Diacenopentalene dicarboximides as new n-type organic semiconductors for field-effect transistors†
Received
23rd June 2016
, Accepted 23rd August 2016
First published on 24th August 2016
Abstract
N,N′-Dihexyl-dibenzopentalene dicarboximide (DBPDI) and N,N′-dioctyl-dinaphthopentalene dicarboximide (DNPDI) were successfully synthesized as new n-type semiconducting materials. They have good solubility in common organic solvents. DBPDI and DNPDI are thermally stable, with decomposition temperatures at 419 and 460 °C, respectively, based on thermogravimetric analysis, and melt at 354 and 387 °C as measured by differential scanning calorimetry. Their optical and electrochemical properties were studied by UV-vis absorption and cyclic voltammetry measurements. Time-dependent density functional theory calculations (TDDFT) were used to explain their unique electronic absorption spectra. DBPDI and DNPDI have relatively low-lying LUMO energy levels at −3.76 eV and −3.45 eV and HOMO energy levels at −5.84 eV and −5.72 eV, respectively. Their application in organic field-effect transistors (FETs) was investigated. Both DBPDI and DNPDI showed n-type field-effect transistor behavior. The DBPDI device obtained by solution-processing technique displayed an average electron mobility of up to 0.06 cm2 V−1 s−1 with an Ion/Ioff ratio of 2.9 × 106.
Introduction
In the last decade, interest in the synthesis of cyclic π-conjugated systems incorporated with an antiaromatic unit has increased. Pentalene, as one of the important antiaromatic systems, has attracted much attention.1 Parent pentalene, with its 8π electron framework, is unstable and easily dimerizes above −196 °C. Dibenzopentalene (DBP) has been synthesized by different methods, and it is a fairly stable compound with a planar structure and 4nπ electron periphery.2 Incorporation of the antiaromatic pentalene unit into the acene framework is expected to be an efficient approach to stabilizing the acene moieties due to the intramolecular donor–acceptor interactions. As expected, the extended dinaphthopentalene (DNP)2o,q,3 and dianthracenopentalene (DAP)4 derivatives were reported, and they can be used as p-type organic semiconductors with good stability. Other pentalene-containing molecules have also been studied for their applications in organic electronics.5 The pentalene unit can be regarded as a weak electron-withdrawing moiety that can stabilize the electron-rich moieties such as acenes but cannot convert them into n-type materials such as DNP,3 DAP,4 DBP-based polymers,5a pyrrole-fused DBP,5c and thienoacene-fused pentalenes,5d which all show hole transport properties. To prepare n-type pentalene derivatives, other electron-deficient groups need to be incorporated. For example, dibenzo[a,e]pentalene-embedded thienoquinoidals with dicyanomethylene termini were prepared in 2015 by Takimiya's group.5b They have low-lying LUMO energy levels (−4.2 to −4.3 eV) and showed n-type field-effect transistor (FET) behaviour. Under ambient conditions, the devices produced by solution-processing technique showed electron mobility of up to 4.5 × 10−2 cm2 V−1 s−1. Besides this work, to the best of our knowledge, no more n-type pentalene-based materials have been reported.
Generally, to obtain the electron-transporting materials, the electron-deficient imide group is used to lower the energy level and stabilize the radical anions. Therefore, aromatic diimides such as pyromellitic diimide,6 naphthalene diimides,7 perylene diimides,8 longer acene diimide9–12 and large ovalene diimide13 were reported to exhibit relatively low energy levels, high electron mobilities, and excellent chemical and thermal stabilities. Following the above design strategies, we expect that incorporation of the imide groups into the diacenopentalene framework would result in new electron-transporting materials with low-lying LUMO energy levels. In this context, two diacenopentalene dicarboximides (DBPDI and DNPDI, Scheme 1) were synthesized, and their performance in organic field-effect transistors (OFETs) was evaluated. Both compounds showed n-type FET behaviour with moderate electron mobilities.
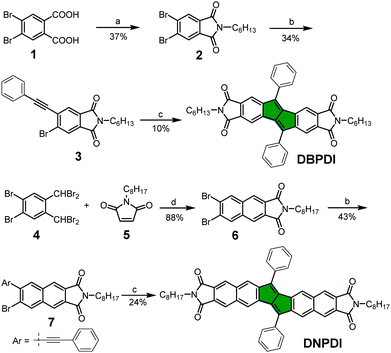 |
| | Scheme 1 Reagents and conditions: (a) 1. SOCl2, DCM; 2. C6H13NH2, AcOH, reflux, 12 h; (b) phenylacetylene, PdCl2(PPh3)2, CuI, Et3N, THF, 75 °C; (c) Pd2(dba)3, P(2-furyl)3, Cs2CO3, CsF, hydroquinone, 1,4-dioxane, 135 °C; (d) NaI, DMF, 80 °C, 12 h; DCM: dichloromethane, THF: tetrahydrofuran, dba: dibenzylideneacetone, DMF: N,N-dimethylformamide. | |
Results and discussion
Synthesis
The synthetic route of compounds DBPDI and DNPDI is shown in Scheme 1. Bromination of o-xylene and subsequent oxidation of the as-formed 1,2-dibromo-4,5-xylene gave the 4,5-dibromophthalic acid 1.14 Compound 1 was treated with SOCl2 to give the corresponding acyl chloride. After removing the excess SOCl2, the acyl chloride reacted with hexylamine (C6H13NH2) in acetic acid under reflux for 12 h to produce the imide 2 in 37% yield. In the following step, compound 2 was reacted with phenylacetylene through Sonogashira cross-coupling reaction to give the mono-substituted intermediate 3 in 34% yield. Eventually, palladium catalysed the cyclodimerization of 3, providing the target molecule DBPDI in 10% yield. DNPDI was prepared by starting from 1,2-dibromo-4,5-bis(dibromomethyl)-benzene 415 and 1-octyl-1H-pyrrole-2,5-dione 5.16 Compounds 4 and 5 underwent a modified Diels–Alder addition reaction to obtain the intermediate 6 in 88% yield.9b Sonogashira cross-coupling reaction between 6 and phenylacetylene gave the mono-substituted compound 7 in 43% yield, and then similar palladium-catalysed cyclodimerization of 7 produced DNPDI in 24% yield. Both DBPDI and DNPDI showed good stability and solubility in normal organic solvents (e.g., chloroform, tetrahydrofuran), and their structures were confirmed by NMR and mass spectrometry (see ESI†).
Optical properties
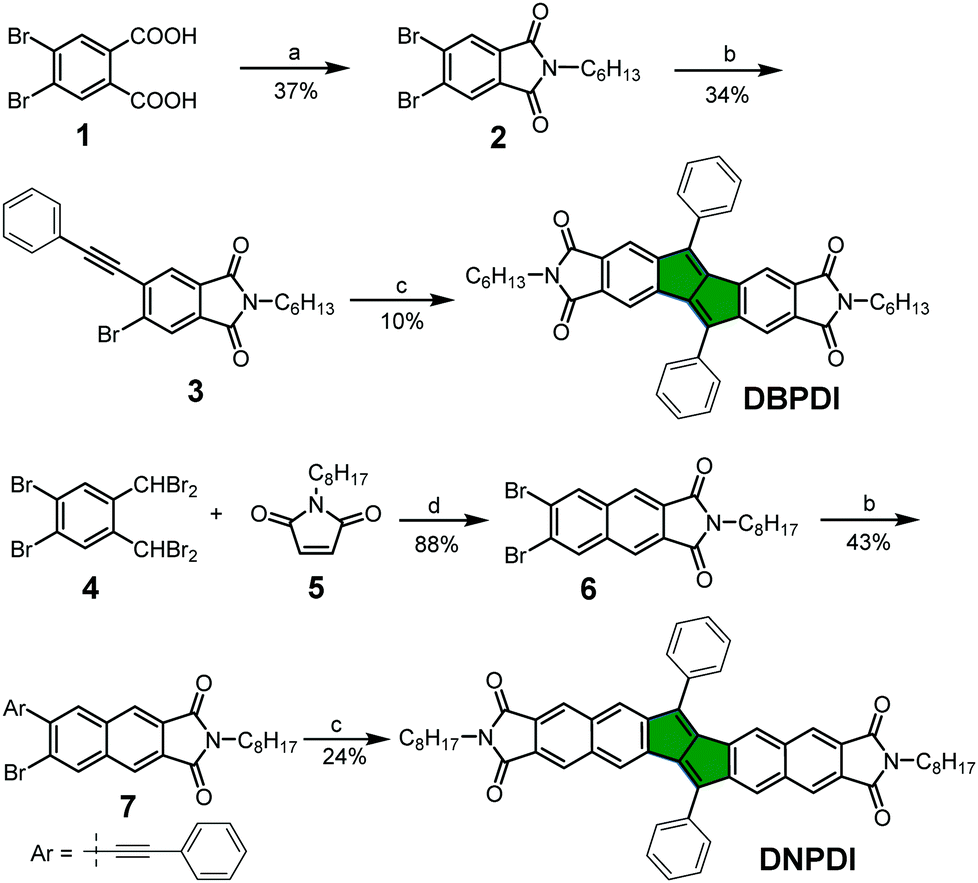
The UV-vis absorption spectra of DBPDI and DNPDI were measured in chloroform solution. The spectra are shown in Fig. 1a, and the relevant data are summarized in Table 1. Two broad absorption bands located in the visible region were observed for both compounds. The maximum absorption wavelengths of DBPDI and DNPDI in the high-energy region are located at 296 and 328 nm, with molar absorption coefficients of 93![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 and 88600 M−1 cm−1, respectively. Time-dependent density functional theory (TDDFT) calculations (B3LYP/6-31G*; see Fig. S1, S2 and Tables S1, S2 in ESI†) indicate that this band originates from a combination of multiple HOMO−n → LUMO+m transitions. The bands in the low-energy region with maxima at 485 and 518 nm are observed for DBPDI and DNPDI, respectively, and they mainly come from the HOMO−1 → LUMO transition (DBPDI: λ = 488.2 nm, oscillator strength f = 0.3379) or HOMO → LUMO transition (DNPDI: λ = 512.3 nm, f = 0.5057). The optical energy band gap (EOptg) of 2.42 and 2.25 eV can be calculated based on the lowest energy of absorption edge in solution (see Table 1). Compared to DBPDI, the absorption maximum of DNPDI in solution is red-shifted 33 nm due to its more extended π-conjugation.
500 and 88600 M−1 cm−1, respectively. Time-dependent density functional theory (TDDFT) calculations (B3LYP/6-31G*; see Fig. S1, S2 and Tables S1, S2 in ESI†) indicate that this band originates from a combination of multiple HOMO−n → LUMO+m transitions. The bands in the low-energy region with maxima at 485 and 518 nm are observed for DBPDI and DNPDI, respectively, and they mainly come from the HOMO−1 → LUMO transition (DBPDI: λ = 488.2 nm, oscillator strength f = 0.3379) or HOMO → LUMO transition (DNPDI: λ = 512.3 nm, f = 0.5057). The optical energy band gap (EOptg) of 2.42 and 2.25 eV can be calculated based on the lowest energy of absorption edge in solution (see Table 1). Compared to DBPDI, the absorption maximum of DNPDI in solution is red-shifted 33 nm due to its more extended π-conjugation.
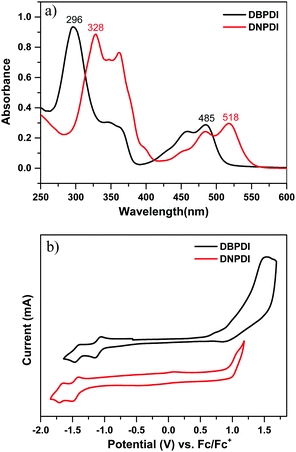 |
| | Fig. 1 (a) UV-vis absorption spectra of DBPDI and DNPDI in chloroform (10−5 M); (b) cyclic voltammograms (CV) of DBPDI and DNPDI measured in dry dichloromethane. | |
Table 1 Summary of photophysical properties and electrochemical data of DBPDI and DNPDI
| |
λ
max (abs) [nm] in solution |
logεmax [M−1 cm−1] |
E
ox1/2 (V) |
E
red1/2 (V) |
HOMO [eV] |
LUMO [eV] |
E
Optg [eV] |
|
DBPDI
|
296 |
4.97 |
1.36 |
−1.46; −1.11 |
−5.84 |
−3.76 |
2.42 |
|
DNPDI
|
328 |
4.95 |
1.03 |
−1.71; −1.47 |
−5.72 |
−3.45 |
2.25 |
Electrochemical properties
Cyclic voltammetry (CV) and differential pulse voltammetry (DPV) were used to study the electrochemical properties of compounds DBPDI and DNPDI (Fig. 1b, Table 1 and Fig. S3 in ESI†). The potential was externally calibrated against the ferrocene/ferrocenium couple. Compound DBPDI shows one oxidation wave with half-wave potential (Eox1/2) of 1.36 V and two reversible reduction waves with half-wave potentials Ered1/2 at −1.46 and −1.11 V. Meanwhile, one oxidation wave with Eox1/2 at 1.03 V and two reversible reduction waves with Ered1/2 at −1.71 and −1.47 V were observed for molecule DNPDI. The HOMO and LUMO energy levels were calculated using the following equations: HOMO = −[Eonsetox + 4.8] eV, LUMO = −[Eonsetred + 4.8] eV, where Eonsetox and Eonsetred are the onset of the first oxidation and reduction wave, respectively.17 The HOMO/LUMO energy levels are calculated to be −5.84/−3.76 eV for DBPDI and −5.72/−3.45 eV for DNPDI. The corresponding electrochemical energy gaps EECg (LUMO–HOMO) are estimated to be 2.08 eV and 2.27 eV for DBPDI and DNPDI, which are close to their optical band gaps EOptg.
DFT calculations
DFT calculations were conducted to better understand the electronic properties of DBPDI and DNPDI (Fig. 2 and Fig. S1, S2 and Tables S1, S2 in ESI†). The coefficients for HOMO of DBPDI and HOMO−1 of DNPDI are distributed along the diacenopentalene framework, while the phenyl rings are nearly nonconjugated to the backbone. The HOMO−1 of DBPDI and HOMO of DNPDI coefficients are mainly distributed along the zigzag edges of diacenopentane including the phenyl rings. The LUMO coefficient is mainly located on the central pentalene unit and the imide moieties. The HOMO/LUMO energy levels were calculated to be −6.05/−3.31 eV for DBPDI and −5.69/−2.98 eV for DNPDI. It is worth noting that the HOMO and HOMO−1 (−6.15 eV) energy levels of DBPDI are quite close. TDDFT calculations predicted the longest wavelength for HOMO−1 → LUMO transition at 488.2 nm for DBPDI and for HOMO → LUMO transition at 512.3 nm for DNPDI (Fig. S1 and S2 in the ESI†), which is consistent with the experimental data.
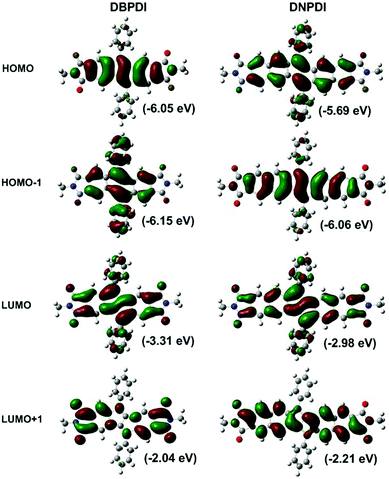 |
| | Fig. 2 The calculated HOMO/LUMO profiles and energy levels of DBPDI and DNPDI (B3LYP/6-31G*). | |
Thermal properties
Thermal stability is one of the key requirements for the practical application of organic electronic materials. Compounds DBPDI and DNPDI showed good thermal stability, with decomposition temperatures (Td, corresponding to a 5% weight loss in thermogravimetric analysis (TGA) curves) at 419 and 460 °C, respectively (Fig. S4a and c, ESI†). The melting points of DBPDI and DNPDI were observed at 354 and 387 °C, and the corresponding crystallization temperatures were observed at 337 and 369 °C, respectively, from differential scanning calorimetry (DSC) curves (Fig. S4b and d, ESI†).
Organic field effect transistors
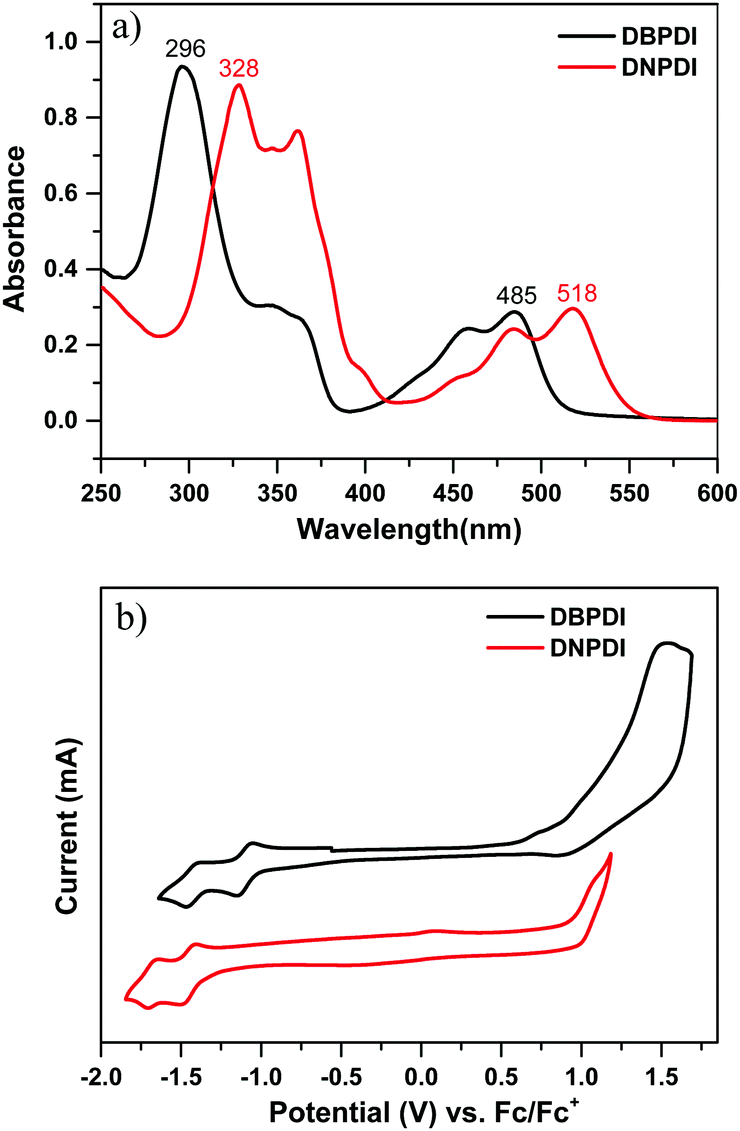
To probe the charge transport properties of compounds DBPDI and DNPDI, we fabricated field-effect transistor (FET) devices for both compounds by solution-processing method. The bottom-gate, top-contact FETs were fabricated on p+-Si/SiO2 substrates by spin-coating 1 wt% chloroform (CHCl3) solutions onto 1-octyltrichlorosilane (OTS) or hexamethyldisilazane (HMDS)-treated substrates.18 The thin films were then annealed at selected temperatures for 10 min in a N2 glovebox. Au source/drain electrodes (100 nm) were patterned on the organic layer through a shadow mask to produce the devices. The typical transfer and output curves measured in N2 are shown in Fig. 3. Well-defined saturation characteristics were observed for both. The OFET device parameters for the thin films of DBPDI and DNPDI under different conditions measured under nitrogen are summarized in Table 2. The device parameters are averaged based on over 36 devices in two different batches. The devices showed good uniformity and reproducibility.
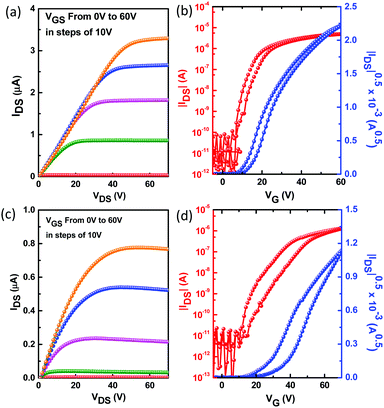 |
| | Fig. 3 Output and transfer characteristics of the thin films DBPDI (a and b) and DNPDI (c and d) spin-coated on OTS-modified substrates. | |
Table 2 OFET characteristics of DBPDI- and DNPDI-based devices fabricated on Si/SiO2 substrates with different surface treatments and different annealing temperatures
| |
Surface treatment |
Annealing temp. |
μ [cm2 V−1 s−1] |
V
T [V] |
On/off |
|
DBPDI
|
HMDS |
XA |
3.0 × 10−3 |
2–4 |
9.6 × 105 |
| HMDS |
A120 |
4.6 × 10−3 |
2–4 |
7.8 × 105 |
| OTS-C8 |
XA |
0.05 |
7–10 |
2.6 × 106 |
| OTS-C8 |
A120 |
0.06 |
5–8 |
2.9 × 106 |
|
DNPDI
|
HMDS |
XA |
4.7 × 10−3 |
28–30 |
3.2 × 106 |
| HMDS |
A120 |
0.011 |
24–26 |
2.0 × 106 |
| HMDS |
A160 |
6.8 × 10−3 |
23–25 |
2.1 × 105 |
| OTS-C8 |
XA |
0.025 |
16–18 |
1.7 × 106 |
| OTS-C8 |
A120 |
0.034 |
15–17 |
1.1 × 106 |
Both compounds exhibited n-type FET behaviour. For compound DBPDI, the devices revealed an average electron mobility of 4.6 × 10−3 cm2 V−1 s−1 (Ion/Ioff = 106) and 0.06 cm2 V−1 s−1 (Ion/Ioff = 106) for HMDS- and OTS-treated substrates, respectively. The device based on DNPDI displayed an average saturation mobility of 0.011 cm2 V−1 s−1 (Ion/Ioff = 106) and 0.034 cm2 V−1 s−1 (Ion/Ioff = 106) for HMDS- and OTS-treated substrates, respectively. The thermal annealing has a lower effect on the charge carrier mobilities, indicating that these two materials are good choices for annealing-free device fabrication in the plastic electronics. The DBPDI showed slightly higher charge carrier mobilities and less hysteresis, indicating that less traps exist in the thin film due to better film crystallinity and less grain boundaries, which is explained in the following section. Both DBPDI and DNPDI showed decreased charge carrier mobility when their devices were operated in ambient conditions.
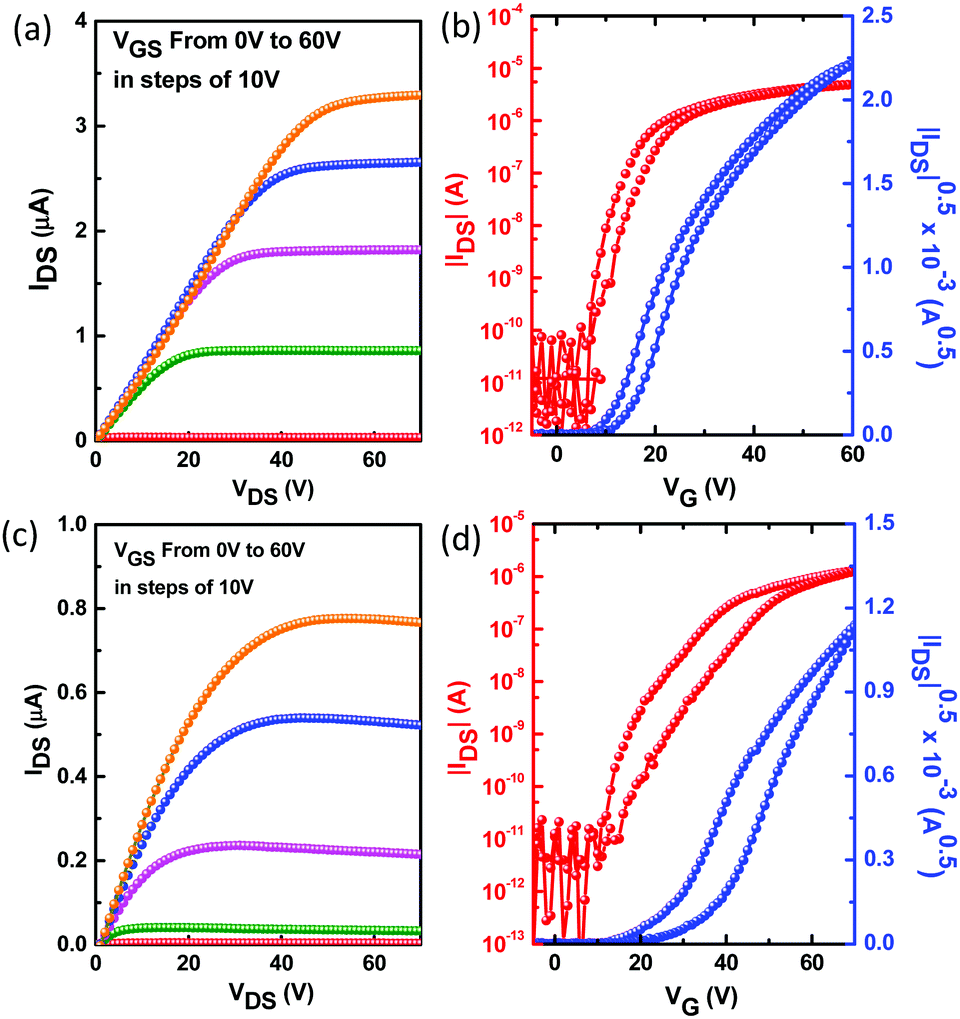
Thin-film morphology and solid-state microstructure were characterized by tapping-mode atomic force microscopy (AFM) and 2D X-ray diffraction (XRD). The thin films exhibited ribbon-like crystals for DBPDI and fibre-like crystals for DNPDI (Fig. 4). The root mean square (RMS) values of surface roughness are 29.1 nm and 30.3 nm for DBPDI before and after annealing, respectively. For DNPDI thin film, the RMS values are 7.7 nm and 8.6 nm before and after annealing, respectively. The larger surface roughness of DBPDI thin film is ascribed to the larger crystal size, which reduces the grain boundaries. The XRD measurements of the thin films on OTS-treated substrates exhibited primary peaks at 2θ = 5.8° and 4.5°, which correspond to d-spacing of 15.2 Å and 19.6 Å for DBPDI and DNPDI, respectively (Fig. 5). These are quite close to their molecular lengths of 15.7 Å and 20.6 Å, indicating that both of them are packed in a layer-like structure. In addition, the intensity of the diffraction peak of DBPDI is much stronger than that of DNPDI, although they have a similar film thickness (60 to 70 nm). The better film crystallinity and larger crystal size of DBPDI thin films ensure a lower amount of traps due to the reduced grain boundaries and give efficient charge transport between the source and drain electrodes.
 |
| | Fig. 4 The AFM images of the thin film spin-coated from CHCl3 solution onto OTS: DBPDI (a: before annealing; b: annealing at 120 °C) and DNPDI (c: before annealing; d: annealing at 120 °C); e–h are the corresponding phase images. | |
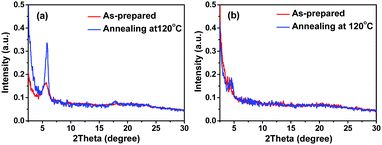 |
| | Fig. 5 XRD patterns of DBPDI (a) and DNPDI (b) thin films on OTS-modified substrates. | |
Conclusion
In conclusion, two diacenopentalene diimides, DBPDI and DNPDI, have been successfully synthesized as new π-conjugated organic semiconducting materials for OFETs. They have good solubility in common organic solvents and show high thermal stability, with decomposition temperatures above 400 °C. DBPDI and DNPDI have relatively low-lying LUMO energy levels at −3.76 and −3.45 eV, respectively. Their thin-film transistor test indicated that they are n-type semiconductors. DBPDI and DNPDI devices produced by solution processing technique displayed average electron mobilities of up to 0.06 cm2 V−1 s−1 with Ion/Ioff ratio of 2.9 × 106 and 0.034 cm2 V−1 s−1 with Ion/Ioff ratio of 1.1 × 106 in N2, respectively. More effort will be carried out to modify this kind of molecule and optimize fabrication conditions19 to improve device performance.
Experiment section
Materials
All reagents were purchased from commercial sources without further purification. Anhydrous dichloromethane (DCM) was distilled from CaH2. 1,4-Dioxane and THF were distilled from sodium-benzophenone immediately prior to use. 4,5-Dibromophthalic acid 1, 1,2-dibromo-4,5-bis(dibromomethyl)benzene 4 and 1-octyl-1H-pyrrole-2,5-dione 5 were prepared by following literature procedures.
General characterization method
1H and 13C NMR spectra were recorded using an Avance 500 MHz Bruker spectrometer in CDCl3 with tetramethylsilane (TMS) as the internal standard. The chemical shift was recorded in ppm, and the following abbreviations are used to explain the multiplicities: s = singlet, d = doublet, t = triplet, m = multiplet, br = broad. Column chromatography was performed on silica gel 60 (Merck 40–60 nm, 230–400 mesh). EI mass spectra were recorded on an Agilent 5975 C DIP/MS mass spectrometer. UV-vis absorption was recorded on a Shimadzu UV-1700 spectrophotometer. Cyclic voltammetry and differential pulse voltammetry measurements were performed in HPLC-grade dry dichloromethane on a CHI 620C electrochemical analyzer with a three-electrode cell, using 0.1 M Bu4NPF6 as the supporting electrolyte, AgCl/Ag as the reference electrode, a gold disk as the working electrode, and Pt wire as the counter electrode, at a scan rate of 50 mV s−1. The potential was externally calibrated against the ferrocene/ferrocenium couple. Differential scanning calorimetry (DSC) was performed on a TA instrument 2920 at a heating/cooling rate of 10 °C min−1 under nitrogen flow.
Synthesis
Compound 2 5,6-dibromo-2-hexylisoindoline-1,3-dione.
A mixture of 1 (4.0 g, 12.35 mmol) and thionyl chloride (6 mL) in 30 mL of dry dichloromethane was stirred overnight at reflux temperature under argon atmosphere. After removing excess thionyl chloride and solvent, the yellowish solid was directly dissolved in 30 mL of dry acetic acid and used in the next reaction. The hexyl amine (1.3 g, 13.58 mmol) was slowly added, and the mixture was heated to 135 °C overnight under argon. The mixture was cooled to room temperature and extracted with EtAc (80 mL × 2). The combined organic phase was washed with brine (100 mL × 2) and saturated NaHCO3 solution (100 mL) and dried over anhydrous Na2SO4. The organic solvent was removed under reduced pressure, and the residue was purified by column chromatography (silica gel, DCM/hexane = 1:5) to afford compound 2 (1.75 g) in 37% yield. 1H NMR (500 MHz, CDCl3, ppm) δ = 8.07 (s, 2H), 3.66 (t, J = 7.5 Hz, 2H), 1.68–1.60 (m, 2H), 1.33–1.25 (m, 6H), 0.87 (t, J = 6.8 Hz, 3H). 13C NMR (CDCl3, 125 MHz): δ = 166.47, 131.87, 131.23, 128.33, 38.52, 31.28, 28.37, 26.46, 22.47, 13.95. HR MS (EI): calcd for C14H15Br2NO2 (M+), 386.9470; found, 386.9458 (error: −3.10 ppm).
Compound 3 5-bromo-2-hexyl-6-(phenylethynyl)isoindoline-1,3-dione.
A suspension of Pd(PPh3)2Cl2 (135 mg, 5 mol%), CuI (36 mg, 5 mol%) and 2 (1.50 g, 3.88 mmol) were purged with argon. After dissolving the mixture with 20 mL of tetrahydrofuran (THF) and 20 mL of anhydrous triethylamine (Et3N), phenyl acetylene (0.435 g, 4.26 mmol) was slowly added to the solution, then the mixture was stirred at 75 °C overnight. After cooling down to room temperature, the mixture was poured into EtOAc (60 mL × 2) and washed with 10% hydrochloride solution (60 mL × 2). The organic layer was dried on Na2SO4 and concentrated under vacuum. Finally, the residue was purified carefully by column chromatography (silica gel, DCM/hexane = 1:10) to give compound 3 (530 mg) in 34% yield. 1H NMR (500 MHz, CDCl3, ppm) δ = 8.07 (s, 1H), 7.97 (s, 1H), 7.63–7.59 (m, 2H), 7.43–7.38 (m, 3H), 3.67 (t, J = 7.3 Hz, 2H), 1.70–1.62 (m, 2H), 1.34–1.25 (m, 6H), 0.88 (t, J = 7.0 Hz, 3H). 13C NMR (CDCl3, 125 MHz): δ = 167.15, 166.69, 131.96, 131.63, 131.58, 131.07, 130.82, 129.57, 128.56, 127.34, 127.26, 121.92, 98.69, 87.20, 38.44, 31.31, 28.44, 26.49, 22.48, 13.95. HR MS (EI): calcd for C22H20BrNO2 (M+), 409.0677; found, 409.0669 (error: −1.96 ppm).
Compound DBPDI.
A reaction flask was charged with hydroquinone (0.113 g, 1.20 mmol), Cs2CO3 (0.334 g, 1.02 mmol), CsF (0.171 g, 1.13 mmol), P(2-furyl)3 (15 mg, 0.08 mmol), and Pd2(dba)3 (15 mg, 0.02 mmol) and purged with argon. A solution of 3 (0.200 g, 0.50 mmol) in 1,4-dioxane (15 mL) was injected into the catalyst mixture with a syringe. The suspension was immediately heated to 135 °C. After heating for 24 h, the reaction mixture was diluted with CH2Cl2 (20 mL), filtered through celite, and then concentrated to yield raw solid. After purification via column chromatography (silica gel, DCM/hexane = 1:4), the product was dissolved in CHCl3, precipitated in methanol/acetone = 3:2 and filtered. This procedure was repeated three times to give the purer product DBPDI (12 mg) in 10% yield for characterization. 1H NMR (500 MHz, CDCl3, ppm) δ = 7.65–7.57 (m, 10H), 7.56 (s, 2H), 7.49 (s, 2H), 3.61 (t, J = 7.3 Hz, 4H), 1.66–1.58 (m, 4H), 1.29–1.25 (m, 12H), 0.86 (t, J = 6.8 Hz, 6H). 13C NMR (CDCl3, 125 MHz): δ = 168.17, 168.05, 154.70, 143.98, 143.44, 140.43, 132.51, 132.45, 131.77, 130.42, 129.47, 128.31, 117.70, 116.41, 38.11, 31.34, 28.50, 26.46, 22.48, 13.98. HR MS (EI): calcd for C44H40N2O4 (M+), 660.2988; found, 660.2994 (error: 0.91 ppm).
Compound 6 6,7-dibromo-2-octyl-1H-benzo[f]isoindole-1,3(2H)-dione.
NaI (1.55 g, 10.5 mmol) was added with stirring to a solution of 4 (1.20 g, 2.1 mmol) and N-octylmaleimide 5 (0.50 g, 2.39 mmol) in N,N-dimethylacetamide (20 mL). The mixture was heated at 80 °C for 12 h. The product was collected from the dark solution by filtration and washed with water, methanol and acetone. The residue was purified by flash column chromatography (silica gel, DCM/hexane = 1:1) to provide compound 6 (0.85 g) in 88% yield. 1H NMR (500 MHz, CDCl3, ppm) δ = 8.33 (s, 2H), 8.20 (s, 2H), 3.74 (t, J = 7.5 Hz, 2H), 1.73–1.68 (m, 2H), 1.37–1.20 (m, 10H), 0.86 (t, J = 7.0 Hz, 3H). 13C NMR (CDCl3, 125 MHz): δ = 167.36, 134.78, 134.18, 129.12, 126.15, 123.27, 38.55, 31.75, 29.13, 28.47, 26.90, 22.60, 14.04. HR MS (EI): calcd for C20H21Br2NO2 (M+), 464.9939; found, 464.9951 (error: 2.58 ppm).
Compound 7 6-bromo-2-octyl-7-(phenylethynyl)-1H-benzo[f]isoindole-1,3(2H)-dione.
A suspension of Pd(PPh3)2Cl2 (65 mg, 5 mol%), CuI (15 mg, 5 mol%) and 6 (850 mg, 1.8 mmol) was purged with argon. Dissolving the mixture with 15 mL of anhydrous tetrahydrofuran (THF) and 15 mL anhydrous triethylamine (Et3N), phenyl acetylene (180 mg, 1.8 mmol) was slowly added to the solution, then the mixture was stirred at 75 °C overnight. After cooling down to room temperature, the mixture was poured into EtOAc (100 mL) and extracted with diluted hydrochloride (60 mL × 3). The organic layer was dried on Na2SO4 and concentrated under vacuum. Finally, the residue was purified carefully by column chromatography (silica gel, DCM/hexane = 1:10) to give compound 7 (380 mg) in 43% yield. 1H NMR (500 MHz, CDCl3, ppm) δ = 8.32 (s, 1H), 8.26 (s, 1H), 8.24 (s, 1H), 8.21 (s, 1H), 7.66–7.62 (m, 2H), 7.43–7.40 (m, 3H), 3.74 (t, J = 7.5 Hz, 2H), 1.74–1.67 (m, 2H), 1.40–1.20 (m, 10H), 0.87 (t, J = 6.8 Hz, 3H). 13C NMR (CDCl3, 125 MHz): δ = 167.58, 167.49, 135.25, 134.28, 133.79, 133.16, 131.88, 129.24, 129.22, 128.82, 128.52, 126.37, 125.95, 123.94, 123.19, 122.30, 96.38, 87.42, 38.50, 31.76, 29.14, 28.50, 26.91, 22.61, 14.05. HR MS (EI): calcd for C28H26BrNO2 (M+), 487.1147; found, 487.1136 (error: −2.26 ppm).
Compound DNPDI.
A reaction flask was charged with hydroquinone (0.172 g, 1.83 mmol), Cs2CO3 (0.508 g, 1.55 mmol), CsF (0.260 g, 1.72 mmol), P(2-furyl)3 (23 mg, 0.12 mmol), and Pd2(dba)3 (23 mg, 0.026 mmol) and purged with argon. A solution of 7 (0.372 g, 0.76 mmol) in 1,4-dioxane (20 mL) was injected into the catalyst mixture with a syringe. The suspension was immediately heated to 135 °C. After heating for 24 h, the reaction mixture was diluted with CH2Cl2 (25 mL), filtered through celite, and then concentrated under vacuum to yield raw solid. After purification via column chromatography (silica gel, DCM/hexane = 1:4), the product was dissolved in CHCl3, precipitated in methanol/acetone = 3:2 and filtered. This procedure was repeated three times to give the purer product DNPDI (75 mg) in 24% yield for characterization. 1H NMR (500 MHz, CDCl3, ppm) δ = 8.08 (s, 2H), 8.05 (s, 2H), 7.98 (s, 2H), 7.83 (d, J = 7.0 Hz, 4H), 7.73 (s, 2H), 7.69 (t, J = 7.5 Hz, 4H), 7.66–7.61 (m, 2H), 3.67 (t, J = 7.0 Hz, 4H), 1.68 (br, 4H), 1.45–1.20 (m, 20H), 0.86 (t, J = 6.5 Hz, 6H). 13C NMR (CDCl3, 125 MHz): δ = 167.85, 150.29, 144.83, 139.78, 136.67, 136.01, 134.77, 133.19, 129.80, 129.34, 129.03, 128.77, 128.72, 124.86, 124.70, 123.45, 123.33, 38.41, 31.80, 29.17, 29.15, 28.58, 27.00, 22.59, 13.93. HR MS (EI): calcd for C56H52N2O4 (M+), 816.3927; found, 816.3908 (error: −2.33 ppm).
Fabrication and characterization of OFET devices
Top-contact, bottom-gate TFTs were prepared. A heavily p+-doped silicon wafer (100, Silicon Quest International, resistivity <0.005 Ω cm−1) with 200 nm thermal silicon dioxide (SiO2) was used as the substrate/gate electrode, with the SiO2 layer serving as the gate dielectric. The SiO2/Si substrate was cleaned with acetone and isopropanol (IPA). It was then immersed in a piranha solution (V(H2SO4):V(H2O2) = 2:1) for 20 minutes, followed by rinsing with deionized water, and then was re-immersed in 0.1 M OTS or HMDS in anhydrous toluene at 60 °C for 20 minutes. It was then rinsed with toluene, and then blow-dried with nitrogen gas. The semiconductor layer was deposited on top of the OTS- or HMDS-modified substrates by spin-coating the 1.0 wt% solution in chloroform, then thermal annealing at selected temperatures for 20 min. Subsequently, gold source/drain electrode pairs were deposited by thermal evaporation through a metal shadow mask to create a series of OFETs with various channel length (L = 100/150 mm) and width (W = 1 mm) dimensions. The OFET devices were then characterized using a Keithley SCS-4200 probe station under N2 conditions in the dark.
Acknowledgements
G. Dai thanks financial support from China Postdoctoral Science Foundation 2016M590498. C. Chi acknowledges financial support from the MOE Tier 1 grant (R-143-000-573-112), Tier 2 grant (MOE2014-T2-1-080) and Tier 3 programme (MOE2014-T3-1-004).
References
- H. Hopf, Angew. Chem., Int. Ed., 2013, 52, 12224 CrossRef CAS PubMed.
-
(a) K. Brand, Ber. Dtsch. Chem. Ges., 1912, 45, 3071 CrossRef;
(b) W. C. Lothrop, J. Am. Chem. Soc., 1941, 63, 1187 CrossRef CAS;
(c) R. F. C. Brown, N. Choi, K. J. Coulston, F. W. Eastwood, U. E. Wiersum and L. W. Jenneskens, Tetrahedron Lett., 1994, 35, 4405 CrossRef CAS;
(d) M. Chakraborty, C. A. Tessier and W. J. Youngs, J. Org. Chem., 1999, 64, 2947 CrossRef CAS PubMed;
(e) M. Saito, M. Nakamura and T. Tajima, Chem. – Eur. J., 2008, 14, 6062 CrossRef CAS PubMed;
(f) T. Kawase, A. Konishi, Y. Hirao, K. Matsumoto, H. Kurata and T. Kubo, Chem. – Eur. J., 2009, 15, 2653 CrossRef CAS PubMed;
(g) Z. U. Levi and T. D. Tilley, J. Am. Chem. Soc., 2009, 131, 2796 CrossRef CAS PubMed;
(h) H. Zhang, T. Karasawa, H. Amada, A. Wakamiya and S. Yamaguchi, Org. Lett., 2009, 11, 3076 CrossRef CAS PubMed;
(i) Z. U. Levi and T. D. Tilley, J. Am. Chem. Soc., 2010, 132, 11012 CrossRef CAS PubMed;
(j) F. Xu, L. Peng, A. Orita and J. Otera, Org. Lett., 2012, 14, 3970 CrossRef CAS PubMed;
(k) T. Maekawa, Y. Segawa and K. Itami, Chem. Sci., 2013, 4, 2369 RSC;
(l) H. Li, B. Wei, L. Xu, W.-X. Zhang and Z. Xi, Angew. Chem., Int. Ed., 2013, 52, 10822 CrossRef CAS PubMed;
(m) C. Chen, M. Harhausen, R. Liedtke, K. Bussmann, A. Fukazawa, S. Yamaguchi, J. L. Petersen, C. D. Daniliuc, R. Fröhlich, G. Kehr and G. Erker, Angew. Chem., Int. Ed., 2013, 52, 5992 CrossRef CAS PubMed;
(n) J. Zhao, K. Oniwa, N. Asao, Y. Yamamoto and T. Jin, J. Am. Chem. Soc., 2013, 135, 10222 CrossRef CAS PubMed;
(o) J. Shen, D. Yuan, Y. Qiao, X. Shen, Z. Zhang, Y. Zhong, Y. Yi and X. Zhu, Org. Lett., 2014, 16, 4924 CrossRef CAS PubMed;
(p) A. Konishi, T. Fujiwara, N. Ogawa, Y. Hirao, K. Matsumoto, H. Kurata, T. Kubo, C. Kitamura and T. Kawase, Chem. Lett., 2010, 39, 300 CrossRef CAS;
(q) F. Xu, L. Peng, K. Shinohara, T. Nishida, K. Wakamatsu, M. Uejima, T. Sato, K. Tanaka, N. Machida, H. Akashi, A. Orita and J. Otera, Org. Lett., 2015, 17, 3014 CrossRef CAS PubMed;
(r) J.-J. Shen, J.-Y. Shao, X. Zhu and Y.-W. Zhong, Org. Lett., 2016, 18, 256 CrossRef CAS PubMed.
- T. Kawase, T. Fujiwara, C. Kitamura, A. Konishi, Y. Hirao, K. Matsumoto, H. Kurata, T. Kubo, S. Shinamura, H. Mori, E. Miyazaki and K. Takimiya, Angew. Chem., Int. Ed., 2010, 49, 7728 CrossRef CAS PubMed.
-
(a) G. Dai, J. Chang, W. Zhang, S. Bai, K.-W. Huang, J. Xu and C. Chi, Chem. Commun., 2015, 51, 503 RSC;
(b) C. Liu, S. Xu, W. Zhu, X. Zhu, W. Hu, Z. Li and Z. Wang, Chem. – Eur. J., 2015, 21, 17016 CrossRef CAS PubMed.
-
(a) M. Nakano, I. Osaka, K. Takimiya and T. Koganezawa, J. Mater. Chem. C, 2014, 2, 64 RSC;
(b) M. Nakano, I. Osaka and K. Takimiya, J. Mater. Chem. C, 2015, 3, 283 RSC;
(c) C. Li, C. Liu, Y. Li, X. Zhu and Z. Wang, Chem. Commun., 2015, 51, 693 RSC;
(d) G. Dai, J. Chang, X. Shi, W. Zhang, B. Zheng, K.-W. Huang and C. Chi, Chem. – Eur. J., 2015, 21, 2019 CrossRef CAS PubMed.
-
(a) Q. Zheng, J. Huang, A. Sarjeant and H. E. Katz, J. Am. Chem. Soc., 2008, 130, 14410 CrossRef CAS PubMed;
(b) J. Shao, J. Chang, G. Dai and C. Chi, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 2454 CrossRef CAS.
-
(a) D. Shukla, S. F. Nelson, D. C. Freeman, M. Rajeswaran, W. G. Ahearn, D. M. Meyer and J. T. Carey, Chem. Mater., 2008, 20, 7486 CrossRef CAS;
(b) J. Chang, Q. Ye, K. W. Huang, J. Zhang, Z. K. Chen, J. Wu and C. Chi, Org. Lett., 2012, 14, 2964 CrossRef CAS PubMed;
(c) J. Chang, J. Shao, J. Zhang, J. Wu and C. Chi, RSC Adv., 2013, 3, 6775 RSC;
(d) J. Shao, J. Chang and C. Chi, Chem. – Asian J., 2014, 9, 253 CrossRef CAS PubMed;
(e) L. Liu, Z. Ren, C. Xiao, B. He, H. Dong, S. Yan, W. Hu and Z. Wang, Chem. Commun., 2016, 52, 4902 RSC.
-
(a) R. J. Chesterfield, J. C. McKeen, C. R. Newman, P. C. Ewbank, D. A. Da Silva Filho, J. L. Brédas, L. L. Miller, K. R. Mann and C. D. Frisbie, J. Phys. Chem. B, 2004, 108, 19281 CrossRef CAS;
(b) T. Zhang, D. Sun, X. Ren, L. Liu, G. Wen, Z. Ren, H. Li and S. Yan, Soft Matter, 2013, 9, 10739 RSC.
-
(a) Z. Wang, C. Kim, A. Facchetti and T. J. Marks, J. Am. Chem. Soc., 2007, 129, 13362 CrossRef CAS PubMed;
(b) H. Usta, C. Kim, Z. Wang, S. Lu, H. Huang, A. Facchetti and T. J. Marks, J. Mater. Chem., 2012, 22, 4459 RSC.
-
(a) Y.-C. Lin, C.-H. Lin, C.-Y. Chen, S.-S. Sun and B. Pal, Org. Biomol. Chem., 2011, 9, 4507 RSC;
(b) Q. Ye, J. Chang, K. W. Huang and C. Chi, Org. Lett., 2011, 13, 5960 CrossRef CAS PubMed;
(c) S. Katsuta, K. Tanaka, Y. Maruya, S. Mori, S. Masuo, T. Okujima, H. Uno, K.-I. Nakayama and H. Yamada, Chem. Commun., 2011, 47, 10112 RSC.
-
(a) H. Qu, W. Cui, J. Li, J. Shao and C. Chi, Org. Lett., 2011, 13, 924 CrossRef CAS PubMed;
(b) J.-J. Chang, H. Qu, Z.-E. Ooi, J. Zhang, Z.-K. Chen, J. Wu and C. Chi, J. Mater. Chem. C, 2013, 1, 456 RSC.
- J. Shao, J. Chang and C. Chi, Org. Biomol. Chem., 2012, 10, 7045 CAS.
-
(a) J. Li, J.-J. Chang, H. S. Tan, H. Jiang, X. Chen, Z. Chen, J. Zhang and J. Wu, Chem. Sci., 2012, 3, 846 RSC;
(b) J. Chang, J. Li, K. L. Chang, J. Zhang and J. Wu, RSC Adv., 2013, 3, 8721 RSC.
- G. Dai, J. Chang, J. Wu and C. Chi, J. Mater. Chem., 2012, 22, 21201 RSC.
- K. M. Psutka, K. J. A. Bozek and K. E. Maly, Org. Lett., 2014, 16, 5442–5445 CrossRef CAS PubMed.
- A. F. Uchoa, K. T. De Oliveira, M. S. Baptista, A. J. Bortoluzzi, Y. Iamamoto and O. A. Serra, J. Org. Chem., 2011, 76, 8824 CrossRef CAS PubMed.
-
(a)
A. J. Bard and L. R. Faulkner, Electrochemical Methods: Fundamentals and Applications, Wiley, New York, 1984 Search PubMed;
(b) J. Pommerehne, H. Vestweber, W. Guss, R. F. Mahrt, H. Bassler, M. Porsch and J. Daub, Adv. Mater., 1995, 7, 551 CrossRef CAS;
(c) C. Chi and G. Wegner, Macromol. Rapid Commun., 2005, 26, 1532 CrossRef CAS.
- J. Chang, C. Chi, J. Zhang and J. Wu, Adv. Mater., 2013, 25, 6442 CrossRef CAS PubMed.
- L. Liu, Z. Ren, C. Xiao, H. Dong, S. Yan, W. Hu and Z. Wang, Org. Electron., 2016, 35, 186 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6tc02601j |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  b,
Linzhi
Jing
b and
Chunyan
Chi
*b
b,
Linzhi
Jing
b and
Chunyan
Chi
*b