
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A dual-phase architecture for efficient amplified spontaneous emission in lead iodide perovskites†
Stefanie
Neutzner
ab,
Ajay Ram
Srimath Kandada
*a,
Guglielmo
Lanzani
ab and
Annamaria
Petrozza
*a
aCenter for Nano Science and Technology@PoliMi, Istituto Italiano di Tecnologia, via Giovanni Pascoli 70/3, 20133, Milan, Italy. E-mail: srinivasa.srimath@iit.it; annamaria.petrozza@iit.it
bDipartimento di Fisica, Politecnico di Milano, Piazza L. da Vinci, 32, 20133 Milano, Italy
First published on 5th May 2016
Abstract
Multi-particle Auger interactions are one of the major intrinsic limiting factors for lasing applications of lead halide perovskites. In this communication, we report a possible way to tackle this problem by investigating the transfer of the photo-excited population to radiative defect inclusions using transient absorption (TA) spectroscopy. We demonstrate a reduction of the threshold of amplified spontaneous emission (ASE) in the presence of an efficient transfer.
1. Introduction
Hybrid lead halide perovskites have recently emerged as exceptional materials for different optoelectronic applications. Beyond the very good performances demonstrated when embodied in photovoltaic (PV) devices,3,4 high photoluminescence quantum yields, long carrier lifetimes5–7 and wavelength tunability8,9 place them as good candidates for lasing applications. Recently, amplified spontaneous emission (ASE) as well as lasing have been demonstrated in a variety of sample morphologies ranging from polycrystalline thin films,10 single crystals,11 nanowires12,13 to films made from colloidal nanocrystals.14 The reported gain thresholds are very promising; however, they have been achieved under ultra-short (fs/ns)2 pulse excitation. Therefore operation under technologically relevant continuous wave (cw) conditions or under electrical injection remains a major challenge in the field.Within the high excitation density regime needed for population inversion in semiconductors, non-radiative Auger recombination leads to a population loss, heating of the material and higher gain threshold.1 One of the established routes implemented in quantum well lasers to counter this process is to create a controlled phase-architecture of spatially separated domains for pumping and population inversion. Such engineered material systems enable spatial redistribution of the excited state population such that inter-particle Auger interactions are lowered, thus leading to an overall reduction in the gain threshold.15–17
Here, we report a proof of principle for the implementation of such an architecture by the exploitation of radiative defect inclusions in polycrystalline thin films of methyl-ammonium lead iodide (MAPbI3) perovskites. These defects form after the phase transition of the semiconductor, from the room temperature (RT) tetragonal structure to the orthorhombic one, stable at temperatures below 160 K.18,19 We use transient absorption spectroscopy to show an efficient transfer of carriers from the dominant orthorhombic phase of the semiconductor to these defects, which intrinsically represents the spatial separation of pumping and emission domains. We demonstrate that this leads to a reduction of the gain threshold and allows us to retrieve design parameters for an optimized architecture. This result provides an interesting route to the defect engineering of these self-assembled materials for cw lasing applications.
2. Results
Fig. 1a shows the optical absorption spectra of a poly-crystalline film of MAPbI3 deposited on glass, taken at different temperatures (see Fig. S1, ESI† for structural data). As previously reported,20,21 the orthorhombic phase, stable at low temperature (<160 K), has a larger gap compared to that of the RT phase. The optical edges are placed at 740 nm and 760 nm at 150 K and 290 K respectively, which then red-shift as the temperature is lowered as a result of continuous expansion/shrinkage of the lattice.22 In a single crystal, the phase transition occurs at 160 K.23 Importantly, it has been shown often that polycrystalline films do not exhibit a sharp transition, at least with regard to the optical signature of the phases.24–26 This is evidenced in Fig. 1a by the existence of the spectral signature of the tetragonal phase in the absorption spectra even down to 140 K. Similar trends are observed also in the photoluminescence spectra shown in Fig. 1b. The lower energy emission peak initially blue-shifts in the temperature range of 170 to 130 K, passing through a maximum in emission intensity and thereafter red-shifts. The nature of the lower energy emission feature in the anomalous temperature range has already been debated in previous reports and assigned either to a bound-exciton next to free-exciton emission,10,23 therefore belonging to the same structural phase, or to a coexistence of the tetragonal and orthorhombic phases.24,25 Recently, Kong et al. have shown temperature dependent X-ray diffraction studies of the film,25 demonstrating the coexistence of the tetragonal and orthorhombic phases within a range of 30 K around the phase transition temperature. However, a very low density of such intrusions can still be optically perceived at temperatures well below such range (i.e. 77 K, see the ESI† of this manuscript). Thus, Kong et al. suggested, following the theoretical prediction of Yin et al.,27 that these are inter- band gap states of the orthorhombic phase due to iodide and lead vacancies, which form radiative recombination centres. We can also speculate that these correspond to linear defect sites, which form in between crystalline grains of the orthorhombic phase. Though, there is a need for a detailed investigation to identify the nature of these defects, from now on, we will generally indicate them as colour centres.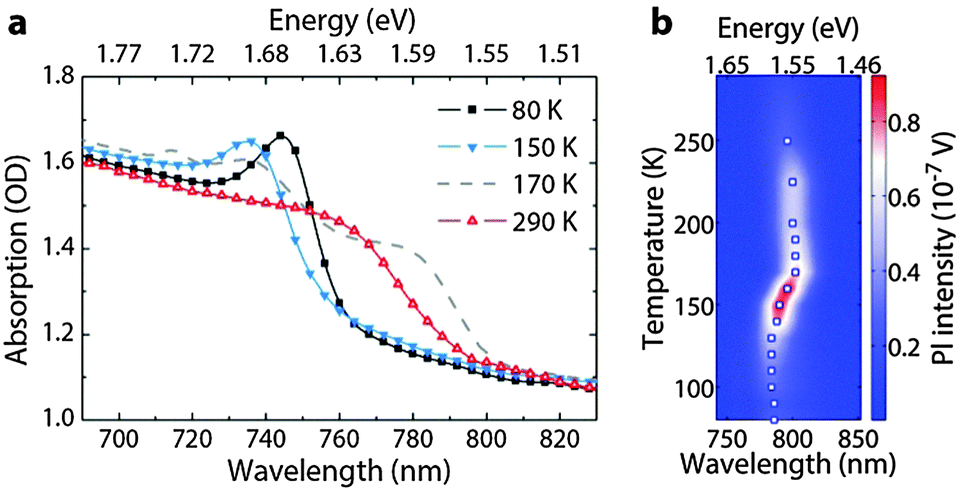 | ||
| Fig. 1 (a) Temperature dependence of the absorption edge. (b) Spectral evolution of the low energy photoluminescence (PL) peak as a function of temperature. The white squares mark the position of the PL maximum as a guide to the eye. | ||
To quantitatively assess the photo-carrier transfer from the semiconductor bandgap to the colour centres, we performed fs-TA spectroscopy at different temperatures with the pump at 530 nm. In Fig. 2, we show the TA spectra of MAPbI3 taken at 150 K (for other temperatures, compare Fig. S2, ESI†). The main features are the photo-bleach (PB) bands due to state filling by the photo-excited carriers.21 In particular, we observe two PB bands, peaking at 730 nm (labeled PBO) and 760 nm (PBD), corresponding to the orthorhombic phase and the defect phase respectively. The time evolution of the spectra shows quenching of PBO and a concomitant growth in the PBD band in tens of picoseconds (see dynamics in Fig. 2c). Fitting the dynamics at 150 K of the two peaks with simple exponential decays, including one term accounting for the transfer time, delivers a time constant of about 13 ± 2 ps for the transfer (Fig. 2c).
 | ||
| Fig. 2 (a) Top: Spectral evolution of the TA signal within 50 ps. Bottom: TA-spectra representing three different time steps. (b) Sketch illustrating the observed processes. (c) Time evolution and corresponding fits for the two PB signals at 150 K. | ||
Note that such a transfer can be observed even at lower temperatures, down to 77 K, albeit with a lower amplitude and not only at the phase transition, as claimed earlier.28 Since the efficiency of the process decreases at lower temperatures (the transfer rate and the strength of the transfer component decrease with lowering of the temperature, compare Fig. S3b, ESI†), we assume that this is related to a change in the density of colour centres.
To further substantiate our hypothesis of population transfer versus other explanations, such as slow de-trapping and to exclude contribution from overlapping TA features, we performed a TA experiment with two different pump wavelengths, above and below the orthorhombic phase energy. Pumping at 400 nm, very similar to the previous case, we photo-excite both the phases and we observe the increase of PBD due to the transfer of population in the ps timescale (Fig. S4, ESI†). Upon pumping at 760 nm, we do not observe any PBO feature and we just observe the recombination dynamics of PBD. This unambiguously proves our assignment of carrier transfer and supports the presence of a defined dual domain structure for pumping and emission.
In order to test the effectiveness of this system for lowering the threshold of lasing, we performed steady state emission measurements with fs-excitation at different temperatures to analyze the ASE thresholds. Since our aim here is to investigate the possible effect of population transfer, we performed the measurements at 140 K, 150 K and 160 K, all within the phase transition regime, as shown in Fig. 3. This small range guarantees immunity of the observed behavior to temperature induced effects and ensures a critical density of colour centres to observe a perceivable change. As shown in Fig. 3, we find that the ASE thresholds at 140 K and 160 K are at 1.45 × 1019 and 1.8 × 1019 respectively, while it is lowered to 7 × 1018 at 150 K. This is supported by the fact that the transfer is most efficient at 150 K as observed in the TA experiment (see Fig. S3, ESI†).
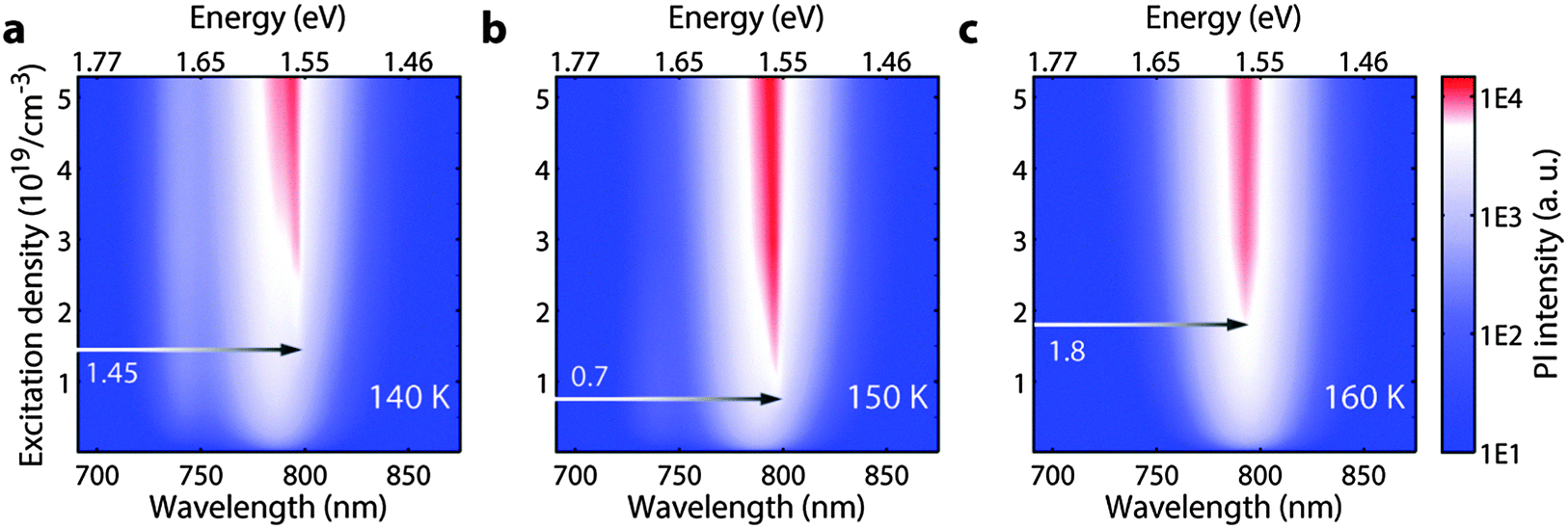 | ||
| Fig. 3 Steady state PL measurements as a function of excitation density for 140 K (a) 150 K (b) and 160 K (c). The corresponding ASE threshold is marked for each temperature. | ||
3. Discussion
Our result points to the possibility of energy relocation from the carrier plasma to radiative defects states, thereby reducing the effect of non-radiative Auger losses. This occurs by exploiting a dual phase architecture that is naturally engineered within the crystalline semiconductor. To optimize such a process, with the aim of transferring the acquired know-how to a system which can operate at RT, we need to deduce design principles. The important parameter here is the transfer rate, along with the relative defect density. The main purpose of the transfer process is to replenish any loss in the population due to Auger, thus mitigating the loss in population inversion needed for the gain mechanism. If the transfer time were much faster than the Auger mechanism within the low energy phase, this would result in the loss of even the transferred population. On the other hand, it should be faster than the recombination within the high energy phase. Thus a precise engineering of the transfer rate is extremely important.We observe that the transfer rate is diffusion limited and thus correlated with the orthorhombic domain size and the density of the colour centres. This is evident from the temperature dependence of the rate, with the transfer becoming slower at lower temperatures along with a concomitant reduction in the amplitude of the transfer component of the PB dynamics (see comparison of the transfer dynamics in Fig. S3, ESI†). Assuming a mobility (μ) of 50 cm2 V−1 s−1 reported for these materials at 150 K,29 and with the observed transfer time (τtrans) of 13 ps, we can estimate the effective length travelled by the carriers before they are quenched,  to be around 29 nm. This represents the effective length-scale of the orthorhombic phase within the materials. At lower temperatures, at 120 K, where the relative concentration of colour centres is lower (thus larger domains of the orthorhombic phase), we observe a transfer time of around 60 ps, corresponding to an effective diffusion of about 60 nm before the carriers can be transferred. Moreover, in a sample that is composed of smaller crystallites and hence is prone to a higher density of grain boundaries, the transfer times are much faster due to the reduction of domain size. Thus effective domain sizes have to be suitably engineered to obtain an efficient transfer process.
to be around 29 nm. This represents the effective length-scale of the orthorhombic phase within the materials. At lower temperatures, at 120 K, where the relative concentration of colour centres is lower (thus larger domains of the orthorhombic phase), we observe a transfer time of around 60 ps, corresponding to an effective diffusion of about 60 nm before the carriers can be transferred. Moreover, in a sample that is composed of smaller crystallites and hence is prone to a higher density of grain boundaries, the transfer times are much faster due to the reduction of domain size. Thus effective domain sizes have to be suitably engineered to obtain an efficient transfer process.
4. Conclusions
In summary, we have investigated the interaction between the orthorhombic phase and radiative defect states in MAPbI3 below the structural phase transition temperature using TA spectroscopy. We unambiguously assign the previously observed abnormal emission behaviour to transfer between the orthorhombic phase and defect sites and demonstrate that an efficient transfer can lower the threshold for ASE. Our results suggest fundamental design principles for mixed phase architectures for lasing applications, based on the interplay between Auger and transfer rates as well as the distribution and size of the respective domains.Acknowledgements
The authors thank Marina Gandini for the preparation of the samples. Funding from the EU Horizon 2020 Research and Innovation program under the grant agreement No. 643238 (SYNCHRONICS) and Fondazione Cariplo (IPER-LUCE, 2015-0080) is acknowledged.Notes and references
- M. Saba, M. Cadelano, D. Marongiu, F. Chen, V. Sarritzu, N. Sestu, C. Figus, M. Aresti, R. Piras, A. Geddo Lehmann, C. Cannas, A. Musinu, F. Quochi, A. Mura and G. Bongiovanni, Nat. Commun., 2014, 5, 5049 CrossRef CAS PubMed.
- M. Cadelano, V. Sarritzu, N. Sestu, D. Marongiu, F. Chen, R. Piras, R. Corpino, C. M. Carbonaro, F. Quochi, M. Saba, A. Mura and G. Bongiovanni, Adv. Opt. Mater., 2015, 3, 1557–1564 CrossRef CAS.
- H. Zhou, Q. Chen, G. Li, S. Luo, T.-b. Song, H.-S. Duan, Z. Hong, J. You, Y. Liu and Y. Yang, Science, 2014, 345, 542–546 CrossRef CAS PubMed.
- N. J. Jeon, J. H. Noh, W. S. Yang, Y. C. Kim, S. Ryu, J. Seo and S. Il Seok, Nature, 2015, 517, 476–480 CrossRef CAS PubMed.
- C. Wehrenfennig, G. E. Eperon, M. B. Johnston, H. J. Snaith and L. M. Herz, Adv. Mater., 2014, 26, 1584–1589 CrossRef CAS PubMed.
- S. D. Stranks, G. E. Eperon, G. Grancini, C. Menelaou, M. J. P. Alcocer, T. Leijtens, L. M. Herz, A. Petrozza and H. J. Snaith, Science, 2013, 342, 341–344 CrossRef CAS PubMed.
- V. D'Innocenzo, A. R. Srimath Kandada, M. De Bastiani, M. Gandini and A. Petrozza, J. Am. Chem. Soc., 2014, 136, 17730–17733 CrossRef PubMed.
- E. T. Hoke, D. J. Slotcavage, E. R. Dohner, A. R. Bowring, H. I. Karunadasa and M. D. McGehee, Chem. Sci., 2015, 6, 613–617 RSC.
- S. Pathak, N. Sakai, F. Wisnivesky Rocca Rivarola, S. D. Stranks, J. Liu, G. E. Eperon, C. Ducati, K. Wojciechowski, J. T. Griffiths, A. A. Haghighirad, A. Pellaroque, R. H. Friend and H. J. Snaith, Chem. Mater., 2015, 27, 8066–8075 CrossRef CAS.
- G. Xing, N. Mathews, S. S. Lim, N. Yantara, X. Liu, D. Sabba, M. Grätzel, S. Mhaisalkar and T. C. Sum, Nat. Mater., 2014, 13, 476–480 CrossRef CAS PubMed.
- Q. Zhang, S. T. Ha, X. Liu, T. C. Sum and Q. Xiong, Nano Lett., 2014, 14, 5995–6001 CrossRef CAS PubMed.
- Y. Fu, H. Zhu, A. W. Schrader, D. Liang, Q. Ding, P. Joshi, L. Hwang, X.-Y. Zhu and S. Jin, Nano Lett., 2016, 16, 1000–1008 CrossRef CAS PubMed.
- H. Zhu, Y. Fu, F. Meng, X. Wu, Z. Gong, Q. Ding, M. V Gustafsson, M. T. Trinh, S. Jin and X. Zhu, Nat. Mater., 2015, 14, 636–642 CrossRef CAS PubMed.
- S. Yakunin, L. Protesescu, F. Krieg, M. I. Bodnarchuk, G. Nedelcu, M. Humer, G. De Luca, M. Fiebig, W. Heiss and M. V Kovalenko, Nat. Commun., 2015, 6, 8056 CrossRef CAS PubMed.
- A. V. Uskov, Y. Boucher, J. Le Bihan and J. McInerney, Appl. Phys. Lett., 1998, 73, 1499–1501 CrossRef CAS.
- O. Gauthier-Lafaye, P. Boucaud, F. H. Julien, S. Sauvage, S. Cabaret, J.-M. Lourtioz, V. Thierry-Mieg and R. Planel, Appl. Phys. Lett., 1997, 71, 3619 CrossRef CAS.
- F. H. Julien, A. Saár and J. Wang, Electron. Lett., 1995, 31, 838–839 CrossRef.
- C. C. Stoumpos, C. D. Malliakas and M. G. Kanatzidis, Inorg. Chem., 2013, 52, 9019–9038 CrossRef CAS PubMed.
- T. Baikie, Y. Fang, J. M. Kadro, M. Schreyer, F. Wei, S. G. Mhaisalkar, M. Gratzel and T. J. White, J. Mater. Chem. A, 2013, 1, 5628 CAS.
- V. D'Innocenzo, G. Grancini, M. J. P. Alcocer, A. R. S. Kandada, S. D. Stranks, M. M. Lee, G. Lanzani, H. J. Snaith and A. Petrozza, Nat. Commun., 2014, 5, 3586 Search PubMed.
- G. Grancini, A. R. Srimath Kandada, J. M. Frost, A. J. Barker, M. De Bastiani, M. Gandini, S. Marras, G. Lanzani, A. Walsh and A. Petrozza, Nat. Photonics, 2015, 9, 695–701 CrossRef CAS PubMed.
- Y. P. Varshni, Physica, 1967, 34, 149–154 CrossRef CAS.
- H. H. Fang, R. Raissa, M. Abdu-Aguye, S. Adjokatse, G. R. Blake, J. Even and M. A. Loi, Adv. Funct. Mater., 2015, 25, 2378–2385 CrossRef CAS.
- C. Wehrenfennig, M. Liu, H. J. Snaith, M. B. Johnston and L. M. Herz, APL Mater., 2014, 2, 081513 CrossRef.
- W. Kong, Z. Ye, Z. Qi, B. Zhang, M. Wang, A. Rahimi-Iman and H. Wu, Phys. Chem. Chem. Phys., 2015, 17, 16405–16411 RSC.
- K. Wu, A. Bera, C. Ma, Y. Du, Y. Yang, L. Li and T. Wu, Phys. Chem. Chem. Phys., 2014, 16, 22476–22481 RSC.
- W.-J. Yin, T. Shi and Y. Yan, Appl. Phys. Lett., 2014, 104, 063903 CrossRef.
- H. Wang, L. Whittaker-Brooks and G. R. Fleming, J. Phys. Chem. C, 2015, 119, 19590–19595 CAS.
- R. L. Milot, G. E. Eperon, H. J. Snaith, M. B. Johnston and L. M. Herz, Adv. Funct. Mater., 2015, 25, 6218–6227 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6tc01360k |
| This journal is © The Royal Society of Chemistry 2016 |