
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ultraviolet and blue cathodoluminescence from cubic Y2O3 and Y2O3:Eu3+ generated in a transmission electron microscope
D.
den Engelsen
a,
G. R.
Fern
a,
T. G.
Ireland
*a,
P. G.
Harris
a,
P. R.
Hobson
b,
A.
Lipman
a,
R.
Dhillon
a,
P. J.
Marsh
a and
J.
Silver
a
aCentre for Phosphor and Display Materials, Wolfson Centre for Materials Processing, Brunel University London, Uxbridge, Middlesex, UB8 3PH, UK. E-mail: Terry.ireland@brunel.ac.uk
bDepartment of Electronic and Computer Engineering, Brunel University London, Uxbridge, Middlesex, UB8 3PH, UK
First published on 8th July 2016
Abstract
Herein we describe the investigation of cubic spherical submicron particles of non-doped Y2O3 and Y2O3 doped with Eu3+ in a transmission electron microscope (TEM) equipped with a spectrometer to detect cathodoluminescence from individual particles. Each submicron particle was made up of nanometre sized crystals. We found that these crystals showed a broad emission band at 353 nm upon bombardment with 200 keV or 80 keV electrons. Upon increasing the Eu3+ concentration from 0 to 2 mol% this UV/blue emission was gradually quenched: at Eu3+ concentrations >2 mol% no UV/blue emission was detected, only the well-known cathodoluminescence (CL) spectrum of Y2O3:Eu3+ could be recorded. This UV/blue emission has been attributed to the intrinsic luminescence of Y2O3 caused by self-trapped excitons. We found that the UV/blue luminescence was strongly temperature dependent and that the trap depth of the self-trapped excitons was 0.14 eV. The ratios of the spectral radiances of 5D1 → 7FJ and 5D0 → 7FJ (J = 0, 1…6) Eu3+ transitions in the CL-TEM spectra of Y2O3:Eu3+ at low Eu3+ concentrations was about a factor of 10 larger than those recorded at 15 keV. This phenomenon has been explained by absorption of the intrinsic luminescence of Y2O3 by Eu3+.
1. Introduction
Yttrium oxide (Y2O3:Eu3+) is a well-known phosphor from its application as the red emitting phosphor in fluorescent lamps and projection cathode ray tubes (CRTs).1–3 Due to the industrial applications and the attractive properties of Y2O3 as host lattice, Y2O3 doped with Eu3+ and other rare earth ions has been well studied and documented. The expected application of Y2O3:Eu3+ in low-voltage field emission displays has stimulated intensive research in the synthesis and characterisation of nanosized phosphors, recently reviewed by Li and Lin.4 In our laboratory we have studied the enhancement of cathodoluminescence by applying double layers of ZnO:Zn and Y2O3:Eu3+.5 Non-doped Y2O3 also yields photoluminescence when excited with UV radiation. Although this has been known for more than 50 years,6 the luminescence of non-doped Y2O3 has not been studied in much detail until recently. The reason for this recent interest is the possible application of non-doped Y2O3 and Y2O3 doped with Eu3+, Tb3+, Nd3+ or Tm3+ as a scintillation material.7–15 When non-doped Y2O3 is excited by α-particles,7 UV-radiation (207 nm)8 or X-rays,9–13 a broad luminescence band is observed between 340 nm and 500 nm. Table 1 summarizes the wavelength at maximum (λmax) of this broad luminescence band recorded in the literature. The values for λmax listed in Table 1 vary substantially. This erratic behaviour of the luminescence of Y2O3 elicited the following statement from Fukabori et al.7 “Light yields of Y2O3 ceramics are different from specimen to specimen. Nature of this phenomenon is not clear yet”. Konrad et al.8 found that the size of nanoparticles plays an important role and might explain a variation of about 30 nm in λmax; however, this does not explain the variation shown in Table 1. Fukabori et al.7 found a relation between the scintillation light output and crystal size in their Y2O3 samples: the larger the crystallites, the higher the light output.Wood and Hayes10 reported that the 364 nm emission band is particularly strong upon X-ray excitation at 1.6 K; Tanner et al.12 also found that the 344 nm band in Y2O3:Eu3+ with either 0.1 mol% or 1 mol% Eu3+ is strongly temperature dependent.
Besides the position of λmax there is another interesting characteristic of the intrinsic luminescence of Y2O3: the quenching of this luminescence upon doping with rare earth ions. This was first noticed by Wickersheim and Lefever6 and later by Jacobsohn et al.9 and Tanner et al.12 Jacobson et al. observed a strong reduction of the intrinsic Y2O3 luminescence upon doping with 20 ppm Tb3+, while the intrinsic Y2O3 luminescence virtually disappeared at doping with 0.08 at% Tb3+. Hayes et al. mention the weak intrinsic luminescence of Y2O3 upon doping with Eu3+.14 This phenomenon has been observed by other workers as well in studies of Y2O3 doped with other rare earth ions, although without comment.15,16 Fukabori et al.15 measured a weak intrinsic band at 350 nm upon doping Y2O3 with 1 mol% Nd, while Fujimoto et al.16 measured a weak emission band at 360 nm upon doping with 0.15 mol% Tm3+. These latter authors assigned this band to the emission of Tm3+; however, we believe that the measured emission band is too broad for an emission line of Tm3+. Upon exciting Y2O3:Eu3+ (5 mol% Eu3+) and Y2O3:Tb3+ (0.5 mol% Tb3+) with α-particles, Cress et al.17 could not detect any broad emission band in the near UV-blue. This indicates that at these rare earth concentrations the intrinsic Y2O3 emission has been quenched completely. Comparing this result with the observation of Hayes et al.,14 it may be concluded that Tb3+ is more effectively quenching the intrinsic Y2O3 luminescence than Eu3+. Ato et al.18 reported a strong thermo-luminescence peak of Y2O3:Eu3+ upon irradiation with γ-rays from a Co60 source at low and high dose rates. They did not suggest at that time that Eu3+ was being reduced by γ-rays: this mechanism was suggested nine years later by Ozawa,19 a co-author of Ato.18
The intrinsic luminescence of Y2O3 has been explained by three different mechanisms: (1) oxygen vacancies,9 (2) self-trapped excitons (STE)7,8,10,12,14,20,21 and (3) ligand-to-metal charge-transfer involving the empty 3d orbitals of the Y3+ ion.13 The latter two mechanisms are more plausible, because luminescence due to STEs14 or charge-transfer generates broad bands, while Hayes et al.14 also concluded that the STE-mechanism is likely from their measurements of optical detection of magnetic resonance (ODMR). No explanation is apparent in the literature for the quenching of the intrinsic luminescence of Y2O3 upon doping with rare earth ions.
The current density in a projection CRT at normal operating conditions has a maximum at about 0.25 A m−2. There have been no reports of finding blue CL from Y2O3:Eu3+ in such CRTs. In a scanning electron microscope the beam current at the specimen is normally between 0.01 nA and 1 nA, dependent on the settings of the condenser lenses,22 and the spot size is often of the order of 1.5 nm, leading to current densities between 6 × 106 A cm−2 and 6 × 108 A cm−2, which are 7 to 9 orders of magnitude larger than in a CRT. If the electron transmission at 200 keV is about 99.99% in 200 nm crystals of Y2O3, then the effective current density is still 3 to 5 orders of magnitude higher. This difference in effective current density could explain why blue emission was never observed before or previously reported. Another reason is the quenching of this emission at Eu3+ concentrations greater than 2 mol% (which is typical for CRTs and fluorescent lamps red phosphors). In our laboratories we are currently involved in cathodoluminescence (CL) and photoluminescence (PL) studies of nanosized and submicron Y2O3 doped with Eu3+,Tb3+ and other rare earth ions.23–26 Yttrium oxide is a stable compound and when doped with Eu3+, nobody (to our knowledge) has observed broad emission bands between 350 and 500 nm as mentioned above. It was therefore a surprise when investigating Y2O3:Eu3+ samples in our transmission electron microscope (TEM) equipped with an optical spectrometer that we observed a broad emission band at 350 nm. Preliminary results of this observation have been presented at the 22nd International Display Workshops in Japan.27 Herein we describe further measurements and present an analysis of the intrinsic emission of non-doped Y2O3 and Y2O3:Eu3+ in the electron beam of a TEM.
2. Materials and methods
Materials and synthesis
Yttrium oxide (99.99%, Ampere Industrie, France) and europium(III) oxide (99.99%, Neo Performance Materials, UK) were used to prepare the europium-doped yttrium nitrate stock solutions. Urea, nitric acid, oxalic acid and isopropanol (IPA) were supplied by (Fisher Scientific, UK). All chemicals were used as received.The synthesis of sub-micrometre spherical Y2O3:Eu3+ precursor particles via a homogeneous precipitation route utilising the hydrothermal decomposition of urea method, followed by annealing the precursor particles at 980 °C resulting in cubic Y2O3:Eu3+ has been described extensively in our earlier work.23–26 The concentration of Eu3+ in Y2O3 was adjusted to 0.1, 0.5, 1, 1.5, 4, 20, 60 and 100 mol%. For comparison reasons three samples of micrometre sized Y2O3:Eu3+ precursor particles were prepared via an oxalate precipitation route.1,3 The first sample was made by co-precipitation of Eu3+-oxalate (2%) and Y3+-oxalate. The second and third samples were made by separately precipitating Y3+-oxalate and Eu3+-oxalate (6% and 2%) and then slurry-mixing (SM) these precipitates before annealing. These samples were then annealed for four hours at 980 °C in air.
Transmission electron microscope
The submicron spherical Y2O3 and Y2O3:Eu3+ samples were investigated with a TEM (2100F, JEOL, Japan) equipped with a Schottky-type field emission gun. When operated in scanning mode (STEM), the spot size of the e-beam at the specimen was adjusted to 0.2 nm or 1.5 nm. Initial work demonstrated the need to reduce the X-rays in the column generated from the condenser lens aperture, which were found to significantly contribute to disperse excitation of phosphor samples. These X-rays excited the phosphor and caused the emission of visible light when the electron beam was not on the sample, leading to unwanted interference and a loss of resolution. To reduce this X-ray excitation of the sample, a hard X-ray aperture was inserted into the column, which reduced the background noise in CL imaging and spectroscopy modes considerably. The TEM was equipped with a Vulcan™ CL detector, Gatan, USA, for imaging and spectroscopic purposes. This system used a Czerny–Turner spectrometer with back-illuminated CCD and a grating with 1200 lines per mm (blazed at 500 nm) for collection of CL emission spectra. Light was collected from the sample using a mirror above and below the sample, which enabled a solid angle of about 5 sr, which is almost half of a sphere. This high solid angle made light collection highly efficient and enabled the collection of CL at low intensity. Unfortunately, the cooled detector of this spectrometer did not allow the recording of spectra at λ < 380 nm. In the subsequent sections spectra recorded with this spectrometer will be represented at λ > 400 nm. Spectra between 200 and 400 nm were recorded with the Black Comet spectrometer of StellarNet Inc. (USA) for undoped Y2O3. This spectrometer had an uncooled detector and the spectra were therefore much noisier.By collecting the visible light with the Vulcan system simultaneously with JEOL's high-angle annular dark-field (HAADF) detector, it was possible to observe the visible light that was emitted from the particles. A small cryostat connected to the sample holder enabled cooling of the samples in the TEM down to 102 K (−171 °C); adjustment of the sample temperature anywhere between 102 K and 303 K could be made. A Gatan electron energy loss spectrometer (EELS) was used to map the position of europium ions at the surface of the nanocrystals.
X-ray powder diffraction
The crystalline phases of the products were determined by X-ray powder diffraction (XRPD) using a Bruker D8 Advance X-ray powder diffractometer fitted with a nickel-filtered copper source and a LynxEye™ silicon strip detector. Data were recorded from 5 to 100 2θ degrees at 25 °C. The diffractometer was previously calibrated using an aluminium oxide line position standard from Bruker and a LaB6 NIST SRM 660a line profile standard. Diffractograms were collected using the annealed powders in a conventional holder. The emission of the nickel filtered Cu source and hence the instrumental line broadening was determined by fitting the NIST standard using Bruker Topas version 3. Phases in the combusted products were identified from the XRD patterns by peak search matching using the ICCD PDF-2 data files.3. Results
Transmission electron microscope analysis
Fig. 1a is a TEM image of the urea-precipitated cubic spherical submicron Y2O3:Eu3+ phosphor particles with diameters between 200 nm to 300 nm. Fig. 1b presents a TEM image of a single particle, which is composed of a number of tessellated nanocrystals from 40 nm to 80 nm. | ||
| Fig. 1 TEM images, (a) and (b), of Y2O3:Eu3+ particles: (a) urea-precipitated route, (b) higher magnification image of a single particle from (a). (c) HAADF image oxalate-precipitated particles. | ||
In Fig. 1c the oxalate-precipitated cubic micrometre sized Y2O3:Eu3+ phosphor particles are presented. These have an irregular morphology with a size range between 1 and 3 micrometres.
X-ray diffraction
X-ray diffraction was only used for the analysis of the oxalate-precipitated samples of Y2O3:Eu3+, because urea-precipitated samples of Y2O3 and Y2O3:Eu3+ after annealing at 980 °C for 4 hours are known to consist of the cubic phase for 100%.23–26Fig. 2 shows the diffractogram for oxalate-precipitated Y2O3:Eu3+. This diagram proved that this material after annealing for 4 hours at 980 °C also consisted for >99% of the cubic phase.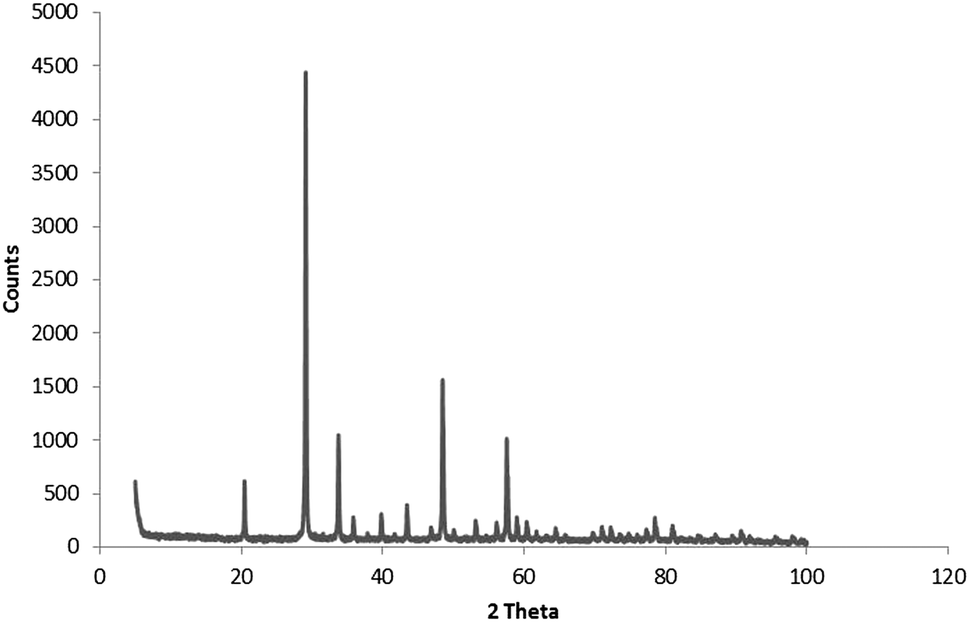 | ||
| Fig. 2 Powder X-ray diffractogram of Y2O3:Eu3+ (2%) oxalate method co-precipitated. Fired at 980 °C for 4 hours. | ||
UV/blue emission band
Fig. 3 shows a spectrum of undoped Y2O3 excited with an electron beam of 200 keV at −120 °C. The as-recorded spectrum is the noisy curve (1), which is represented by a Gaussian profile (3) corrected for the background (2). The Gaussian profile has been fitted to the spectrum with a least squares algorithm using Microsoft's Excel solver. | ||
| Fig. 3 Spectrum of undoped Y2O3 excited with an electron beam of 200 keV and spot size 1.5 nm at −120 °C (1). Recorded with the StellarNet spectrometer with an integration of 16 s. (2): Background. (3): Gaussian profile fitted to the data points and corrected for background. | ||
By averaging the values for λmax and the full width at half maximum (FWHM) from spectra recorded at various temperatures, we determined that λmax = 353 nm (28![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300 cm−1) and FWHM = 5735 cm−1. This value of λmax is identical to the value published by Fukabori et al.7 and close to the values published by Konrad et al.,8 Wood and Hayes10 and Tanner et al.12 Fukabori et al. found that in some of their samples the emission extended much further in the visible region. This has not been confirmed in our measurements, neither in those of the other workers.8–10,12,13 Nevertheless, the broad UV emission band represented in Fig. 4 extends substantially into the visible part of electromagnetic radiation. At 405 nm the spectral radiance is almost 5 times smaller than at 353 nm; however, for comparing the intrinsic Y2O3 luminescence with the Eu3+ emission lines for the doped samples, we used the Gatan spectrometer with the cutoff at about 390 nm, basically because of the detector limitation of the StellarNet spectrometer.
300 cm−1) and FWHM = 5735 cm−1. This value of λmax is identical to the value published by Fukabori et al.7 and close to the values published by Konrad et al.,8 Wood and Hayes10 and Tanner et al.12 Fukabori et al. found that in some of their samples the emission extended much further in the visible region. This has not been confirmed in our measurements, neither in those of the other workers.8–10,12,13 Nevertheless, the broad UV emission band represented in Fig. 4 extends substantially into the visible part of electromagnetic radiation. At 405 nm the spectral radiance is almost 5 times smaller than at 353 nm; however, for comparing the intrinsic Y2O3 luminescence with the Eu3+ emission lines for the doped samples, we used the Gatan spectrometer with the cutoff at about 390 nm, basically because of the detector limitation of the StellarNet spectrometer.
 | ||
| Fig. 4 CL spectra of Y2O3:Eu3+ recorded at −171 °C, 200 keV beam voltage and spot size of 1.5 nm. (A) non-doped Y2O3, inset: spectrum at larger scale between 600 nm and 900 nm; (B) 0.1 mol% Eu3+; (C) 0.5 mol% Eu3+; (D) 1 mol% Eu3+. The sharp lines in the spectrum are Y2O3:Eu3+ transitions; the strongest is the 5D0 → 7F2 Eu3+ transition at 611 nm. | ||
Fig. 4 shows how we were able to turn the red-emitting phosphor Y2O3:Eu3+ into a UV/blue-emitting phosphor in the TEM: the radiance of the UV/blue luminescence is dwarfing the (area) radiance of the well-known emission lines of Eu3+ in Y2O3 at 0.1 mol% Eu3+. Important conditions for observing the broad UV luminescence are: low temperature of the sample: −171 °C, large spot size of the e-beam of 1.5 nm, high beam current by maximising the condenser lens aperture and a specimen thickness of at least 100 nm. The energy of the electron beam does not seem paramount: we observed the UV/blue luminescence both at 200 keV at 80 keV. In Fig. 4A the sharp emission lines of Eu3+ are not present: whereas the strongest transition in Fig. 4B–D is that due to the 5D0 → 7F2 Eu3+ transition at 611 nm. The vertical scales of the graphs in Fig. 4 cannot be compared due to the different integration times during spectra recording and different specimen thicknesses. It can be seen that the spectral radiance (normalised to the spectral radiance at 611 nm) of the blue emission at 400 nm decreases strongly when the concentration of Eu3+ is increased from 0.1 to 1 mol%. At Eu3+ concentrations ≥2 mol% in urea-precipitated Y2O3:Eu3+ we could not detect any UV/blue emission at 400 nm. Beside the strong UV/blue emission at 400 nm, two very weak long wavelength bands can be observed (inset of Fig. 4A), one at about 675 nm and the other at about 770 nm. Electron bombardment of the carbon-coated Cu-grid without Y2O3:Eu3+ particles did not show any CL; hence, interference from the Cu-grid holder material can be excluded.
The luminescence spectrum between 400 nm and 500 nm in Fig. 4A (and the weak long wavelength bands) did not noticeably change its shape upon reducing the energy of the electron beam from 200 keV to 80 keV. This therefore excludes Cherenkov radiation being the origin of the observed emission bands at 353 nm, 675 nm and 770 nm.28 At 400 nm the refractive index of Y2O3 is 1.98: to calculate the refractive index of Y2O3 as a function of λ, use was made of the dispersion formula given by Nigara.29 The condition for Cherenkov radiation in a medium with a refractive index of 1.98 is satisfied when the electron velocity >1.52 × 108 m s−1, which starts at an electron energy of 80 keV. So, at 80 keV there cannot be Cherenkov radiation in Y2O3 at wavelengths >∼405 nm. From this straightforward calculation it can be concluded that any light at wavelengths >405 nm that is emitted upon bombarding non-doped, transparent Y2O3 crystals with electrons at 80 keV cannot be ascribed to Cherenkov radiation. Since the position and shape of the blue emission did not change upon increasing the electron energy to 200 keV, we can reasonably exclude Cherenkov radiation for 200 keV electrons as well. Additional evidence for this conclusion is the strong temperature dependence of the UV/blue and red emission, as we shall discuss in the following paragraphs. Cherenkov radiation is virtually not affected by temperature as long as the refractive index and the density of the specimen do not change substantially. In ref. 27 it was supposed that the blue emission of Y2O3:Eu3+ at 400 nm was caused by reduction of Eu3+ to Eu2+. From Fig. 4A it must be concluded that this is not the case and that the blue emission in all graphs of Fig. 4 is intrinsic luminescence of Y2O3. In other words, it is concluded that the UV/blue luminescence observed in the TEM is identical to the blue/UV emission from undoped Y2O3 and Y2O3 doped with small amounts of rare earth ions upon excitation by other sources of ionizing radiation.7–16
In order to quantify the quenching of the intrinsic luminescence of Y2O3 upon increasing the Eu3+ concentration we define a quenching factor η:
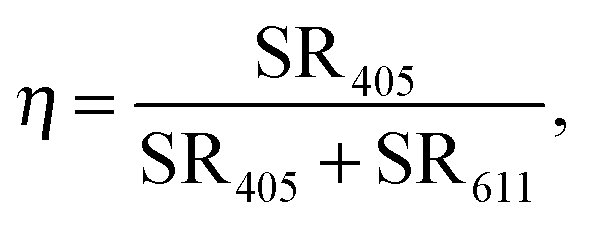 | (1) |
Fig. 4B–D refer to Y2O3 doped with 0.1, 0.5 and 1 mol% Eu3+. Apart from the diminishing intrinsic luminescence of the host material there is another interesting phenomenon, viz. the changing ratios between the spectral radiance of the strongest Eu3+ transition at 611 nm and the spectral radiances of Eu3+ transitions at λ < 611 nm. Furthermore, these latter transitions are about two orders of magnitude stronger than the corresponding transitions in the photoluminescence (PL) spectrum of Y2O3:Eu3+ as represented in Ozawa's book on page 165.3 We shall discuss these phenomena together with the intrinsic luminescence of Y2O3 in the next section.
Fig. 5 is a HAADF image of urea-precipitated Y2O3:Eu3+ (0.1 mol% Eu3+) at −168 °C and 200 keV. The image illustrates the effect of positioning of the e-beam on η. The square denoted by “Spatial Drift” indicates the image that was used to compensate the thermal drift of the sample holder during cooling and warming up. This feature enabled a stable position of the electron beam on the nanocrystal during recording of the spectra, which required more than 2 minutes in some cases. The sites SI.1 to SI.5 in Fig. 5 refer to spots with different particle thickness: SI.1 is a very small particle, whereas SI.2 and SI.3 refer to spots with two particles on top of each other.
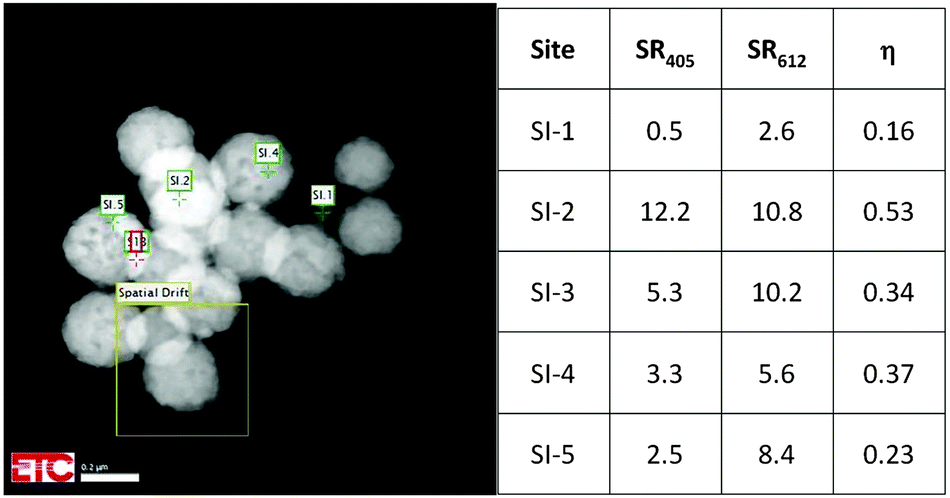 | ||
| Fig. 5 Quenching factor η at various sites for urea-precipitated Y2O3:Eu3+ (0.1 mol% Eu3+) at −168 °C and 200 keV. | ||
This is reflected in the rather high value for the Eu3+ emission. In the other urea-precipitated samples of Y2O3:Eu3+ containing larger Eu3+ concentrations, the variation of the quenching factor η was less than a factor of two. Since some Y2O3:Eu3+ particles are partially hollow, it is impossible to determine a relation between R405 and R611 and specimen thickness from Fig. 5. It should be kept in mind that the radiance Rb of the Y2O3 emission band, defined as
 | (2) |
Fig. 6 shows the effect of temperature on the spectral radiance of the intrinsic emission of undoped Y2O3: the lower the temperature, the stronger the UV/blue luminescence. The drift corrector facility, indicated in Fig. 5, guaranteed that the spectra shown in Fig. 6 were recorded at the same spot. Since the time for recording the spectra (integration time) was also kept constant, the spectra for different temperatures can be compared directly: i.e. there is no effect of thickness. The intrinsic blue luminescence in Y2O3:Eu3+ also increased by more than a factor of 10 upon decreasing the temperature from 31 °C to −171 °C.
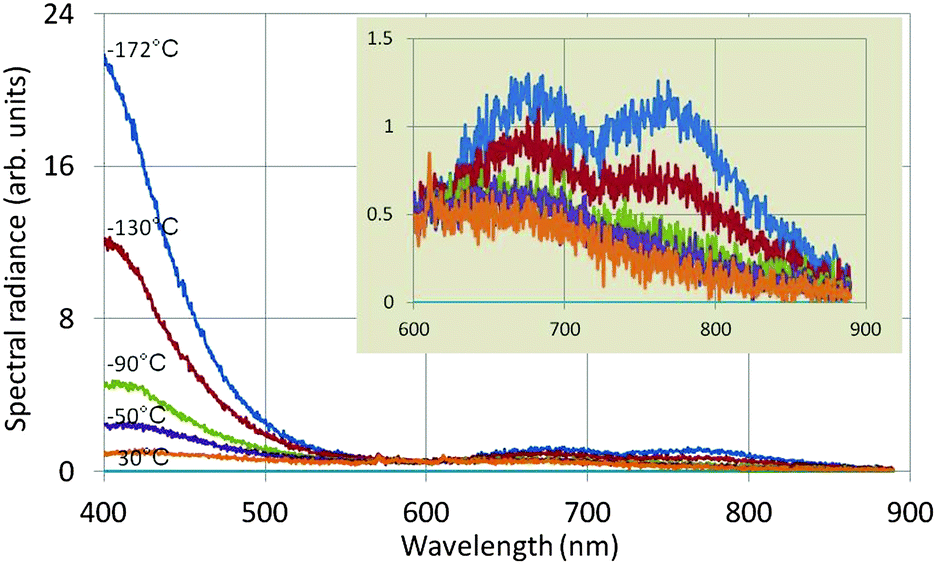 | ||
| Fig. 6 Spectra of undoped Y2O3 recorded at 80 keV and various temperatures. The inset shows the spectra between 600 and 900 nm at a different vertical scale. | ||
The two long wavelength bands shown in the inset of Fig. 6 are much weaker than the UV/blue emission and are also stronger at −172 °C than at 30 °C. It can be seen that the temperature behaviour of the band at 675 nm deviates from that of the other band at 770 nm and the UV/blue band. Upon excitation of Y2O3 or Y2O3:Eu3+ by X-rays or α-particles no emission was observed from these bands by other workers.7,9–11,13–16 These bands also disappeared when the Eu3+ concentration was increased. The origin of these two bands is unknown.
Fig. 7 is an Arrhenius plot of the spectral radiance (SR) measured at 353 nm with the StellarNet spectrometer (data points 1) and SRs measured at 405 nm with the Gatan spectrometer (data points 2 and 3). The data points of (1) refer to undoped Y2O3 and 200 keV beam energy, the data points of (2) refer also to undoped Y2O3 but at 80 keV and the data points of (3) refer to Y2O3:Eu3+ with 0.1 mol% Eu3+ and 200 keV. The drift corrector facility enabled us to stay at the same point during recording of the spectra and after changing the temperature. In some cases a tiny shift was applied to deal with beam degradation. However, the measured effect of beam degradation on spectral radiance was <2%, which was smaller than the noise level in the spectra, especially in the StellarNet spectra, as shown in Fig. 3. The curves in Fig. 7 have been fitted to the data points with eqn (4) that describes the temperature-dependent effect of STEs. This will be discussed in the next section. The trap depth that describes the experimental data for non-doped Y2O3 is 0.13 eV and it is 0.16 eV for Y2O3:Eu3+ with 0.1 mol% Eu3+. For Y2O3:Eu3+ with other Eu3+ concentrations the Arrhenius plots are similar to that of 0.1 mol% Eu3+ as indicated in Fig. 7. In view of the spread in the data we consider that the trap depth of the STEs in undoped and doped Y2O3 is identical and has a value of 0.14 eV.
 | ||
| Fig. 7 Arrhenius plot of spectral radiance versus 1000 K. (1): SR353 of undoped Y2O3 recorded at 200 keV; (2): SR405 of undoped Y2O3 recorded at 80 keV; (3): SR405 of Y2O3:Eu3+ with 0.1 mol% Eu3+ recorded at 200 keV. The dashed curves have been fitted to the experimental data with eqn (4). | ||
In Fig. 8 we have summarized the quenching factor η (for the blue band) as a function of Eu3+ concentration for all samples investigated in the TEM. There are three types of samples collected in Fig. 8, viz. urea-precipitated (without a number), oxalate co-precipitated (no. 1) and SM-oxalate precipitated (no. 2). As mentioned above, blue luminescence could not be observed for the urea-precipitated samples at Eu3+ concentrations >1.5 mol%, whereas the SM-oxalate samples showed very large blue luminescence for 2 and 6 mol% Eu3+. The spread in η was about a factor of 3 in the SM-oxalate samples; in the urea-precipitated samples it was less.
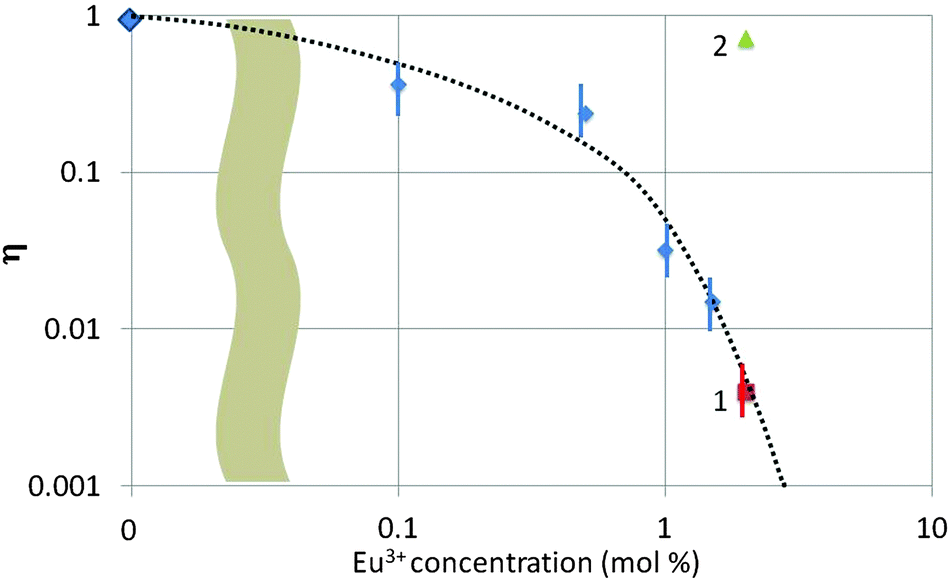 | ||
| Fig. 8 Quenching factor η as a function of Eu3+ concentration in Y2O3. (1): oxalate-co-precipitated Y2O3:Eu3+, (2) SM-oxalate-precipitated Y2O3:Eu3+. Other points refer to urea-precipitated samples. The abscissa is quasi-logarithmic, because the point with 0 mol% Eu3+ dope has been indicated. | ||
Fig. 8 illustrates the different behaviour of the SM-oxalate-precipitated and urea-precipitated samples, whereas the oxalate co-precipitated sample (point 1 in Fig. 8) with molecular mixing of the Eu3+ and Y3+ ions indicates that this sample behaves as the urea-precipitated samples. The deviating behaviour of the SM-oxalate-precipitated sample is explained by its inhomogeneity, in which Eu3+-rich areas in the crystals are alternating with areas with very low Eu3+ concentration. The penetration depth of 200 keV electrons in crystalline Y2O3 is about 75 μm,5,30 which is much larger than the size of the oxalate-precipitated crystals. The Eu3+ rich areas in the crystal hit by the e-beam do not contribute to the build-up of the blue band because of concentration quenching, whereas the areas with very low Eu3+ concentration take care of the strong blue band signal. This hypothesis was confirmed by the very low Eu3+ signal in the EELS (not shown) at various spots. From this consideration it can be concluded that the SM-oxalate precipitated sample should be inserted at a much lower concentration in Fig. 8. Since the effective Eu3+ concentration is unknown, the best we could do is plotting the result at the as-made Eu3+ concentration.
Finally we would like to mention that we also observed blue luminescence in the TEM when bombarding undoped monoclinic Y2O3 crystals with 200 keV electrons at 1.5 nm spot size. The monoclinic material was unstable under these electron bombardment conditions, decomposing partly into the more stable cubic form, and therefore we do not reproduce any results of these measurements in this article.
Discussion of blue luminescence in Y2O3 and transition ratios
When an electron beam of 200 keV hits an Y2O3 crystal it produces besides defects, backscattered and secondary electrons, X-rays and holes by inelastic scattering processes. Electrons and holes can combine to form free excitons and STEs. Herein we adopt the model that STEs are responsible for the blue luminescence in undoped Y2O3 and Y2O3:Eu3+ as indicated in the literature.7,8,10,12,14,20,21 According to Mikhailik and Kraus31 and Blasse32 the radiance B of a scintillator upon excitation can be written as:| B = αNe–hSQ | (3) |
 | (4) |
It should be kept in mind that eqn (3) for self-activated Y2O3 refers to the radiance of the blue emission band. When Y2O3 is doped with Eu3+, energy will be transferred from the host lattice to the Eu3+ luminescent centres: hence, the CL spectrum of Y2O3:Eu3+ can be detected. The CL of Y2O3:Eu3+ can also be described with an energy transfer probability S in eqn (3), which is 0 for undoped Y2O3 and has a maximum value, albeit < 1, for a dopant concentration of 2 mol% and larger.
As mentioned above, Fig. 4 shows that the spectral radiance of Eu3+ transitions at λ < 580 nm decreases in the sequence Fig. 4B–D. We have plotted this behaviour for some transitions in Fig. 9.
 | ||
| Fig. 9 Ratio of spectral radiance between 5D1 → 7F1 (533 nm) and 5D0 → 7F2 (611 nm): R1 and ratio of spectral radiance between 5D0 → 7F4 (713 nm) and 5D0 → 7F2 (611 nm): R2. For R1 the measurements at −170 °C (cold) and room temperature (RT) gave different results. | ||
Fig. 9 represents the ratios R1 and R2, which are the ratios of the spectral radiances 5D1 → 7F1 (533 nm) and 5D0 → 7F2 (611 nm) and that of 5D0 → 7F4 (713 nm) and 5D0 → 7F2 (611 nm) respectively. It can be seen that R1 decreases almost by a factor of 10 upon increasing the dopant concentration from 0.1 mol% to 4%, whereas R2 is constant, both at cryogenic and room temperature. The latter ratio is constant, because it refers to 5D0 transitions. The transitions of the 5D2 and 5D3 states to the 7FJ manifold show a similar behaviour as the 5D1 → 7F1 transition; however, due to the lower spectral radiances the error bar in the graphs is larger and therefore these will not be reproduced here. The partial concentration quenching of the 5D1 transition of the Eu3+ ion represented in Fig. 9 is well known and it has been described by Blasse and Grabmaier2 and Klaassen et al.33 Blasse and Grabmaier explained the behaviour of R1 in terms of the following cross relaxation in Eu3+:
| 5D1 + 7F0 → 5D0 + 7F3. | (5) |
For a qualitative explanation we shall make use of the energy diagram depicted in Fig. 10. Most levels indicated in Fig. 10 have been reported by Wen et al.,34 only the lowest energy level of the manifolds is indicated. The energy level of the intrinsic Y2O3 emission band has been centred at 28300 cm−1 above the ground level 7F0 of Eu3+, based on the spectrum depicted in Fig. 4. The level of the STE Y2O3 emission band overlaps with several Eu3+ levels; hence, it seems obvious that energy can radiationless be transferred from an STE in Y2O3 to the 5D4 level of Eu3+, which is only slightly lower. This process, which takes place at Eu3+ concentrations >2 mol%, is indicated by arrow 4. From the 5D4 level energy will radiationless trickle down to the 5D3, 5D2, 5D1 and 5D0 levels, indicated by arrows 7. As mentioned above, the decay times of transitions from 5DJ levels with J > 0 are about 10 times shorter than these from 5D0,33 which enhances efficient energy transfer and hence rather fast quenching of the Y2O3 emission upon increasing the Eu3+ concentration.2,35 In terms of the probability S in eqn (3) it means that energy from an e–h pair is transferred to a Eu3+ ion.
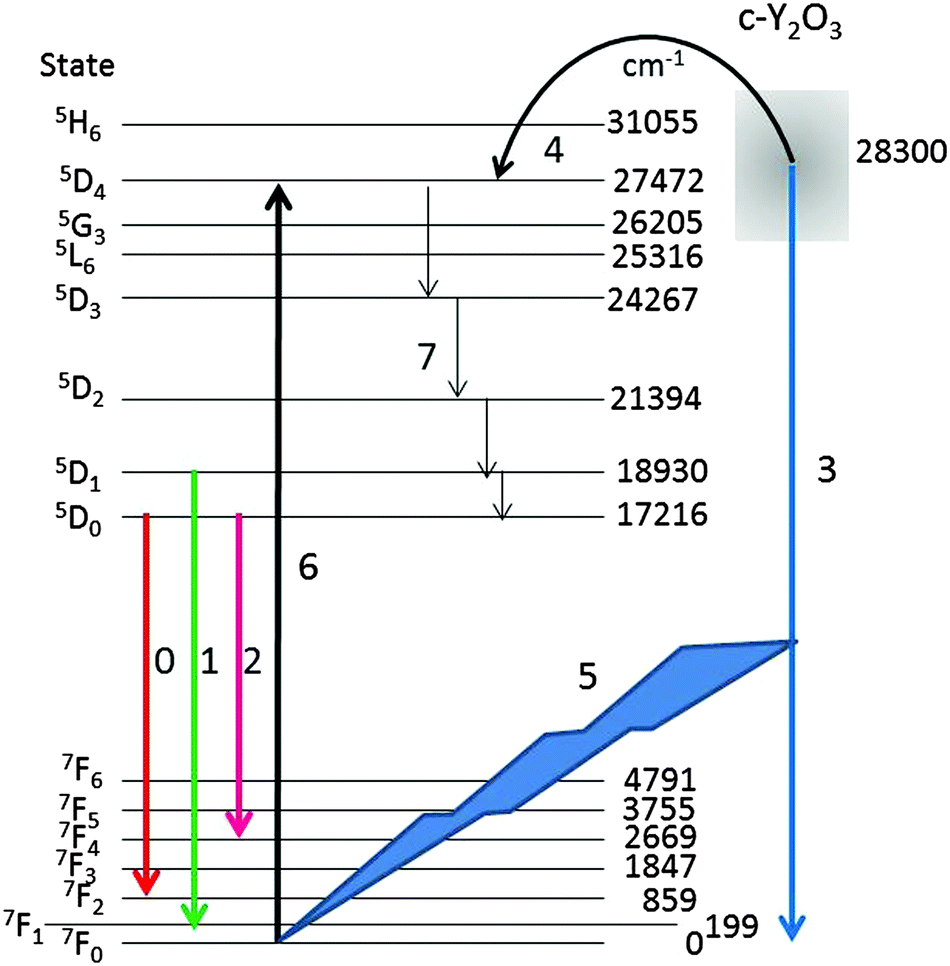 | ||
| Fig. 10 Energy levels of Eu3+ in Y2O3. The broad intrinsic luminescence of Y2O3 has been represented at 28300 cm−1 by arrow (3). The arrows (0), (1) and (2) refer to the 5D0 → 7F2, 5D1 → 7F1 and 5D0 → 7F4 Eu3+ transitions respectively, while (4) indicates the radiationless energy transfer from Y2O3 to Eu3+. For arrows (5), (6) and (7) we refer to text. | ||
At low Eu3+ concentrations <2 mol% the strong intrinsic emission at 28300 cm−1 may be absorbed by Eu3+ ions in the lattice: this process is indicated by arrows (5) and (6) in Fig. 10. It has been indicated in this figure that the Y2O3 UV radiation is absorbed from the ground level 7F0; hence, the 5D4 Eu3+ level will be populated more densely. As mentioned above, from 5D4 energy will trickle down to the lower 5DJ levels. The process indicated by arrows (5) and (6) will stop when the intrinsic luminescence of Y2O3 has quenched at large Eu3+ concentration. The latter process explains the high value of R1 in Fig. 9 and similarly the rather strong 5D1 → 7FJ and 5D2 → 7FJ transitions in Fig. 4B–D. The foregoing consideration also explains why the ratio R1 in Fig. 9 at low temperature is higher than at room temperature: the process indicated by the arrows (5) and (6) is more dominant at low temperature because of the much stronger UV luminescence of Y2O3.
4. Conclusions
In the preceding sections we have described the cathodoluminescence of undoped Y2O3 and Y2O3:Eu3+ crystals in the TEM by high-energy electron bombardment. At low temperatures we observed a broad emission band at about 353 nm, which has been ascribed to the migration of excitons to luminescence centres in Y2O3. We found that the UV/blue luminescence is strongly temperature dependent and that at concentrations >2 mol% Eu3+ no blue light could be detected.The temperature behaviour of the intrinsic luminescence of Y2O3 has been explained with a model for the self-trapped excitons. The depth of these traps was found to be 0.14 eV. The concentration dependence of the UV/blue luminescence has been explained by the good overlap between the level of the blue Y2O3 band and the 5D4 level of the Eu3+ ion. The strong radiance of 5D1 → 7FJ and 5D2 → 7FJ Eu3+ transitions in the spectra excited at 200 keV in Y2O3:Eu3+ with low Eu3+ concentration has been explained by absorption of the intrinsic Y2O3 radiation by Eu3+ ions. Although we have presented a detailed explanation of the concentration and temperature behaviour of the UV/blue emission, we have also generated new questions. The most important is about the nature of the red emission bands at 675 nm and 770 nm.
Based on the results described in this article we would like to make a suggestion to other scientists working with electron microscopes in the field of biomedical imaging with phosphors. This fast growing technology has recently been reviewed by Gai et al.36 Since undoped Y2O3 or Y2O3:Eu3+ with low Eu3+ concentration will manifest the strong intrinsic luminescence of Y2O3 in the e-beam of a TEM (and likely also in a SEM) at low temperatures, we think that this work opens new perspectives for labelling (biological) samples with nanocrystals of Y2O3:Eu3+.
Acknowledgements
We are grateful to the EPSRC and the Technology Strategy Board (TSB) for funding the PURPOSE (TP11/MFE/6/I/AA129F; EPSRC TS/G000271/1), CONVERTED (JeS no. TS/1003053/1) and PRISM (EP/N508974/1) programs. We are also grateful to the TSB for funding the CONVERT program.References
- Phosphor Handbook, ed. W. Yen, S. Shionoya and H. Yamamoto, CRC Press, Boca Raton, 2nd edn, 2007 Search PubMed.
- G. Blasse and B. C. Grabmaier, Luminescent Materials, Springer-Verlag, Berlin, 1994 Search PubMed.
- L. Ozawa, Cathodoluminescence, Theory and Applications, Kodansha & VCH, Verlag, Tokyo, 1990 Search PubMed.
- G. Li and J. Lin, Chem. Soc. Rev., 2014, 43, 7099 RSC.
- D. den Engelsen, P. G. Harris, T. G. Ireland and J. Silver, ECS J. Solid State Sci. Technol., 2014, 3, R53 CrossRef CAS.
- K. A. Wickersheim and R. A. Lefever, J. Electrochem. Soc., 1964, 111, 47 CrossRef CAS.
- A. Fukabori, L. An, A. Ito, V. Chani, K. Kamada, A. Yoshikawa, T. Ikegami and T. Goto, Ceram. Int., 2012, 38, 2119 CrossRef CAS.
- A. Konrad, U. Herr, R. Tidecks, F. Kummer and K. Samwer, J. Appl. Phys., 2001, 90, 3516 CrossRef CAS.
- L. G. Jacobsohn, B. L. Bennett, R. E. Muenchausen, J. F. Smith and D. W. Cooke, Proc. SPIE, 2006, 6321, 63210J CrossRef.
- R. L. Wood and W. Hayes, J. Phys. C: Solid State Phys., 1982, 15, 7209 CrossRef CAS.
- T. C. de Oliveira, M. Souza da Silva, L. Menezes de Jesus, D. Vieira Sampaio, J. C. Alves dos Santos, N. R. da Silva Souza and R. Santos da Silva, Ceram. Int., 2014, 40, 16209 CrossRef.
- P. A. Tanner, L. Fu and B. M. Cheng, J. Phys. Chem. C, 2009, 113, 10773 CAS.
- G. Blasse and L. H. Brixner, Eur. J. Solid State Inorg. Chem., 1991, 28, 767 CAS.
- W. Hayes, M. J. Kane, O. Salminen and A. I. Kuznetsov, J. Phys. C: Solid State Phys., 1984, 17, L383 CrossRef CAS.
- A. Fukabori, V. Chani, J. Pejchal, K. Kamada, A. Yoshikawa and T. Ikega, Opt. Mater., 2011, 34, 452 CrossRef CAS.
- Y. Fujimoto, T. Yanagida, Y. Yokota, A. Ikesue and A. Yoshikawa, Opt. Mater., 2011, 34, 448 CrossRef CAS.
- C. D. Cress, C. S. Redino, B. J. Landi and R. P. Raffaelle, J. Solid State Chem., 2008, 181, 2041 CrossRef CAS.
- Y. Ato, R. Huzimura and L. Ozawa, Jpn. J. Appl. Phys., 1968, 7, 1497 CrossRef CAS.
- L. Ozawa, Appl. Phys. Lett., 1977, 31, 694 CrossRef CAS.
- W. Hayes, J. Lumin., 1984, 31, 99 CrossRef.
- A. Lushchik, M. Kirm, Ch. Lushchik, I. Martinson and G. Zimmerer, J. Lumin., 2000, 87–89, 232 CrossRef CAS.
- D. B. Williams and C. B. Carter, Transmission Electron Microscopy, A Textbook for Materials Science, Springer, New York, 2009 Search PubMed.
- D. den Engelsen, P. G. Harris, T. G. Ireland and J. Silver, ECS J. Solid State Sci. Technol., 2015, 4, R1 CrossRef CAS.
- D. den Engelsen, P. G. Harris, T. G. Ireland, R. Withnall and J. Silver, ECS J. Solid State Sci. Technol., 2013, 2, R201 CrossRef CAS.
- X. Jing, T. Ireland, C. Gibbons, D. J. Barber, J. Silver, A. Vecht, G. Fern, P. Trogwa and D. C. Morton, J. Electrochem. Soc., 1999, 146, 4654 CrossRef CAS.
- J. Silver, T. G. Ireland and R. Withnall, J. Electrochem. Soc., 2004, 151, H66 CrossRef CAS.
- G. R. Fern, A. Lipman, J. Silver, A. Howkins, T. G. Ireland, P. Marsh, D. den Engelsen, Proceedings of 22nd International Display Workshops, December 9–11, Otsu, Japan, 2015.
- J. V. Jelley, Čerenkov Radiation and its applications, Pergamon Press, London, 1958 Search PubMed.
- Y. Nigara, Jpn. J. Appl. Phys., 1968, 7, 404 CrossRef CAS.
- D. den Engelsen, P. G. Harris, T. G. Ireland and J. Silver, ECS J. Solid State Sci. Technol., 2015, 4, R1 CrossRef CAS.
- V. B. Mikhailik and H. Kraus, Phys. Status Solidi B, 2010, 247, 1583 CrossRef CAS.
- G. Blasse, J. Lumin., 1994, 60–61, 930 CrossRef CAS.
- D. B. M. Klaasen, R. A. M. van Ham and T. G. M. van Rijn, J. Lumin., 1989, 43, 261 CrossRef.
- J. Wen, L. Hu, M. Yin and S. Xia, Current Appl. Phys., 2012, 12, 732 CrossRef.
- D. L. Dexter, J. Chem. Phys., 1953, 21, 836 CrossRef CAS.
- S. Gai, C. Li, P. Yang and J. Lin, Chem. Rev., 2014, 114, 2342 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2016 |