
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Single-step synthesis of hyperbranched, luminescent Mn2+-doped ZnSe1−xSx nanocrystals using dichalcogenide precursors†
Mohammad S.
Yazdanparast
,
Matthew T.
Webb
and
Emily J.
McLaurin
*
Department of Chemistry, 213 Chemistry and Biochemistry Building, Kansas State University, Manhattan, KS 66506, USA. E-mail: mclaurin@k-state.edu
First published on 20th June 2016
Abstract
Taking advantage of dichalcogenide precursors, a simple, single-step heat-up method for obtaining Mn2+-doped ZnSe1−xSx nanocrystals (NCs) is presented. Tuning the ratios of diselendide and disulfide precursors results in alloys with varying shape, size and composition. When diphenyldiselenide (Ph2Se2) is used, highly branched networks of small NCs form. Dimethydisulfide (Me2S2) induces formation of larger NCs with less branching. Mixtures of the two exhibit branching proportional to the amount of Ph2Se2vs. Me2S2, allowing formation of a series of branched Mn2+-doped NCs. Interestingly, these NCs exhibit photoluminescence (PL) characteristic of Mn2+, despite the large number of defects and unusual shapes they possess. This demonstrates Mn2+ can be successfully doped into NCs with several degrees of branching, in which it acts as an efficient radiative trap. Addition of thiol to the NCs led to a large enhancement in the Mn2+ PL. In organic solution, the sensitivity to thiol varied with the degree of NC branching, with hyperbranching NCs giving the most sensitive response. After transfer to aqueous solution, the Mn2+ PL of the hyperbranched NCs increased dramatically in response to μM concentrations of dithiothreitol (DTT).
Introduction
Mn2+-doped semiconductors are widely used in many photo- and electroluminescent applications.1,2 The availability of luminescent colloidal nanocrystals (NCs) of these materials expanded their use to bioimaging and sensing,3–5 and solar energy,6–8 among other applications.9,10 The nature of the Mn2+ dopants can provide a probe of local environment, elucidating NC structural information via their optical and magnetic properties.11–14 Applications involving Mn2+ phosphorescence involve doping of wide bandgap semiconductors.15 The fixed 4T1 energy level of the Mn2+ provides a trap for energy absorbed by the NC. This energy is released radiatively as an orange luminescence. This is only observed when the conduction band energy level is above the energy of the Mn2+ 4T1, and thus frequently the zinc chalcogenides are employed as host materials.3,13,15–18Dichalcogenides, namely diorganyl dichalcogenides, have recently been revived as synthons in colloidal NC syntheses.19–22 They are touted for their ability to provide access to unfavourable crystal phases,19 resulting in a myriad of NC shapes, compositions, and properties.20–22 Their reactivity can be tuned by changing the organic substituents, allowing formation of quantum dots, tetrapods, nanorods, and other asymmetric, hyperbranched NCs from nearly identical reaction conditions.20
Hyperbranched and asymmetric semiconductor nanocrystals have large surface-area-to-volume ratios often resulting in a large number of surface defects.23,24 Both of these characteristics garner interest for use of these NCs for selective, efficient catalysis,25 gas adsorption and sensing,26 and defect-related luminescence.27
Luminescent Mn2+-doped chalcogenide NCs are readily synthesized using colloidal methods.15,16 Protocols using binary clusters,28,29 multi-step shell additions,12 and post-synthetic diffusion30 have been established. Each method provides advantages and disadvantages, such that the synthetic method can be chosen depending on the application requirements. Overall, these methods have multiple steps often involving reactive, expensive precursors, and the products of these syntheses have been limited in scope. This is ascribed to the difficulty in doping transition metal ions into anisotropic NCs.31,32 Although there are numerous reports of emissive Mn2+-doped spherical NCs and even nanorods,33–39 there are only two reports of luminescent asymmetric Mn2+-doped NCs.32,40 In the detailed work of Wu & Warner, luminescent Mn2+-doped ZnSe NCs were synthesized with spherical and pod-like structures. The branching is ascribed to polymorphism, resulting in tetrapods, with average sizes of 15 nm.24,40 A sophisticated, successful effort to obtain asymmetric luminescent Mn2+-doped NCs was presented by Yao et al. in which a one-pot synthesis generated elongated doped NCs exhibiting Mn2+ emission. The worm-like shapes and defects support an oriented-attachment-type growth of these 4–10 nm long particles.32,41,42
Here we report the first example, to the best of our knowledge, of luminescent Mn2+-doped hyperbranched NCs. Zinc chalcogenides were chosen to exploit the characteristic luminescence observed from the Mn2+ 4T1 → 6A1 transition. Changing the dichalcogenide precursors resulted in different morphologies, with compositions varying with precursor ratios. An electron paramagnetic resonance (EPR) signal is observed for the NCs, providing additional evidence for doping and information about the NC structure. This simple procedure provides a new synthetic method for successful incorporation of Mn2+ in wide bandgap ZnSe1−xSx semiconductor NCs using dichalcogenide precursors. The incorporation of a radiative defect allows luminescence to be observed from these materials.
Experimental section
Materials
All the following chemicals are commercially available and were used without further purification; manganese(II) acetate tetrahydrate (Mn(OAc)2·4H2O, ≥99%), manganese(II) chloride tetrahydrate (MnCl2·4H2O, 99.99%), oleylamine (OLA, ≥98%), stearic acid (SA, ≥95%), dodecane thiol (DDT, ≥98%), selenium (≥99.5%), tetramethylammonium chloride (NMe4Cl, ≥98%), triethylamine (TEA), cyclohexane, toluene, water, phosphate buffered saline (PBS), and chloroform from Aldrich, diphenyl diselenide (Ph2Se2, 99%), dimethyl disulfide (Me2S2, 99%), phenylselenol (PhSeH, 98%), and methanol (MeOH) from Acros, zinc nitrate hexahydrate (Zn(NO3)2·6H2O) from J. T. Baker, acetone, dithiothreitol (DTT), and L-lysine from Fisher, and zinc undecylenate (Zn(Un)2) from Gelest.Preparation of manganese stearate (Mn(St)2)
A mixture of Mn(OAc)2·4H2O (1.000 g, 4.08 mmol) and stearic acid (5.226 g, 18.37 mmol) were degassed under vacuum at 100 °C and then under inert atmosphere, the temperature was increased to 180 °C and held for 20 minutes. After cooling the reaction mixture to 100 °C, the reaction mixture was degassed for another 60 minutes. The flask was cooled down to room temperature and a white solid was obtained.Hyperbranched Mn2+:ZnSe (1)
In a three neck flask Mn(St)2 (∼0.003 g, 5 μmol), zinc undecylenate (0.0865 g, 0.200 mmol), Ph2Se2 (0.1000 g, 0.32 mmol) and OLA (5.00 g, 18.7 mmol) were degassed at 100 °C for 90 minutes and then under nitrogen atmosphere, the temperature was increased to 280 °C and held for 10–60 minutes. Next, the heating mantle was removed, and the reaction mixture was cooled down to 65 °C immediately using a water bath. Finally, 2 ml of toluene was added to the reaction mixture followed by 5 ml of methanol. The particles were separated by centrifugation, resuspended in toluene, and precipitated with methanol again. The final sample was dispersed in cyclohexane. Mn2+:ZnSe NCs were also synthesized using Mn(OAc)2·4H2O and MnCl2·4H2O under analogous conditions.Mn2+:ZnSe1−xSx (2–5)
Samples were prepared using a procedure similar to that for 1, but with mixtures of Ph2Se2 and Me2S2 or Ph2S2. The total amount of dichalcogenide was kept near ∼0.33 mmol. Ratios of Ph2Se2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :R2S2 were approximately 3:1 (2), 1:1 (3a), 4:5 (3b), and 1:3 (4). In the case of Mn2+:ZnS (5), Me2S2 (31 μl, 0.35 mmol) was used instead of Ph2Se2 and the reaction was carried out at 300 °C. Because of the lower boiling point of Me2S2, it must be injected into the reaction mixture after the degassing step.
:R2S2 were approximately 3:1 (2), 1:1 (3a), 4:5 (3b), and 1:3 (4). In the case of Mn2+:ZnS (5), Me2S2 (31 μl, 0.35 mmol) was used instead of Ph2Se2 and the reaction was carried out at 300 °C. Because of the lower boiling point of Me2S2, it must be injected into the reaction mixture after the degassing step.
[NMe4]2[Zn4(SePh)10]
The cluster precursor was prepared by adapting previous methods.43,44 Three separate solutions were prepared and degassed for 30–60 minutes by bubbling with N2: (1) Zn(NO3)2·6H2O (7.318 g) in 35 ml MeOH, (2) Me4NCl (4.42 g) in 20 ml MeOH, and (3) TEA (9.5 g) in 20 ml MeOH. Under a N2 overpressure, PhSeH (10 g) was added to solution 3, followed by stirring for 20 minutes. Solution 1 was then transferred to solution 3 dropwise via cannula, forming a cloudy solution. Next, solution 2 was cannula transferred to solution 3 over a period of 10–30 min, followed by cooling in an ice bath for 30 minutes. The solid was then filtered and rinsed thoroughly with MeOH and toluene, yielding a white powder.Mn2+:ZnSe (6)
Unbranched Mn2+:ZnSe NCs were synthesized by adapting previously described methods.28,44 Briefly, OLA (10 g) and MnCl2·4H2O (0.0012 g, 6.1 μmol) were combined in a flask and degassed at 100 °C under vacuum for 120 minutes. After cooling below 80 °C, the cluster precursor (0.2 g, 0.073 mmol) and Se (0.0108 g, 0.137 mmol) were added under N2 overpressure. After briefly degassing the solution under vacuum at 100 °C, the reaction was heated to 280 °C under N2 and the NCs were grown at this temperature for 15 minutes. After cooling below 80 °C, the NCs were isolated by precipitation and resuspension with MeOH and toluene, respectively.Water-soluble hyperbranched Mn2+:ZnSe
A simple ligand-exchange method was used to suspend the NCs in aqueous solution. Previously prepared NCs were dispersed in toluene, precipitated out using methanol, and isolated by centrifugation. This procedure was repeated 3–4 times. Then, the NCs were dispersed in chloroform, precipitated out after adding acetone, and isolated by centrifugation. This process was repeated twice. Next, the NCs were dispersed in chloroform (4 ml) and an aqueous solution of L-lysine (0.03 g in 10 ml water) was added to it and stirred vigorously overnight. Then, the NCs were precipitated out by centrifugation and dispersed in PBS.Thiol titrations
Thiol titration experiments were performed using a PTI Quanta Master 400 fluorometer. The samples were prepared in a 10 mm septum-sealed screw-top cuvette (Starna Cells, Inc.) with a stir bar and were diluted to have absorbance ∼0.3 at 350 nm. A syringe pump (New Era Pump Systems, Inc.) was used for the titration with injection of thiol via a syringe with NanoTight™ adapters (Idex Health & Science, LLC) and 250 μm diameter flexible fused silica capillary tubing (Polymicro Technologies) which was inserted through the cuvette septum. While stirring, spectra were taken at intervals of 2–30 minutes, depending on sample brightness and sensitivity. Titrations were done until sample saturation.Physical measurements
In this study a Cary 500 UV-Vis-NIR spectrophotometer was used for recording UV-Vis absorption spectra. A PTI Quanta Master 400 fluorometer was used to record photoluminescence and photoluminescence excitation spectra. For transmission electron microscopy (TEM) analysis, a drop of sample was dried on a copper grid. TEM images were recorded using a FEI Tecnai G2 Spirit BioTWIN microscope. The EPR spectrum was recorded using a Bruker EMXplus X-band EPR spectrometer. Powder X-ray diffraction (XRD) patterns were recorded using a PANalytical Empyrean multi-purpose X-ray diffractometer.Results and discussion
Formation of Mn2+:ZnSe1−xSx nanocrystals
This procedure is related to the synthesis of Mn2+-doped II–VI NCs using a tetramer ([Zn4(EPh)10]2−) or decamer ([Zn10Se4(EPh)16]4−) cluster precursor. Both methods nucleate and grow NCs in a single, heat-up step and the cluster contains organyl chalcogenide ligands (phenylthiols and phenylselenols) similar to the dichalcogenides used here.28,29 The most widely used cluster has E = S, likely because of the high cost of the selenol precursor (vs. thiol). However, NCs synthesized with this precursor are known to contain sulphur impurities.29 The key advantages of the dichalcogenide-doping method presented here are the simplified procedure, as preparation of cluster precursor(s) is not required, and the air-stable diselenide precursor is substantially less expensive than the phenylselenol precursor required for metal-selenide cluster synthesis.The procedure used to obtain the NCs is shown in Fig. 1. Generally, a Zn2+ precursor (zinc undecylenate), Mn2+ precursor (manganese stearate), and dichalcogenide(s) are combined in a coordinating solvent (oleylamine). After heating at 280 °C in an inert atmosphere for 10–60 minutes, NCs form. The exception was for Mn2+:ZnS (5), which required 300 °C to obtain a significant quantity of NCs. The NCs are isolated by precipitation and resuspension in organic solvent followed by characterization to determine NC photophysical and magnetic properties, and structures.
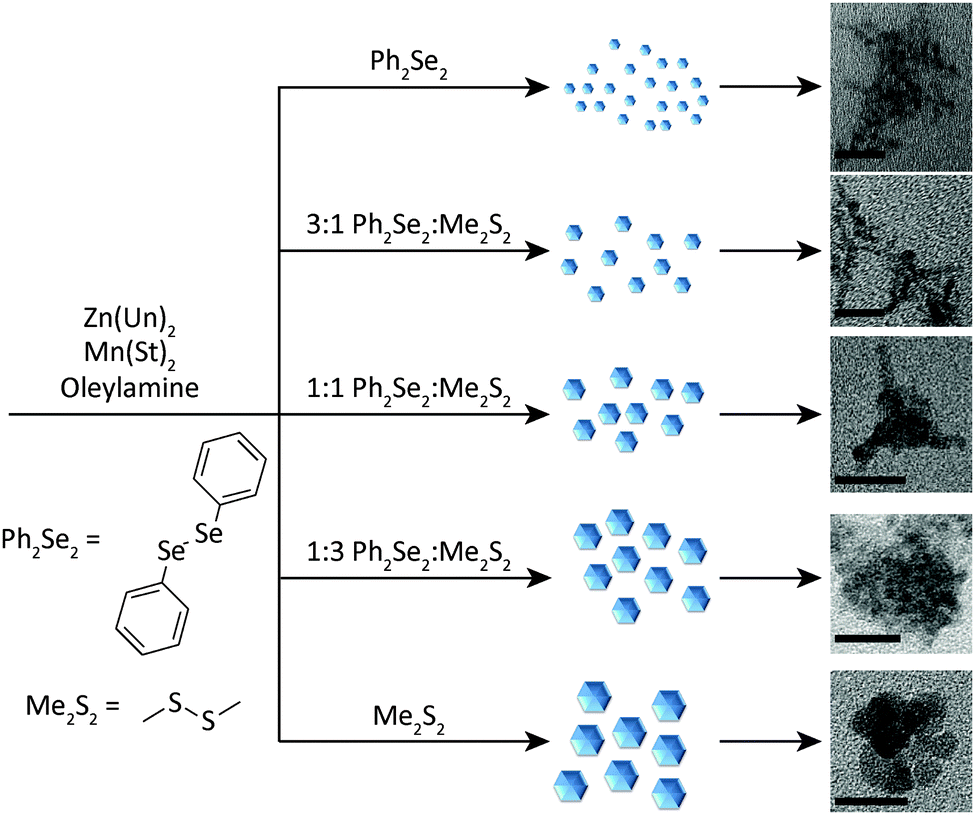 | ||
| Fig. 1 General synthetic scheme for formation of Mn2+-doped ZnSe1−xSx NCs. In the reaction, zinc undecylenate, Zn(Un)2, manganese stearate, Mn(St)2, and a mixture of diphenyl diselenide and dimethyl disulphide (or diphenyl disulphide) are combined and heated to reaction temperature in an alkylamine under an inert atmosphere. Depending on the ratio of diselenide to disulphide, NCs of different shapes and sizes are obtained. Images of samples 1–5 are shown. Scale bars are 20 nm. | ||
Initially, NCs were synthesized using only the Ph2Se2 dichalcogenide precursor (1). These NCs exhibited absorption and PL spectra characteristic of Mn2+:ZnSe NCs (Fig. 2, black line). The absorption feature near 390 nm indicates formation of the ZnSe host NCs. The PL spectrum has a peak centred at 586 nm, indicative of Mn2+ luminescence. The rapid transfer of energy from the NC to the Mn2+ efficiently competes with excitonic recombination, and no blue ZnSe emission is observed. Subsequent reactions combined the Ph2Se2 precursor with a disulphide precursor, to form Mn2+:ZnSe1−xSx NCs of different shapes. Two disulphide precursors were used: Ph2S2 and Me2S2. Starting ratios of diselenide to disulphide were varied from 1:0 (1), 3:1 (2), 1:1 (3), 1:3 (4), and 0:1 (5). The first absorption feature of the NCs blue-shifted from that of 1, indicating likely sulphur incorporation, with a systematic decrease in wavelength with increasing Me2S2.
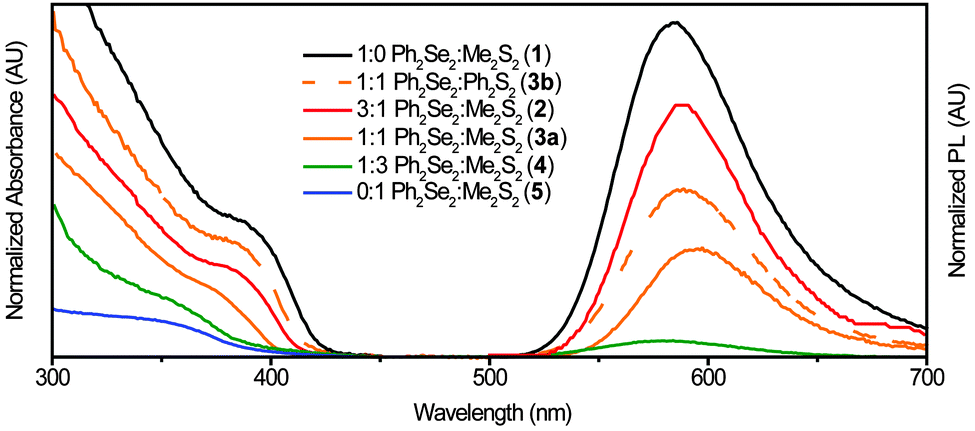 | ||
| Fig. 2 Absorption (left) and PL (right) spectra of Mn2+:ZnSe1−xSx NCs with varying dichalcogenide precursor ratios. The NC first absorption peak shifts from ∼390 nm for Ph2Se2 alone (1) to a broad feature at ∼340 nm for Me2S2 alone (5). The PL peak red-shifts from 586 nm to 589 nm with increasing S. For 4, very little luminescence was detected and for 5 no luminescence was detected. | ||
Comparing absorption and emission spectra for the Me2S2 and Ph2S2 samples with similar ratios (3a and 3b), yields insight into the effect of disulphide precursor on the resulting product. Although similar ratios were used, the absorption of 3a is noticeably blue-shifted from that of 3b, indicating decreased reactivity of Ph2S2vs. Me2S2, as reported previously.20 These NCs (3b) exhibit a PL peak at 588 nm, slightly red-shifted from the PL peak of 1 at 586 nm. For the other NC samples, the PL peaks red-shifted to 589 nm with increasing disulphide (2, 3a). The PL intensity decreased with larger Me2S2 amounts, such that only a broad, very weak peak was observed for 4 and none was detectable in the Me2S2 only sample (5). To confirm the orange PL associated with Mn2+ was coming from the NCs, photoluminescence excitation (PLE) spectra were taken. The resulting spectra (Fig. S1, ESI†) exhibit the characteristic first absorption feature of the NCs with a continued increase at higher energies.
Morphology and structure of Mn2+:ZnSe1−xSx nanocrystals
The key to changing the NC shape and degree of branching is the ratio of diselenide to disulphide precursor. Fig. 3 shows TEM images of NCs prepared with varying ratios of diselenide and disulphide. As the amount of disulphide increases, the particles go from branched networks to rounded, linked platelets. Comparing 3a and 3b (Fig. 3d and b) further illustrates the effect of disulphide on the reaction. The TEM image of 3b more closely resembles that of 1 (Fig. 3a), exhibiting significant branching. This branching appears to stem not only from the centre of a particle, but also from the outward reaching NC arms (Fig. S2–S5, ESI†). The appearance of isolated particles (shown in Fig. 3 insets) within the samples suggests oriented attachment may be occurring as well, creating highly branched networks.41,45,46 This will be discussed further in the discussion of the formation mechanism. TEM images of 2 and 3a (Fig. 3c and d) also exhibit networks of hyperbranched particles. Although these networks are complex, the insets provide key details about the smaller, branched particles forming the larger networks. For both 2 and 3a, smaller particles with more rounded, thicker arms are observed. These characteristics are passed on to the networks that form. Further increases in the S:Se ratio show formation of larger, rounder particles, followed by oriented attachment to form large, spherical clusters of NCs (Fig. 3e and Fig. S6, ESI†). When only Me2S2 is used as the dichalcogenide precursor, large, round platelets form and link together, generating round clusters of these NCs (Fig. 3f and Fig. S7, ESI†). Syntheses of Mn2+:ZnSe with other Mn2+ precursors produced similar hyperbranched NCs (Fig. S8, ESI†).
 | ||
| Fig. 3 TEM images of (a) 1, (b) 3b, (c) 2, (d) 3a, (e) 4, and (f) 5. Large image scale bars are 50 nm. Inset image scale bars are 50 nm for a–c and 20 nm for d–f. Highly branched structures at low S:Se ratios transition to more spherical samples at higher S:Se ratios. Insets highlight particles that are linking together, likely through oriented attachment, to form larger networks and aggregates of the NCs. The shape, size, and structure of these particles is reflected in the larger structure of the resulting nanomaterial. | ||
Additional structural information about the NCs was obtained from XRD. The diffraction patterns in Fig. 4a exhibit a zinc-blende pattern, with highly broadened peaks due to the small domain size of the NCs. With increasing disulphide precursor, the peaks shift to larger angles, characteristic of a transition from ZnSe to ZnS. Peak positions indicate larger sulphur content for sample 3a than 3b, providing additional evidence for the low reactivity of Ph2S2vs. Me2S2. Ultimately, the peak shifts for 3a are small relative to what is expected for a 1:1 ratio of S:Se in the NCs, indicating a decreased reactivity for Me2S2 (vs. Ph2Se2). No Mn diffraction peaks are observed, indicating successful doping of Mn2+ ions into the NCs.47
 | ||
| Fig. 4 (a) XRD patterns of samples 1, 2, 3a, 3b, and 4. The patterns indicate zinc-blend structure and the broadness of the peaks indicates small NC diameters or domains. As the amount of S increases, the peaks shift to larger angles. The black lines at the bottom of the figure represent the positions of bulk ZnSe and the blue lines represent those of bulk ZnS. (b) EPR spectrum of 3b showing a splitting pattern with 6 peaks and a hyperfine coupling constant of 66.2 G. | ||
Additional evidence of successful Mn2+ doping into these NCs is provided by EPR. The EPR spectrum of 3b is shown in Fig. 4b. The 6-peak hyperfine structure characteristic of Mn2+ is observed. In addition, a coupling constant of 66.2 G is estimated, consistent with values found for ZnSe (66.1 G)16 and ZnS (68.4 G).48 The estimated value is substantially smaller than that of surface Mn2+ (∼90 G),13,14,49 further affirming Mn2+ incorporation.
Possible formation mechanisms
The dichalcogenides are known to produce nanomaterials with a variety of morphologies.20–22 Vela and co-workers presented an elegant study in which differences in dichalcogenide bond dissociation energies were correlated with changes in particle size and shape, and found diselenide precursors reacted more quickly than analogous disulphides.20 Although the systems studied differ from those presented here (they used Cd2+, different ligands, lower temperatures, and hot-injection), the results can help explain the observed trend in particle size and shape.The formation of the NCs shown in Fig. 3 can be explained in terms of the precursor reactivity of the disulphide vs. Ph2Se2. Thus, for the faster reacting Ph2Se2-containing reactions, the initially formed particles are relatively small in diameter, and begin branching and attaching to form the more complex networks seen in Fig. 3a–d. This behaviour has been attributed to the introduction of more branching angles due to twinning defects that occur in samples with more rapid growth.23 When larger amounts of the less reactive Me2S2 are present, larger particles form due to slow growth, which leads to less branching. Over time, these particles begin to link together, likely via oriented attachment.24,41 At final reaction times, the reaction product is a network of these linked particles. A cartoon of this possible mechanism is shown in Fig. 1, in which with less Me2S2, smaller particles form and aggregate, forming more highly-branched, polycrystalline networks. With more Me2S2, larger NCs form before coming together to form rounded aggregates of platelets. Thus, there does not appear to be an additional ripening process occurring during the attachment process, and the final sample is characteristic of the initial NC size, as observed for similar systems.20
Application of NCs for thiol sensing
As mentioned in the introduction, an advantage of branched NCs is their large surface area, which makes them useful for applications in sensing, among others.25–27 Thiols are known to influence processes in biology and play key roles in nanodevices.50–54 To explore the potential application of these highly branched Mn2+-doped NCs, the luminescence enhancement in the presence of thiol was studied.DDT was used as a model thiol and Fig. 5a depicts the PL enhancement observed when small amounts of DDT are added to a solution of 3b. The Mn2+ PL increases, until eventually saturating at a concentration of ∼70 mM, as shown in Fig. 5b. Similar, less-sensitive behaviour is observed for the less branched NCs (2) and NCs with no branching (6). This change in sensitivity could be due to the large number of NC defects, which decrease in the order: hyperbranched > branched > unbranched. Addition of DDT may passivate these defects, providing more efficient energy transfer to the Mn2+-dopants, and a brighter Mn2+ luminescence.55–57
 | ||
| Fig. 5 (a) Steady-state PL of 3b in the presence of dodecanethiol (DDT). Increasing the amount of thiol leads to a concomitant increase in Mn2+ PL from the NCs. (b) Scatter plot of the Mn2+ PL intensity vs. [DDT] for NC samples with different degrees of branching. The more branched 3b NCs show the steepest increase in PL intensity with thiol. NCs with less (2) and no (6) branching show decreasing sensitivity to thiol, with smaller increases in slope. | ||
To demonstrate the utility of these NCs for biological sensing, the NCs were transferred to aqueous solution. After transfer, the Mn2+ luminescence of the NCs decreased, but not significantly. The NCs illuminated by a UV lamp before and after water-solubilization are shown in Fig. 6a. Once in aqueous solution, the NCs were titrated with dithiothreitol (DTT), with a corresponding increase in PL (Fig. 6b). DTT is key in the study of biological functions such as protein unfolding, signal transduction, and as a redox mediator.58–61 Here, it acts as a reductant, enhancing the NC PL at concentrations from 0 to ∼40 μM before decreasing again at concentrations >∼60 μM (Fig. 6c). The sensitivity of the NCs to DTT is linear within this range, with an analytical sensitivity of 2.9 μM and a detection limit of 1.0 μM.
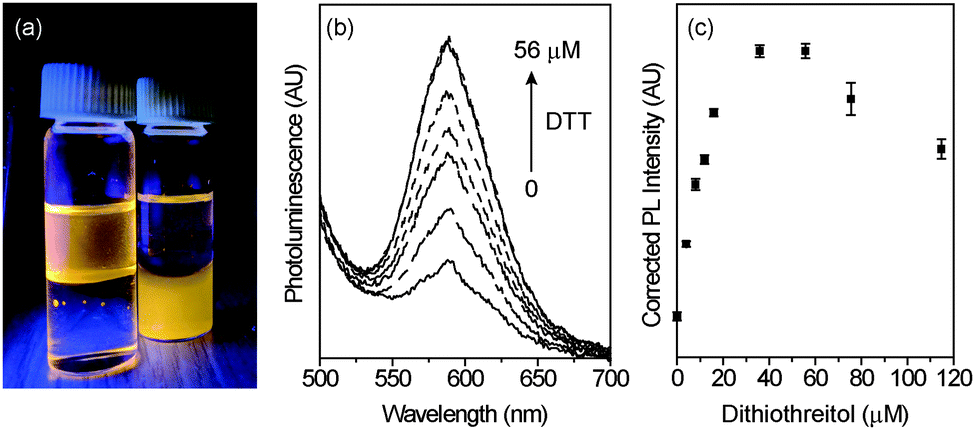 | ||
| Fig. 6 (a) Photograph of Mn2+-doped hyperbranched NCs illuminated by a UV lamp in aqueous solution and in chloroform. (b) Steady-state PL of hyperbranched Mn2+–ZnSe NCs in PBS with addition of a dithiothreitol (DTT) solution. As the concentration of DTT increases from 0 to 56 μM, the Mn2+ PL increases. (c) Scatter plot of the Mn2+ PL intensity vs. [DTT] exhibiting a linear increase in luminescence in the μM range before decreasing in intensity at concentrations >60 μM. | ||
This simple sensor exhibits a large, linear increase in luminescence with thiol, highlighting the advantages of these NCs as sensors. In organic solvent, differences in sensitivity (ΔPL vs. ΔDDT) vary with branching, although additional study is required to explore the complex interplay of structure, branching, defect formation, and dopant incorporation, which all likely play a role in the luminescence response. In aqueous medium, enhancement of PL by DTT is linear at low concentrations, with high sensitivities. The mechanism of PL increase is likely related to passivation of surface traps, providing a pathway for enhanced sensitivity to biological thiols through the increase in surface defects present in these hyperbranched Mn2+-doped materials.
Conclusions
A new synthetic method for obtaining doped ZnSe1−xSx NCs yields luminescent NCs of a variety of structures. Variations in dichalcogenide precursor can be used to tune the NC size and shape. Knowing these trends in reactivity, syntheses can be designed to obtain NCs of desired size, shape, and dopant incorporation. This method provides access to luminescent materials that are not usually emissive due to high defect levels. The resulting emissive, hyperbranched materials decrease losses in PL due to defects by employing Mn2+ as a luminescent trap. These NCs can be used as turn-on thiol sensors, with sensitivity varying with branching in organic solution. A similar response is observed in aqueous solution, with a large increase in PL at low thiol levels. This synthetic procedure is extendable to other NC systems synthesized using dichalcogenides and other dopants, providing a new pathway to luminescent, hyperbranched materials.Acknowledgements
This research was supported by Kansas State University. A KS-USRG provided financial support. TEM images were obtained through the Nanotechnology Innovation Centre of Kansas State (NICKS). The authors thank C. Gura for assistance with XRD (Department of Geology) and J. Douglas for assistance with EPR (University of Kansas).References
- T. Jüstel, in Luminescence, ed. C. Ronda, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2007, ch. 3, pp. 61–73 Search PubMed.
- G. Blasse and B. C. Grabmaier, Luminescent Materials, Springer, Berlin Heidelberg, 1994, ch. 6, pp. 108–133 Search PubMed.
- R. Thakar, Y. Chen and P. T. Snee, Nano Lett., 2007, 7, 3429–3432 CrossRef CAS PubMed.
- S. Santra and D. Dutta, in Nanoparticles in Biomedical Imaging, ed. J. W. M. Bulte and M. M. J. Modo, Springer, New York, 2008, ch. 22, vol. 3, pp. 463–485 Search PubMed.
- P. Wu and X.-P. Yan, Chem. Soc. Rev., 2013, 42, 5489–5521 RSC.
- P. K. Santra and P. V. Kamat, J. Am. Chem. Soc., 2012, 134, 2508–2511 CrossRef CAS PubMed.
- T. Debnath, P. Maity, S. Maiti and H. N. Ghosh, J. Phys. Chem. Lett., 2014, 5, 2836–2842 CrossRef CAS PubMed.
- J. Tian, L. Lv, C. Fei, Y. Wang, X. Liu and G. Cao, J. Mater. Chem. A, 2014, 2, 19653–19659 RSC.
- Z. Hu, S. Xu, X. Xu, Z. Wang, Z. Wang, C. Wang and Y. Cui, Sci. Rep., 2015, 5, 14817 CrossRef CAS PubMed.
- H.-J. Kim, H.-D. Lee, C. S. S. P. Kumar, S. S. Rao, S.-H. Chung and D. Punnoose, New J. Chem., 2015, 39, 4805–4813 RSC.
- S. Ithurria, P. Guyot-Sionnest, B. Mahler and B. Dubertret, Phys. Rev. Lett., 2007, 99, 265501 CrossRef PubMed.
- Y. Yang, O. Chen, A. Angerhofer and Y. C. Cao, J. Am. Chem. Soc., 2008, 130, 15649–15661 CrossRef CAS PubMed.
- S. Acharya, D. D. Sarma, N. R. Jana and N. Pradhan, J. Phys. Chem. Lett., 2010, 1, 485–488 CrossRef CAS.
- W. Zheng, Z. Wang, J. Wright, B. Goundie, N. S. Dalal, R. W. Meulenberg and G. F. Strouse, J. Phys. Chem. C, 2011, 115, 23305–23314 CrossRef CAS.
- R. Beaulac, S. T. Ochsenbein and D. R. Gamelin, in Nanocrystal Quantum Dots, ed. V. I. Klimov, CRC Press, Boca Raton, FL, USA, 2nd edn, 2010, ch. 11, pp. 397–453 Search PubMed.
- D. J. Norris, N. Yao, F. T. Charnock and T. A. Kennedy, Nano Lett., 2001, 1, 3–7 CrossRef CAS.
- R. N. Bhargava, D. Gallagher, X. Hong and A. Nurmikko, Phys. Rev. Lett., 1994, 72, 416–419 CrossRef CAS PubMed.
- J. H. Yu, S.-H. Kwon, Z. Petrášek, O. K. Park, S. W. Jun, K. Shin, M. Choi, Y. I. Park, K. Park, H. B. Na, N. Lee, D. W. Lee, J. H. Kim, P. Schwille and T. Hyeon, Nat. Mater., 2013, 12, 359–366 CrossRef CAS PubMed.
- M. E. Norako, M. A. Franzman and R. L. Brutchey, Chem. Mater., 2009, 21, 4299–4304 CrossRef CAS.
- Y. Guo, S. R. Alvarado, J. D. Barclay and J. Vela, ACS Nano, 2013, 7, 3616–3626 CrossRef CAS PubMed.
- J. Wang, A. Singh, P. Liu, S. Singh, C. Coughlan, Y. Guo and K. M. Ryan, J. Am. Chem. Soc., 2013, 135, 7835–7838 CrossRef CAS PubMed.
- R. L. Brutchey, Acc. Chem. Res., 2015, 48, 2918–2926 CrossRef CAS PubMed.
- A. G. Kanaras, C. Sönnichsen, H. Liu and A. P. Alivisatos, Nano Lett., 2005, 5, 2164–2167 CrossRef CAS PubMed.
- H. Li, A. G. Kanaras and L. Manna, Acc. Chem. Res., 2013, 46, 1387–1396 CrossRef CAS PubMed.
- Z.-Y. Zhou, N. Tian, J.-T. Li, I. Broadwell and S.-G. Sun, Chem. Soc. Rev., 2011, 40, 4167–4185 RSC.
- T.-D. Nguyen, C.-T. Dinh and T.-O. Do, Chem. Commun., 2015, 51, 624–635 RSC.
- K. Nose, Y. Soma, T. Omata and S. Otsuka-Yao-Matsuo, Chem. Mater., 2009, 21, 2607–2613 CrossRef CAS.
- S. L. Cumberland, K. M. Hanif, A. Javier, G. A. Khitrov, G. F. Strouse, S. M. Woessner and C. S. Yun, Chem. Mater., 2002, 14, 1576–1584 CrossRef CAS.
- P. I. Archer, S. A. Santangelo and D. R. Gamelin, J. Am. Chem. Soc., 2007, 129, 9808–9818 CrossRef CAS PubMed.
- V. A. Vlaskin, C. J. Barrows, C. S. Erickson and D. R. Gamelin, J. Am. Chem. Soc., 2013, 135, 14380–14389 CrossRef CAS PubMed.
- J. H. Yu, X. Liu, K. E. Kweon, J. Joo, J. Park, K.-T. Ko, D. W. Lee, S. Shen, K. Tivakornsasithorn, J. S. Son, J.-H. Park, Y.-W. Kim, G. S. Hwang, M. Dobrowolska, J. K. Furdyna and T. Hyeon, Nat. Mater., 2010, 9, 47–53 CrossRef CAS PubMed.
- T. Yao, S. Kou, Y. Sun, Q. Zhao and J. Yang, CrystEngComm, 2012, 14, 8440–8445 RSC.
- J. D. Bryan and D. R. Gamelin, in Prog. Inorg. Chem., ed. K. D. Karlin, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2005, ch. 2, vol. 54, pp. 47–126 Search PubMed.
- S. W. Lu, B. I. Lee, Z. L. Wang, W. Tong, B. K. Wagner, W. Park and C. J. Summers, J. Lumin., 2000, 92, 73–78 CrossRef CAS.
- J. Y. Lee, D. S. Kim, J. H. Kang, S. W. Yoon, H. Lee and J. Park, J. Phys. Chem. B, 2006, 110, 25869–25874 CrossRef CAS PubMed.
- P. T. K. Chin, J. W. Stouwdam and R. A. J. Janssen, Nano Lett., 2009, 9, 745–750 CrossRef CAS PubMed.
- M. Zhang, Y. Lu, J.-F. Chen, T.-K. Zhang, Y.-Y. Liu, Y. Yang, W.-T. Yao and S.-H. Yu, Langmuir, 2010, 26, 12882–12889 CrossRef CAS PubMed.
- S. Acharya, S. Sarkar and N. Pradhan, J. Phys. Chem. C, 2013, 117, 6006–6012 CrossRef CAS.
- C. Q. Jin, C. H. Ge, G. Xu, Y. X. Wei, Q. P. Ding, M. Zhu and H. B. Duan, J. Alloys Compd., 2015, 648, 481–487 CrossRef CAS.
- Y. A. Wu and J. H. Warner, J. Mater. Chem., 2012, 22, 417–424 RSC.
- W. Lv, W. He, X. Wang, Y. Niu, H. Cao, J. H. Dickerson and Z. Wang, Nanoscale, 2014, 6, 2531–2547 RSC.
- X. Xue, R. L. Penn, E. R. Leite, F. Huang and Z. Lin, CrystEngComm, 2014, 16, 1419–1429 RSC.
- I. G. Dance, A. Choy and M. L. Scudder, J. Am. Chem. Soc., 1984, 106, 6285–6295 CrossRef CAS.
- V. A. Vlaskin, R. Beaulac and D. R. Gamelin, Nano Lett., 2009, 9, 4376–4382 CrossRef CAS PubMed.
- R. L. Penn and J. F. Banfield, Science, 1998, 281, 969–971 CrossRef CAS PubMed.
- H. Zhang, R. L. Penn, Z. Lin and H. Cölfen, CrystEngComm, 2014, 16, 1407–1408 RSC.
- B. Yang, X. Shen, H. Zhang, Y. Cui and J. Zhang, J. Phys. Chem. C, 2013, 117, 15829–15834 CrossRef CAS.
- B. J. Park, W. B. Im, W. J. Chung, H. S. Seo, J. T. Ahn and D. Y. Jeon, J. Mater. Res., 2007, 22, 2838–2844 CrossRef CAS.
- L. Zu, A. W. Wills, T. A. Kennedy, E. R. Glaser and D. J. Norris, J. Phys. Chem. C, 2010, 114, 21969–21975 CrossRef CAS.
- Z. J. Donhauser, B. A. Mantooth, K. F. Kelly, L. A. Bumm, J. D. Monnell, J. J. Stapleton, D. W. Price, A. M. Rawlett, D. L. Allara, J. M. Tour and P. S. Weiss, Science, 2001, 292, 2303–2307 CrossRef CAS PubMed.
- D. M. Townsend, K. D. Tew and H. Tapiero, Biomed. Pharmacother., 2003, 57, 145–155 CrossRef CAS.
- Q. Li and J. R. Lancaster, Nitric Oxide-Biol. Chem., 2013, 35, 21–34 CrossRef CAS PubMed.
- E. A. Weiss, Acc. Chem. Res., 2013, 46, 2607–2615 CrossRef CAS PubMed.
- L. Sun, Y. A. Diaz-Fernandez, T. A. Gschneidtner, F. Westerlund, S. Lara-Avila and K. Moth-Poulsen, Chem. Soc. Rev., 2014, 43, 7378–7411 RSC.
- A. L. Weaver and D. R. Gamelin, J. Am. Chem. Soc., 2012, 134, 6819–6825 CrossRef CAS PubMed.
- J. D. Rinehart, A. L. Weaver and D. R. Gamelin, J. Am. Chem. Soc., 2012, 134, 16175–16177 CrossRef CAS PubMed.
- S. Sarkar, B. K. Patra, A. K. Guria and N. Pradhan, J. Phys. Chem. Lett., 2013, 4, 2084–2090 CrossRef CAS PubMed.
- R. E. Feeney, R. B. Yamasaki and K. F. Geoghegan, in Modification of Proteins, ed. R. E. Feeney and J. R. Whitaker, American Chemical Society, Washington, DC, USA, 1982, ch. 1, vol. 198, pp. 3–55 Search PubMed.
- S. García-Santamarina, S. Boronat and E. Hidalgo, Biochemistry, 2014, 53, 2560–2580 CrossRef PubMed.
- W. W. Cleland, Biochemistry, 1964, 3, 480–482 CrossRef CAS PubMed.
- R. P. Szajewski and G. M. Whitesides, J. Am. Chem. Soc., 1980, 102, 2011–2026 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional TEM images and luminescence spectra. See DOI: 10.1039/c6tc01207h |
| This journal is © The Royal Society of Chemistry 2016 |