by blade casting from solutions in neat xylene. | ||
The AFM images of PCBM and OPCBMMB films shown in Fig. 2b and c indicate a considerable smoothing of the surface when casting the films from xylene, with the RMS decreasing from 2.5 to 0.4 nm, and the strong aggregative tendency of PCBM being hindered by OPCBMMB. It is thus apparent that OPCBMMB is of use in preparing more homogenous films than PCBM from solvents of less environmental concern. In addition, ultraviolet photoelectron spectroscopy measurements reveal that the ionization potential as a basic electronic parameter of OPCBMMB is very similar to PCBM (Fig. S6, ESI†). These points we took as an indication that initial donor acceptor intermixing, the opto-electronic and the charge-transfer/transport properties were not strongly affected by the oligomerisation process, even when the oligomers accounted for around 65% of the fullerene content in the blend.
Experiments were conducted to determine the effectiveness of this material as a replacement for PCBM as the electron acceptor in BHJ solar cells. Investigations were made with prototypical P3HT using industrially applicable o-xylene:1-methylnaphthalene (95:5). Very similar initial performances were achieved with the two acceptors as indicated in Fig. 3a (device parameters are detailed in Table S4, ESI†). It should be noted that devices were fabricated in an ambient environment onto flexible plastic substrates yielding large cell areas of 27 mm2. The use of these conditions and materials was deemed appropriate due to their direct relevance to upscaled device fabrication in spite of the their well documented influence on initial device performances.40 Furthermore, previous stabilization studies under thermal stress have almost exclusively focused on glass substrates, even though the choice of the substrate is relevant because of possible substrate-dependent morphological stability.41 Indeed, we observe a greater resistance to thermal stress in our flexible substrates as compared to equivalent devices fabricated on glass (Fig. S7, ESI†). With this in mind, an accelerated thermal degradation study at 120 °C was carried out on these blends in order to determine a possible stabilizing effect of the OPCBMMB on the BHJ morphology and PCEs. The change in PCE over the course of 100 h is shown in Fig. 3b. A fast reduction in PCE is observed in all PCBM samples at very early time scales, followed by a slower reduction at longer timescales. In contrast, incorporation of the OPCBMMB, even with the relatively crystalline P3HT, leads to a degradation profile in which the initial catastrophic drop in performance is suppressed leading to a slower overall performance roll-off. A simple estimation of the T80 values, attained by linear interpolation of the PCEs to either side of the point of interest, allows a quantitative comparison of the relative performance of the two acceptors. PCBM and OPCBMMB samples reached the quasi-T80 point after approximately 60 and over 600 min, respectively, representing an order of magnitude improvement in resistance to thermal stressing at elevated temperatures. This trend in PCE is mainly caused by the loss in JSC, consistent with previous reports for PCBM,11,42 while the VOC and FF remain relatively stable (Fig. S8, ESI†).
 | ||
| Fig. 3 (a) Representative initial J–V curves for P3HT:fullerene (1:0.8) solar cells on plastic substrates incorporating PCBM (red) and OPCBMMB (black); (b) changes in PCE as a function of annealing time at 120 °C for P3HT:PCBM (red) and P3HT:OPCBMMB (black) solar cells; (c) PL quenching (excitation at 450 nm) calculated using PLQ = 100·[1 − PLintegral(sample)/PLintegral(P3HT)], as a function of annealing time at 120 °C for films of P3HT:PCBM (red) and P3HT:OPCBMMB (black); (d) and (e) optical micrographs of corresponding P3HT:PCBM and P3HT:OPCBMMB films, respectively, taken by transmission through spaces between the electrodes of devices on plastic substrates after 100 h of annealing at 120 °C. Scale bars represent 50 μm. | ||
PL quenching yields, defined as the proportional loss of photoluminescence of a system upon addition of a quencher material, can be used as a simple yet powerful experimental tool for investigating the photophysical compatibility and effective mixing of donor and acceptor materials.43–47 Here, measurements were conducted on P3HT:PCBM and P3HT:OPCBMMB films as a function of annealing time to monitor morphologically driven changes in the BHJ photophysics and by inference charge generation efficiency under thermal stress (Fig. 3c). Initial PL quenching yields are in excess of 90% in blends comprising either acceptor, suggesting a well dispersed distribution of the accepting (quenching) species throughout the P3HT matrix in both cases. After thermal stressing at short timescales, a stark reduction in quenching efficiency is observed in PCBM containing blends, while those with OPCBMMB maintain a high level of quenching. As quenching results from the electron transfer process, requiring P3HT and PCBM to be in close proximity, this indicates a rapid migration of PCBM molecules away from regions rich in P3HT and into large aggregates, as further supported by an observed increase in scattering contribution to extinction measurements (Fig. S9, ESI†). Consequently, the volume of purer P3HT (and PCBM) domains increases. Not only are these domains more emissive, they no longer contribute as substantially to the generation of useful current, in perfect agreement with the observed losses in photocurrent and overall solar cell performance. Furthermore, optical micrographs of samples, taken after 100 h of annealing, reveal the concurrent changes in morphology (Fig. 3d and e). Numerous aggregates are observed in the case of both acceptors. While PCBM-based samples produce a greater aggregated surface overall, the aggregates are smaller than those arising in OPCBMMB-based devices. Thus, it would appear that the balance between nucleation and growth, in the evolution of the blend morphology, has been shifted in favour of the growth mechanism, comparable with the observations of Schroeder et al.18b Although it might be intuitively expected that large aggregates compromise the integrity of the layers, it is interesting to note that here no correlation between aggregate size and thermal stability is observed. It may therefore be suggested that the growth of the aggregates themselves are not responsible for the degradation of device performance, but are rather a byproduct of demixing of the BHJ which is itself far more impactful, in concordance with work elsewhere.48
In order to test the generality of the stabilization phenomenon, PCBM and OPCBMMB devices were fabricated using other well-known polymers and subjected to thermally induced degradation. Again, devices were built on plastic substrates. The effects of accelerated thermal ageing, at 120 °C in an inert atmosphere, on the efficiencies of devices incorporating the exemplar polymers Si-PCPDTBT, and PCDTBT are shown in Fig. 4a and b, respectively. These polymers were chosen due to the stark differences in their crystallinity; PCDTBT tending to form amorphous films and Si-PCPDTBT more readily forming crystalline films, a factor that is expected to play a significant role in morphological evolution under thermal stress.49 In both cases, the change in PCE over time strongly correlates with the change in JSC (Fig. S10 and S11, ESI†) and a clear stabilising effect with the use of OPCBMMB is observed. In all Si-PCPDTBT devices a rapid initial decay is observed, having a substantial impact on device performance, followed by a relatively slow loss process representing two distinct degradation phases. In the presence of OPCBMMB the rate of the rapid decay process is reduced; the quasi-T80 point being reached after 284 min of annealing, as opposed to 54 min reached with PCBM. This represents an approximately fivefold increase in stability of the fast loss processes. Conversely, the slow phase degradation behavior exhibited differs markedly. Devices incorporating the OPCBMMB appear even more strongly stabilized against the effects of annealing, with only a negligible loss of a further 10% at 75 h, while those containing PCBM lost a further 40%.
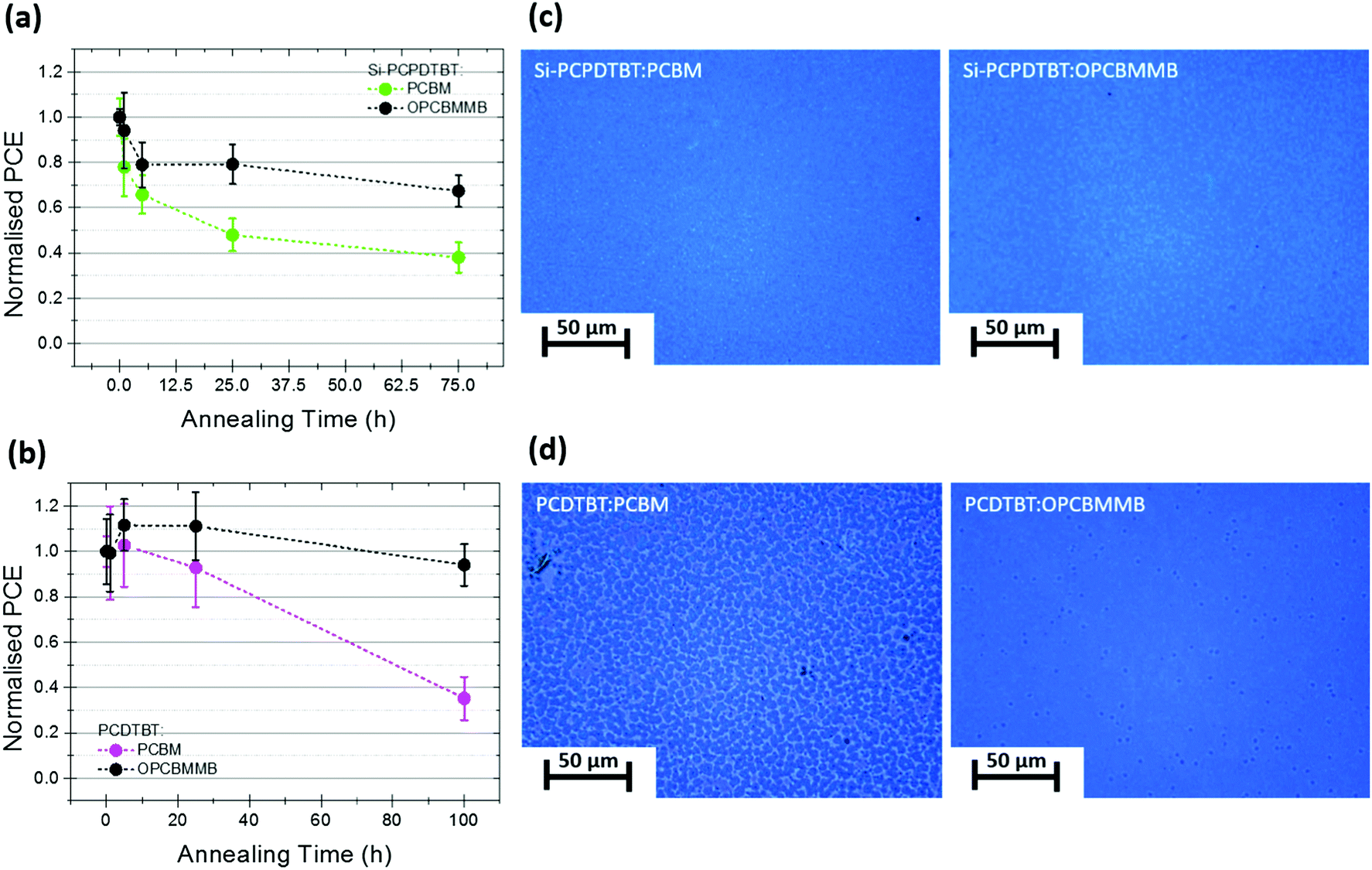 | ||
| Fig. 4 (a and b) Changes in PCE as a function of annealing time at 120 °C for donor:PCBM (green or purple) and donor:OPCBMMB (black) solar cells corresponding to Si-PCPDTBT and PCDTBT, respectively, on plastic substrates. (c) Optical micrographs of corresponding Si-PCPDTBT:PCBM and Si-PCPDTBT:OPCBMMB devices taken by transmission through spaces between the electrodes of the devices on the substrate after 75 h of annealing at 120 °C. (d) Optical micrographs of corresponding PCDTBT:PCBM and PCDTBT:OPCBMMB devices taken by transmission through spaces between the electrodes of the devices on the substrate after 100 h of annealing at 120 °C. The scale bars represent 50 μm. | ||
In the case of devices with PCDTBT, the degradation kinetics appear much slower and a relatively long lasting increase in performance was observed, consistent with other reports on the use of ZnO electron selective layers.41 The relatively impressive stability displayed with PCBM (when compared with the other polymer systems) was again substantially improved upon with OPCBMMB, with less than 10% of the initial device efficiency being lost after 100 hours of thermal stressing (Fig. 4b). The quasi-T80 point was estimated by extrapolation to be reached at 2490 min in the PCBM-based control samples while, in contrast, samples containing OPCBMMB were predicted to last for 8210 min, yielding an approximate tripling of the device thermal stability (at the elevated temperatures).
Optical microscopy was conducted on the degraded samples in order to visualize the extent of aggregation within the devices incorporating the different polymers. The corresponding photomicrographs in Fig. 4c and d clearly show differences in the formation of features in the case of each polymer; aggregation only being clearly visible in the case of PCDTBT:PCBM. Recalling that aggregation was clearly observed within all P3HT:fullerene blends, the apparent differences observed between each polymer system imply strongly that the dominant processes at work are entirely system specific. Recent works in the literature have demonstrated that the rate of fullerene aggregation is strongly dependent on its diffusivity and miscibility as well as the extent of photo-induced dimerisation between fullerenes within a polymer matrix.14,16,17,50,51 These properties are heavily influenced by polymer backbone and side-chain structure and the amorphous/void space fractions of the thin films through which small molecules may more easily traverse.17,52 Therefore, it should follow that acceptor migration and aggregation within polymers containing distinctly different backbones and side-chains, and featuring differences in crystallinity and glass transition temperatures should exhibit dissimilar behavior with respect to morphological development. While differences in the observed degradation kinetics between different polymer-fullerene blends are expected, importantly, across all the systems investigated, the incorporation of OPCBMMB consistently afforded additional thermal stability independent of the polymer donor utilized.
Conclusion
The facile ATRAP reaction selectively results in trans-additions to the PCBM fullerene moiety, yielding symmetrical products with opto-electronic properties extraordinarily similar to PCBM. The molecular weights are low, perhaps due to steric hindrance of the reaction, and this has resulted in a material with a high proportion of PCBM.OPCBMMB has been shown to be an effective replacement for PCBM in devices based on both highly crystalline and amorphous donor polymers, giving similar device performances with enhanced thermal stabilities while retaining PCBM's optoelectronic and processing behavior. Future work will consider the formation of higher molecular weight materials excluding mixtures with PCBM, and on the elucidation of the stabilization mechanism in bulk heterojunctions.
Acknowledgements
The research leading to these results has received funding from the European Union Seventh Framework Program (FP7/2011) under grant agreement ESTABLIS no. 290022 and from the Region Aquitaine under agreement 2011 FULLINC. BAB acknowledges funding from FAPESP (2011/02205-3) and CAPES (BEX 11216-12-3). MS acknowledges primary support from a fellowship by the Portuguese Fundação para a Ciência e a Tecnologia (SFRH/BPD/71816/2010). CJB and AO gratefully acknowledge the Cluster of Excellence ‘Engineering of Advanced Materials at the University of Erlangen-Nürnberg’, which is funded by the German Research Foundation (DFG). Dr M. Pédeutour is warmly thanked for her administrative support.References
- J. Hummelen, B. Knight, F. LePeq, F. Wudl and J. Yao, J. Org. Chem., 1995, 60, 532 CrossRef CAS.
- G. Yu, J. Gao, J. C. Hummelen, F. Wudl and A. J. Heeger, Science, 1995, 270, 1789 CAS.
- S. Gunes, H. Neugebauer and N. Sariciftci, Chem. Rev., 2007, 107, 1324 CrossRef PubMed.
- (a) M. T. Dang, L. Hirsch and G. Wantz, Adv. Mater., 2011, 23, 3597 CrossRef CAS PubMed; (b) R. C. Hiorns, R. De Bettignies, J. Leroy, S. Bailly, M. Firon, C. Sentein, A. Khoukh, H. Preudhomme and C. Dagron-Lartigau, Adv. Funct. Mater., 2006, 16, 2263 CrossRef CAS.
- (a) M. C. Scharber and N. S. Sariciftci, Prog. Polym. Sci., 2013, 38, 1929 CrossRef CAS PubMed; (b) G. Dennler, M. C. Scharber, T. Ameri, P. Denk, K. Forberich, C. Waldauf and C. J. Brabec, Adv. Mater., 2008, 20, 579 CrossRef CAS; (c) M. C. Scharber, D. Mühlbacher, M. Koppe, P. Denk, C. Waldauf, A. J. Heeger and C. J. Brabec, Adv. Mater., 2006, 18, 789 CrossRef CAS.
- J. Nelson, Mater. Today, 2011, 14, 462 CrossRef CAS.
- J. W. Ryan and Y. Matsuo, Sci. Rep., 2015, 5, 8319 CrossRef CAS PubMed.
- (a) F. Guo, H. Azimi, Y. Hou, T. Przybilla, M. Hu, C. Bronnbauer, S. Langner, E. Spiecker, K. Forberich and C. J. Brabec, Nanoscale, 2015, 7, 1642 RSC; (b) C. Tao, S. Neutzner, L. Colella, S. Marras, A. R. Srimath Kandada, M. Gandini, M. De Bastiani, G. Pace, L. Manna, M. Caironi, C. Bertarelli and A. Petrozza, Energy Environ. Sci., 2015, 8, 2365 RSC.
- D. Chen, A. Nakahara, D. Wei, D. Nordlund and T. P. Russell, Nano Lett., 2011, 11, 561 CrossRef CAS PubMed.
- X. Yang, J. Loos, S. Veenstra, W. Verhees, M. M. Wienk and J. M. Kroon, Nano Lett., 2005, 5, 579 CrossRef CAS PubMed.
- M. Campoy-Quiles, T. Ferenczi, T. Agostinelli, P. G. Etchegoin, Y. Kim, T. D. Anthopoulos, P. N. Stavrinou, D. D. C. Bradley and J. Nelson, Nat. Mater., 2008, 7, 158 CrossRef CAS PubMed.
- S. Mauger, L. Chang, S. Friedrich, C. Rochester, D. Huang, P. Wang and A. Moulé, Adv. Funct. Mater., 2013, 23, 1935 CrossRef CAS.
- A. Distler, T. Sauermann, H.-J. Egelhaaf, S. Rodman, D. Waller, K.-S. Cheon, M. Lee and D. M. Guildi, Adv. Energy Mater., 2014, 4, 1300693 CrossRef.
- G. E. Morse, A. Tournebize, A. Rivaton, T. Chassé, C. Taviot-Gueho, N. Blouin, O. R. Lozman and S. Tierney, Phys. Chem. Chem. Phys., 2015, 17, 11884 RSC.
- Z. Li, H. C. Wong, Z. Huang, H. Zhong, C. H. Tan, W. C. Tsoi, J. S. Kim, J. R. Durrant and J. T. Cabral, Nat. Commun., 2013, 4, 2227 Search PubMed.
- F. Piersimoni, G. Degutis, S. Bertho, K. Vandewal, D. Spoltore, T. Vangerven, J. Drijkoningen, M. K. Van Bael, A. Hardy, J. D'Haen, W. Maes, D. Vanderzande, M. Nesladek and J. Manca, J. Polym. Sci., Part B: Polym. Phys., 2013, 51, 1209 CrossRef CAS.
- T. Heumueller, W. R. Mateker, A. Distler, U. F. Fritze, R. Cheacharoen, W. H. Nguyen, M. Biele, M. Salvador, M. von Delius, H.-J. Egelhaaf, M. D. McGehee and C. J. Brabec, Energy Environ. Sci., 2016, 9, 247 CAS.
- (a) C. Lindqvist, J. Bergqvist, C.-C. Feng, S. Gustafsson, O. Bäcke, N. D. Treat, C. Bounioux, P. Henriksson, R. Kroon, E. Wang, A. Sanz-Velasco, P. M. Kristiansen, N. Stingelin, E. Olsson, O. Inganäs, M. R. Andersson and C. Müller, Adv. Energy Mater., 2014, 4, 1301437 CrossRef; (b) B. C. Schroeder, Z. Li, M. A. Brady, G. C. Faria, R. S. Ashraf, C. J. Takacs, J. S. Cowart, D. T. Duong, K. H. Chiu, C.-H. Tan, J. T. Cabral, A. Salleo, M. L. Chabinyc, J. R. Durrant and I. McCulloch, Angew. Chem., Int. Ed., 2014, 53, 12870 CrossRef CAS PubMed; (c) C. Lindqvist, J. Bergqvist, O. Büacke, S. Gustafsson, E. Wang, E. Olsson, O. Inganäs, M. R. Andersson and C. Müller, Appl. Phys. Lett., 2014, 104, 153301 CrossRef.
- (a) C. Lindqvist, A. Sanz-Velasco, E. Wang, O. Bäcke, S. Gustafsson, E. Olsson, M. R. Andersson and C. Müller, J. Mater. Chem. A, 2013, 1, 7174 RSC; (b) J. J. Richards, A. H. Rice, R. D. Nelson, F. S. Kim, S. A. Jenekhe, C. K. Luscombe and D. C. Pozzo, Adv. Funct. Mater., 2013, 23, 514 CrossRef CAS.
- (a) F. Giacalone and N. Martín, Chem. Rev., 2006, 106, 5136 CrossRef CAS PubMed; (b) Main-Chain and Side-Chain C60-Polymers, in Fullerene Polymers. Synthesis, Properties and Applications, ed. N. Martín and F. Giacalone, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2009 Search PubMed.
- B. Gholamkhass, T. J. Peckham and S. Holdcroft, Polym. Chem., 2010, 1, 708 RSC.
- (a) J. W. Rumer and I. McCulloch, Mater. Today, 2015, 18, 425 CrossRef CAS; (b) J. U. Lee, W. Jung, J. W. Jung and W. H. Jo, J. Mater. Chem., 2012, 22, 24265 RSC; (c) L. Derue, O. Dautel, A. Tournebize, M. Drees, H. Pan, S. Berthumeyrie, B. Pavageau, E. Cloutet, S. Chambon, L. Hirsch, A. Rivaton, P. Hudhomme, A. Facchetti and G. Wantz, Adv. Mater., 2014, 26, 5831 CrossRef CAS PubMed; (d) A. Tournebize, A. Rivaton, J.-L. Gardette, C. Lombard, B. Pépin-Donat, S. Beaupré and M. Leclerc, Adv. Energy Mater., 2014, 4, 1301530 CrossRef.
- (a) M. Raïssi, H. Erothu, E. Ibarboure, H. Cramail, L. Vignau, E. Cloutet and R. C. Hiorns, J. Mater. Chem. A, 2015, 3, 18207 RSC; (b) P. D. Topham, A. J. Parnell and R. C. Hiorns, Block Copolymer Strategies for Solar Cells, J. Polym. Sci., Part B: Polym. Phys., 2011, 49, 1131 CrossRef CAS.
- A. Gügel, P. Belik, M. Walter, A. Kraus, E. Harth, M. Wagner, J. Spickermann and K. Müllen, Tetrahedron, 1996, 52, 5007 CrossRef.
- (a) S. Shi, K. C. Khemani, Q. C. Li and F. Wudl, J. Am. Chem. Soc., 1992, 114, 10656 CrossRef CAS; (b) H. Ito, Y. Ishida and K. Saigo, Tetrahedron Lett., 2006, 47, 3095 CrossRef CAS.
- R. C. Hiorns, E. Cloutet, E. Ibarboure, L. Vignau, N. Lemaître, S. Guillerez, C. Absalon and H. Cramail, Macromolecules, 2009, 42, 3549 CrossRef CAS.
- R. C. Hiorns, E. Cloutet, E. Ibarboure, A. Khoukh, H. Bejbouji, L. Vignau and H. Cramail, Macromolecules, 2010, 43, 6033 CrossRef CAS.
- O. F. Pozdnyakov, A. O. Pozdnyakov, B. Schmaltz and C. Mathis, Polymer, 2006, 47, 1028 CrossRef CAS.
- H. Santos Silva, PhD Thesis, Synthesis and Modelling of Polyfullerenes, Université de Pau et Pays de l'Adour, and Universität Tübingen, 2015.
- (a) M. Urien, G. Wantz, E. Cloutet, L. Hirsch, P. Tardy, L. Vignau, H. Cramail and J.-P. Parneix, Org. Electron., 2007, 8, 727 CrossRef CAS; (b) A. Dupuis, P. Wong-Wah-Chung, A. Rivaton and J.-L. Gardette, Polym. Degrad. Stab., 2012, 97, 366 CrossRef CAS.
- F. Audouin, R. Nuffer and C. Mathis, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 3456 CrossRef CAS.
- (a) H. H. Ramanitra, H. Santos Silva, B. A. Bregadiolli, A. Khoukh, C. M. S. Combe, S. A. Dowland, D. Bégué, C. F. O. Graeff, C. Dagron-Lartigau, A. Distler, G. Morse and R. C. Hiorns, Macromolecules, 2016, 49, 1681 CrossRef CAS; (b) M. Stephen, H. H. Ramanitra, H. Santos Silva, S. A. Dowland, D. Bégué, K. Genevičius, K. Arlauskas, G. Juška, G. E. Morse, A. Distler and R. C. Hiorns, Chem. Commun., 2016, 52, 6107 RSC.
- C. S. Johnson Jr., Prog. Nucl. Magn. Reson. Spectrosc., 1999, 34, 203 CrossRef.
- T. T. Ngo, D. N. Nguyen and V. T. Nguyen, Adv. Nat. Sci.: Nanosci. Nanotechnol., 2012, 3, 045001 CrossRef.
- N. T. Tung, H. Lee, Y. Song, N. D. Nghia and D. Sohn, Synth. Met., 2010, 160, 1303 CrossRef CAS.
- F. Neese, The ORCA program system, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73 CrossRef CAS.
- B. Wunderlich and M. Dole, J. Polym. Sci., 1957, 24, 210 CrossRef.
- M. Schnell, ChemPhysChem, 2010, 11, 758 CrossRef CAS PubMed.
- J. Liu, X. Guo, Y. Qin, S. Liang, Z.-X. Guo and Y. Li, J. Mater. Chem., 2012, 22, 1758 RSC.
- (a) H. K. Nguyen, M. Larbadi, S. Capaccioli, M. Lucchesi, P. Rolla and D. Prevosto, Macromolecules, 2013, 46, 555 CrossRef CAS; (b) L. Lucera, P. Kubis, F. W. Fecher, C. Bronnbauer, M. Turbiez, K. Forberich, T. Ameri, H.-J. Egelhaaf and C. J. Brabec, Energy Technol., 2015, 3, 373 CrossRef; (c) F. Machui, L. Lucera, G. D. Spyropoulos, J. Cordero, A. S. Ali, P. Kubis, T. Ameri, M. M. Voigt and C. J. Brabec, Sol. Energy Mater. Sol. Cells, 2014, 128, 441 CrossRef CAS; (d) L. Lucera, F. Machui, P. Kubis, H. D. Schmidt, J. Admas, S. Strohm, T. Ahmad, K. Forberich, H.-J. Egelhaaf and C. J. Brabec, Energy Environ. Sci., 2016, 9, 89 RSC.
- Z. Li, K. H. Chiu, R. S. Ashraf, S. Fearn, R. Dattani, H. C. Wong, C.-H. Tan, J. Wu, J. T. Cabral and J. R. Durrant, Sci. Rep., 2015, 5, 15149 CrossRef CAS PubMed.
- M. T. Dang, L. Hirsch, G. Wantz and J. D. Wuest, Chem. Rev., 2013, 113, 3734 CrossRef CAS PubMed.
- P. E. Schwenn, K. Gui, A. M. Nardes, K. B. Krueger, K. H. Lee, K. Mutkins, H. Rubinstein-Dunlop, P. E. Shaw, N. Kopidakis, P. L. Burn and P. Meredith, Adv. Energy Mater., 2011, 1, 73 CrossRef CAS.
- D. Baran, N. Li, A.-C. Breton, A. Osvet, T. Ameri, M. Leclerc and C. J. Brabec, J. Am. Chem. Soc., 2014, 136, 10949 CrossRef CAS PubMed.
- N. Gasparini, M. Salvador, S. Fladischer, A. Katsouras, A. Avgeropoulos, E. Spiecker, C. L. Chochos, C. J. Brabec and T. Ameri, Adv. Energy Mater., 2015 DOI:10.1002/aenm.201501527.
- A. J. Ward, A. Ruseckas, M. M. Kareem, B. Ebenhoch, L. A. Serrano, M. Al-Eid, B. Fitzpatrick, V. M. Rotello, G. Cooke and I. D. W. Samuel, Adv. Mater., 2015, 27, 2496 CrossRef CAS PubMed.
- G. J. Hedley, A. J. Ward, A. Alekseev, C. T. Howells, E. R. Martins, L. A. Serrano, G. Cooke, A. Ruseckas and I. D. W. Samuel, Nat. Commun., 2013, 4, 2867 Search PubMed.
- J. A. Bartelt, Z. M. Beiley, E. T. Hoke, W. R. Mateker, J. D. Douglas, B. A. Collins, J. R. Tumbleston, K. R. Graham, A. Amassian, H. Ade, J. M. J. Fréchet, M. F. Toney and M. D. McGehee, Adv. Energy Mater., 2013, 3, 364, DOI:10.1002/aenm.201200637.
- (a) A. J. Pearson, T. Wang, A. D. F. Dunbar, H. Yi, D. C. Watters, D. M. Coles, P. A. Staniec, A. Iraqi, R. A. L. Jones and D. G. Lidzey, Adv. Funct. Mater., 2014, 24, 659 CrossRef CAS; (b) R. Lin, M. Wright, B. Puthen-Veettil, X. Wen, M. J. Y. Tayebjee and A. Uddin, Phys. Status Solidi A, 2015, 212, 1931 CrossRef CAS.
- Z. Li, K. H. Chiu, R. S. Ashraf, S. Fearn, R. Dattani, H. C. Wong, C.-H. Tan, J. Wu, J. T. Cabral and J. R. Durrant, Sci. Rep., 2015, 5, 15149 CrossRef CAS PubMed.
- H. C. Wong, Z. Li, C. H. Tan, H. Zhong, Z. Huang, H. Bronstein, I. McCulloch, J. T. Cabral and J. R. Durrant, ACS Nano, 2014, 8, 1297 CrossRef CAS PubMed.
- L. H. Nguyen, H. Hoppe, T. Erb, S. Günes, G. Gobsch and N. S. Sariciftci, Adv. Funct. Mater., 2007, 17, 1071 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Equipment details, XPS results, TGA and DSC thermograms, AM1 level calculations and data, UV solid state spectra, and organic photovoltaic device data. See DOI: 10.1039/c6tc03290g |
| This journal is © The Royal Society of Chemistry 2016 |