
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hysteretic magnetoelectric behavior of CoFe2O4–BaTiO3 composites prepared by reductive sintering and reoxidation
Till
Walther
 a,
Ulrich
Straube
b,
Roberto
Köferstein
a and
Stefan G.
Ebbinghaus
*a
a,
Ulrich
Straube
b,
Roberto
Köferstein
a and
Stefan G.
Ebbinghaus
*a
aInstitute of Chemistry, Inorganic Chemistry, Martin Luther University Halle-Wittenberg, Kurt Mothes Strasse 2, 06120 Halle, Germany. E-mail: stefan.ebbinghaus@chemie.uni-halle.de; Fax: +49-345-5527028; Tel: +49-345-5525870
bInstitute of Physics, Martin Luther University Halle-Wittenberg, von Danckelmann Platz 3, 06120 Halle, Germany
First published on 15th April 2016
Abstract
Magnetoelectric composites (CoFe2O4)x–(BaTiO3)1−x with x = 0.1, 0.2, 0.3, 0.4 and 0.5 were prepared by a polyol mediated synthesis route. The densification step was performed in a reducing forming gas atmosphere at 1250 °C. Under these conditions Co2+ and Fe3+ are reduced to the corresponding metals and thus a reaction of these elements with the BaTiO3 matrix is prevented. Reoxidation in air to CoFe2O4 at a rather low temperature of 800 °C leads to samples free of secondary phases. While the course of the synthesis was monitored by TGA, XRD and photometric analysis, the final composites were characterized via SEM, EDX, impedance spectroscopy and magnetic and magnetoelectric (ME) measurements. All samples show pronounced ME hystereses and αME maxima dependent on the Hdc field sweep direction. The sample with x = 0.4 exhibits the highest maximum αME of 420 μV Oe−1 cm−1.
1. Introduction
Multiferroic composites consist of a combination of compounds, which exhibit at least one ferroic order phenomenon like ferroelectricity and ferro- or ferrimagnetism. The coupling between magnetostrictive and piezoelectric phases allows manipulating the electric polarization by a magnetic field or the magnetization by an electric field. These so-called direct and indirect magnetoelectric (ME) effects promise new applications and devices, such as spintronics and MERAMS.1–4 Several composites have been identified as magnetoelectric materials.5 The first discovered magnetoelectric composite – and still one of the most prominent combinations – is the system consisting of CoFe2O4 (CFO) and BaTiO3 (BTO).6 The hard ferrimagnetic CoFe2O4 shows large magnetostriction while BaTiO3 is ferroelectric with high piezoelectricity. Furthermore, this system is free of resource-critical elements and is predicted to exhibit high ME voltages.7–10 CoFe2O4 and BaTiO3 have already been combined in all three prominent connectivities for composite multiferroics, namely 0–3, 1–3 and 2–2.11,12 The reported αME values for 0–3 composites are typically ranging from a few μV Oe−1 cm−1 to 10 mV Oe−1 cm−1.13–15 However, the theoretically predicted much higher αME values have not yet been achieved.16 This is most often explained by an insufficient interface between the piezoelectric and magnetostrictive phase that prevents mechanical transfers and thus good coupling. One approach to tailor the interface is to build up composites from nanoparticles because of their large surface to volume ratio. One disadvantage of this strategy is that impurities which originate from the reaction of BaTiO3 with CoFe2O4 at the interface, like substituted barium hexaferrite Ba(Co0.5Ti0.5)xFe12−xO19, are favored by large interface areas. While pure BaFe12O19 is multiferroic on its own17,18 and even composites with perovskites have been investigated,19,20 the incorporation of Co2+ and Ti4+ in BaFe12O19 has a significant impact on its magnetic properties.21,22To overcome the formation of impurity phases, we present in this article a polyol mediated synthesis route for magnetoelectric CoFe2O4–BaTiO3 composite ceramics. Using a reductive sintering step followed by reoxidation under mild conditions, the formation of Ba(Co0.5Ti0.5)xFe12−xO19 was successfully suppressed and phase pure samples with relative densities >80% were obtained. Measurements of the magnetoelectric coupling show frequency independent values of the ME coefficient above 300 Hz. All samples exhibit a pronounced ME hysteresis with a maximum of αME at Hdc ≈ ±2500 Oe. Furthermore, we for the first time describe a dependence of the maximum αME values on the direction of the DC-field sweep.
2. Experimental
2.1 Material preparation
All chemicals were used as purchased without further purification, if not stated otherwise. Five samples of (CoFe2O4)x–(BaTiO3)(1−x) composites were synthesized with xCFO = 0.1, 0.2, 0.3, 0.4 and 0.5. A modified polyol mediated process was used to prepare the precursor powders for the ceramics.23,24 The preparation scheme is shown in Fig. 1 and is described as follows. | ||
| Fig. 1 Scheme of sample preparation. | ||
Fe(NO3)3·9H2O (20 mmol, Sigma Aldrich) and Co(NO3)2·6H2O (10 mmol, Sigma Aldrich) were dissolved in deionized water (6 ml). NaOH (80 mmol, Grüssing) and diethylene glycol (250 ml, Carl Roth) were added to the red solution, which was then heated within 45 min to its boiling point (∼160 °C) and refluxed for 1 h. After cooling to room temperature, acetone (250 ml, Overlack) was added, resulting in a brown precipitate. Subsequent centrifugation and washing with acetone led to a brown CoFe2O4 precursor powder. The amount of ferrite was determined by thermogravimetric analysis.
For sample 0.5CFO–0.5BTO, 5 mmol of the presynthesized CoFe2O4 precursor and 5 mmol Ba(OH)2·8H2O (FLUKA) were mixed with diethylene glycol (250 ml, Carl Roth) in an argon flushed flask. Distilled Ti(i-OPr)4 (5 mmol, Alfa Aesar) was added and the reaction mixture was heated within 45 min to the boiling point (∼160 °C) and kept under reflux for 1 h. After cooling to room temperature, 250 ml of acetone (Overlack) were added to the gray suspension and subsequent centrifugation and washing with acetone led to a gray composite precursor. The precursor powders for the other samples were prepared accordingly, using the respective stoichiometric quantities.
The precursor powders were first calcined in static air at 700 °C for 1 h with a heating rate of 10 K min−1. Afterwards, a first reduction step in flowing forming gas (80 ml min−1, 10% H2) at 950 °C for 1 h (heating rate 5 K min−1) was performed and the resulting light gray powders were pressed into disks (100 mg, ∅ = 6 mm) that were sintered in flowing forming gas (80 ml min−1, 10% H2) at 1250 °C for 1 h (heating rate 5 K min−1). Finally, reoxidation in static air at 800 °C for 6 h (heating rate 10 K min−1) led to black ceramic bodies of CoFe2O4–BaTiO3 composites.
2.2 Characterization
Thermogravimetric measurements in flowing synthetic air or forming gas (10% H2) (flow rate 40 ml min−1, heat rate 10 K min−1) were performed using a Netzsch STA 409 system. X-ray diffraction patterns were recorded at room temperature on a Bruker D8 Advance diffractometer operating with CuKα radiation. For the quantitative cobalt ferrite determination, small aliquots of the samples were dissolved in a mixture of hydrochloric acid (≥37%, Sigma Aldrich) and hydrogen peroxide solution (30%, Overlack). After dissolution, residual peroxides were decomposed by heating. The solutions were diluted to an approximated Fe concentration of 1 mg l−1 and Spectroquant Iron Test solution (Merck Millipore) was added. For absorbance measurements at λ = 560 nm a VWR UV-3100PC Spectrophotometer was used. The Fe concentrations and corresponding CoFe2O4 contents were determined by a calibration series. Scanning electron microscopy images in the backscattered electron (BSE) mode and EDX spectra were recorded using a Philips ESEM XL 30 FEG. For impedance measurements an eutectic Ga–In alloy was coated as electrodes on top and bottom surfaces of the ceramic bodies. The temperature and frequency dependent impedance spectra (0 to 180 °C; 100 Hz to 13 MHz) were recorded using a Hewlett-Packard 4192A impedance analyzer. Magnetic measurements were carried out using the ACMS option of a Quantum Design PPMS 9. Hysteresis loops were measured at 300 K with a cycling of the magnetic field between +90 and −90 kOe. For the magnetoelectric investigations, 100 nm thick gold electrodes were sputtered onto the sample surfaces using a Cressington Sputter Coater 108auto. Electric poling was done applying an electric DC field of 4 kV cm−1 to the samples at room temperature. Then the samples were heated to 200 °C for 1 h (heating rate 10 K min−1) and the electric field was dynamically adjusted setting the current limit to 0.1 mA. Due to the increasing conductivity of the samples the field decreased to a few V cm−1 at 200 °C but again reached 4 kV cm−1 during cooling to room temperature. After poling, the samples were short circuited for 10 min. Immediately afterwards, the ME measurements were performed at 300 K in a Quantum Design PPMS 9 using a custom made setup based on the AC-Transport measurement option. A magnetic AC field of Hac = 10 Oe with different frequencies was applied by a solenoid with 1160 loops of copper wire and the ME voltage was measured in dependence of the magnetic DC field upon cycling between +10 and −10 kOe. Magnetic AC and DC fields were aligned parallel to the electric polarization. Raw data were corrected for eddy currents measured on an empty sample holder. The magnetoelectric coefficient was calculated as αME = U/(Hac·h) with h being the sample height.3. Results and discussion
3.1 Thermal analysis
Simultaneous thermogravimetric and differential thermal analyses were carried out on the samples to investigate calcination, sintering and reoxidation behavior. The results are discussed for 0.3CFO–0.7BTO exemplarily as follows.Calcination of the dried composite precursor in air at 700 °C leads to a total weight loss of 45%. This weight change comprises the evaporation of residual solvent and adsorbed water, an exothermal combustion of organic residues and the endothermal decomposition of intermediately formed BaOx(CO3)1−x.25–27
Following the path of preparation and representing the calcination and sintering steps under reducing conditions, an air-calcined sample was heated in the thermobalance in flowing forming gas to 1250 °C and kept at this temperature for 1 h. The corresponding weight change and DTA curves are shown in Fig. 2. The main weight loss of 9.0% occurs between 350 and 1000 °C and is accompanied by two distinct exothermal signals in the DTA curve. During this step, the reduction of CoFe2O4 to an alloy of Co and Fe (expected weight loss: 7.7%), the decomposition of residual BaCO3 and the final formation of BaTiO3 take place. During the dwell time of 1 h at 1250 °C a further slight weight loss of about 2% was observed. This weight loss could be caused by a gradual generation of oxygen defects and the accompanied partial reduction of Ti4+ in the BaTiO3 matrix. Overall a weight loss of 11.0% occurred.
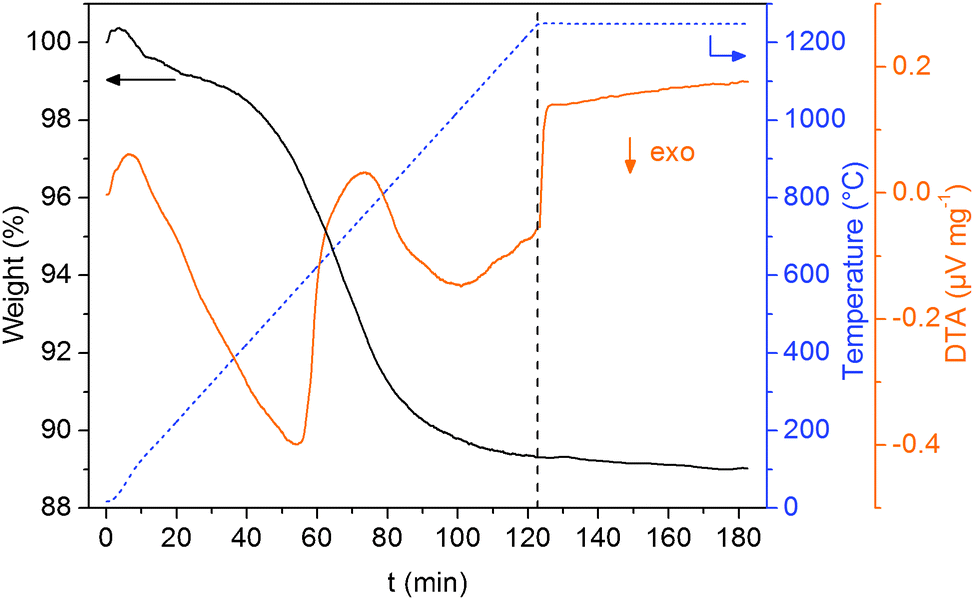 | ||
| Fig. 2 TGA/DTA investigation of 0.3CFO–0.7BTO air-calcined powder under reductive sintering conditions in flowing forming gas. | ||
Afterwards, the resulting CoFe2–BaTiO3 sample was heated in synthetic air up to 800 °C and kept at that temperature for 6 h to investigate the reoxidation behavior. As shown in Fig. 3, a weight gain accompanied by an exothermic DTA signal starts at 430 °C and is finished about 1 h after reaching 800 °C. This process leads to a total weight change of +8.2%. Longer heating at 800 °C did not lead to any significant additional weight change. For the complete oxidation of the CoFe2 alloy in the 0.3CFO–0.7BTO sample, a theoretical weight gain of 8.4% is expected. Thus, the observed change of +8.2% can be assigned completely to the reoxidation of the alloy to CoFe2O4 as additionally supported by the XRD results discussed in the next paragraph.
 | ||
| Fig. 3 TGA/DTA investigation in flowing air of 0.3CoFe2–0.7BTO under reoxidation conditions. | ||
3.2 X-ray diffraction
The course of the synthesis was monitored by X-ray powder diffraction as exemplarily shown for xCFO = 0.5 in Fig. 4. The diffractogram of the precursor obtained by precipitation without further temperature treatment (Fig. 4a) mainly shows crystalline BaCO3 and CoFe2O4 implying that at least one Ti-containing amorphous phase is present. Calcination in air at 700 °C for 1 h leads to the formation of BaTiO3 although some BaCO3 is still detectable in the powder (Fig. 4b). Calcination or sintering in air at temperatures higher than 700 °C leads to reactions between the BaTiO3 matrix and the ferrite, i.e. the formation of impurities such as Ti4+- and Co2+-doped BaFe12O19, hexagonal BaTiO3 as well as increased Co2+ and Fe3+ doping of BaTiO3. To avoid this, an additional calcination step in forming gas (10% H2) at 950 °C for 1 h was performed. Under these conditions residual BaCO3 decomposes and the X-ray diffraction pattern (Fig. 4c) indicates the formation of (pseudo-) cubic modification for BaTiO3. In addition, CoFe2O4 is reduced to an alloy of Co and Fe that does not react with BaTiO3 as the ferrite would. The resulting reduced powder was pressed into disks (∅ = 6 mm, m = 100 mg) and sintered at 1250 °C for 1 h in forming gas. The corresponding X-ray diffraction pattern shows a mixture of tetragonal BaTiO3 and the CoFe2 alloy (Fig. 4d). Afterwards, the ceramic body was reoxidized in air at 800 °C for 6 h resulting in an oxidation of the CoFe2 alloy to CoFe2O4 (Fig. 4e).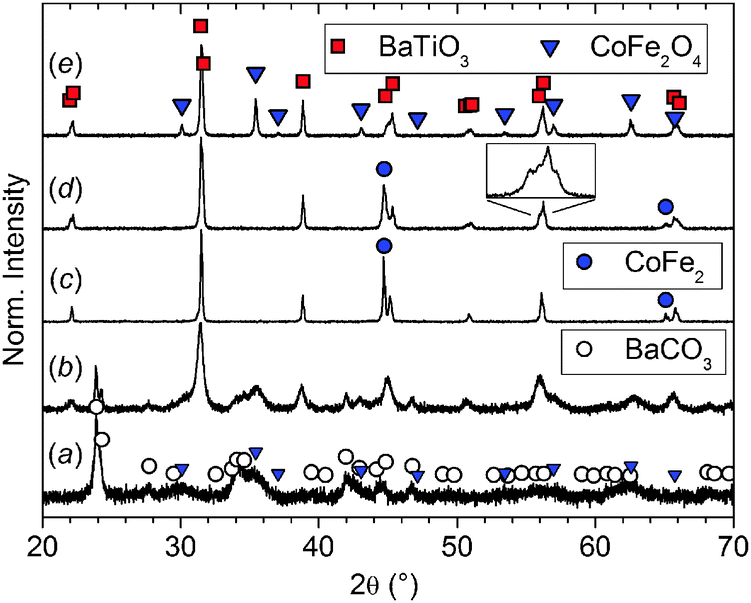 | ||
| Fig. 4 X-ray diffraction patterns of 0.5CFO–0.5BTO: (a) precursor; (b) calcined in air at 700 °C for 1 h; (c) calcined in forming gas at 950 °C for 1 h; (d) sintered in forming gas at 1250 °C for 1 h, zoom: tetragonal splitting of BaTiO3 211 reflex; (e) reoxidized in air at 800 °C for 6 h. | ||
As shown in Fig. 5, phase pure CoFe2O4–BaTiO3 composites were obtained for all investigated compositions with xCFO = 0.1, 0.2, 0.3, 0.4, and 0.5. By Rietveld refinement the cell parameters of the BaTiO3 matrix were determined as a = 3.9997(6) and c = 4.0254(8) for all samples. Indicating a homogeneous composition independent of xCFO. Thus, by reductive sintering and subsequent reoxidation, the formation of impurities as BaFe12O19 can be prevented completely and the reaction of BaTiO3 with Co2+ and Fe3+ is minimized.
 | ||
| Fig. 5 X-ray diffraction patterns of composites with increasing CoFe2O4 content. | ||
It was found that if the disks are too dense after the reductive sintering, the oxidation leads to cracks in the samples due to an increasing volume during the reaction of CoFe2 to CoFe2O4. The optimal relative densities for reoxidizing (65 to 80%) are dependent on xCFO and decrease with higher CoFe2O4 content. For the final samples relative densities, with respect to the (weighted) single crystal values,28 between 80% and 90% were achieved.
3.3 Photometric CoFe2O4 assay
In Table 1 the results of the photometric CoFe2O4 quantifications are listed. The uniform deficiency in the CoFe2O4 content compared to the nominal values is most likely caused by a certain solubility of the Co- and Fe-containing precursors during precipitation and washing of the composite powder. On the other hand the deviation between the expected and measured CoFe2O4 content is below 2 mol% in most cases and for simplification the nominal percentages are used throughout the text for describing the compositions. Nevertheless, for all calculations with respect to xCFO, the experimentally determined contents were used.| Sample | CoFe2O4 mole fraction (mol%) | |
|---|---|---|
| Nominal | Determined | |
| 0.1CFO–0.9BTO | 10 | 8.7 ± 0.4 |
| 0.2CFO–0.8BTO | 20 | 18.4 ± 0.5 |
| 0.3CFO–0.7BTO | 30 | 28.2 ± 0.9 |
| 0.4CFO–0.6BTO | 40 | 37.9 ± 1.4 |
| 0.5CFO–0.5BTO | 50 | 46.3 ± 1.4 |
3.4 SEM and EDX investigations
Scanning electron micrographs were taken from the polished surfaces of sintered samples as shown exemplarily in Fig. 6 for xCFO = 0.1, 0.3 and 0.5. A minor porosity was observed for all samples in accordance with the measured densities of 80% to 90%. The pores are distributed randomly throughout the samples. The BaTiO3 phase was found to consist of grains with sizes of 1–5 μm independent of xCFO. From Fig. 6a it can be seen that for 0.1CFO–0.9BTO isolated CoFe2O4 grains (dark gray) are embedded in the BaTiO3 matrix (light gray) but most of the CoFe2O4 is assembled in centers of partially interconnected grains separated and surrounded by BaTiO3. With higher xCFO values the amount of these CoFe2O4 clusters increases. In 0.3CFO–0.7BTO only a small portion of the ferrite occurs as isolated particles completely surrounded by BaTiO3 (Fig. 6b and d). For xCFO = 0.4 and 0.5 (Fig. 6c), the CoFe2O4 particles form irregular connected structures that permeate through wide areas, from several 10 to 100 μm in diameter. | ||
| Fig. 6 Scanning electron microscopy images (BSE mode) of polished ceramics: (a) xCFO = 0.1; (b) 0.3; (c) 0.5; (d) position markers for EDX spectra of 0.3CFO–0.7BTO, roughly representing the irradiated surface area. | ||
To assign the two distinguishable phases in the BSE mode, EDX measurements were carried out at representative sample areas (Fig. 6d). The EDX spectra confirm the formation of two phases, namely CoFe2O4 and BaTiO3, as can be seen in Fig. 7. The traces of barium and titanium, visible in the CoFe2O4 spectrum, are due to the large interaction volume of the electron beam compared to the grain size.
 | ||
| Fig. 7 EDX spectra of 0.3CFO–0.7BTO at the two positions marked in Fig. 6d. | ||
In 0.5CFO–0.5BTO a third phase can be identified in the BSE images from its medium-gray contrast, although no additional reflexes show up in the corresponding X-ray diffraction pattern. EDX-line scans revealed this phase to be slightly oxygen-deficient BaTiO3−δ.
3.5 Impedance spectroscopy
In Fig. 8 the temperature dependence of the real part of the permittivities (ε′) and the corresponding loss tangents measured at 1 kHz are shown. The ε′ values at 0 °C vary between 476 for xCFO = 0.1 and 232 for xCFO = 0.5. In the measured temperature range the permittivities of xCFO = 0.1 and 0.2 increase continuously, reaching 5610 and 5068 at 180 °C. For the composite with xCFO = 0.3 the formation of a broad shoulder at around 150 °C can be seen and ε′ reaches a value of about 4650 at 180 °C. For xCFO = 0.4 and 0.5, a maximum forms at 150 °C with ε′ values of 4402 and 4447. When measured at frequencies higher than 10 kHz, this maximum at 150 °C is also observed for the samples 0.1CFO–0.9BTO and 0.2CFO–0.8BTO as shown in Fig. 9 for f = 1 MHz. The occurrence of the maximum is accompanied by an increase in the loss tangent and can be assigned to the ferroelectric–paraelectric transition of BaTiO3.29–32 For large-grained BaTiO3 this phase transition occurs at 120 °C. According to Mitoseriu et al. the shift to 150 °C is due to the convolution of extrinsic defect-related dielectric relaxation with the intrinsic ferroelectric component.33 The presence of the phase transition in the impedance data proves the formation of the tetragonal (ferroelectric) BaTiO3 modification in accordance with the XRD results discussed above. The consistent phase transition temperatures hint at a uniform composition of the BaTiO3, indicating that no xCFO-dependent doping of Co2+ or Fe3+ occurred for any of the samples. The loss tangents at room temperature are below 0.5 with the exception of 0.5CFO–0.5BTO and only slightly increase with temperature below the tetragonal–cubic phase transition of BaTiO3. | ||
| Fig. 8 (a) Temperature dependence of the real part of the permittivity and (b) corresponding loss tangents at 1 kHz. | ||
 | ||
| Fig. 9 (a) Temperature dependence of the real part of the permittivity and (b) corresponding loss tangents at 1 MHz. | ||
Concerning the frequency dependence at room temperature, ε′ decreases rather strongly in the range of roughly 100 Hz–10 kHz and remains quite stable for higher frequencies for all samples, as can be seen in Fig. 10. This phenomenon is typical for electrically conducting particles embedded in an insulating matrix and is called Maxwell Wagner polarization.34 A similar behavior has already been reported for various CoFe2O4–BaTiO3 composites.13,35,36
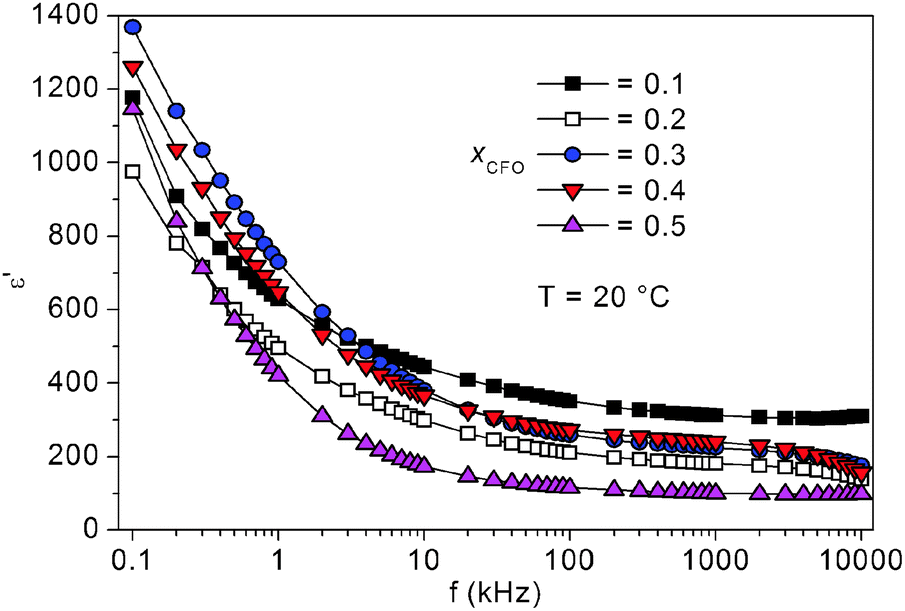 | ||
| Fig. 10 Frequency dependence of the real part of the permittivities at room temperature. | ||
3.6 Magnetic properties
The field dependent magnetization curves of the composites are depicted in Fig. 11. All samples show clear hystereses in accordance with the ferrimagnetism of the spinel component. The saturation magnetization values were determined by a linear extrapolation of the magnetization in the high field ranges (50–90 kOe) to H = 0 Oe. As expected, saturation and remanent magnetization increase with CoFe2O4 content.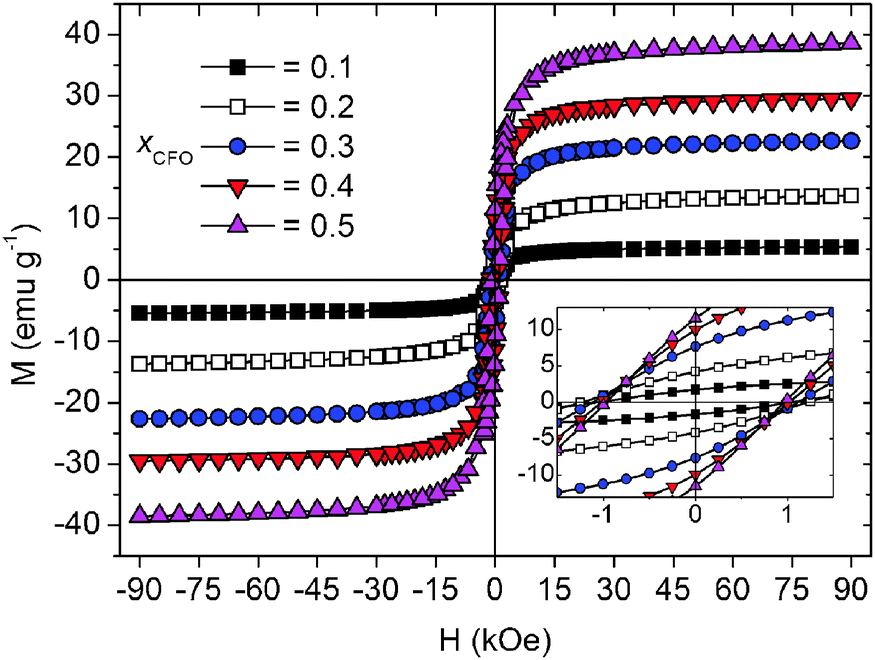 | ||
| Fig. 11 Field dependent magnetization hysteresis curves measured at 300 K. The inset shows a magnification of the low field region. | ||
Normalizing these magnetization values with respect to the ferrite content (i.e. emu g−1 of CoFe2O4), as shown in Fig. 12, reveals a small maximum of MR with a value of 27.0 emu g−1 for xCFO = 0.3, while the MS data show a general slight increase with higher CoFe2O4 contents, reflecting the increasing ferrite particle sizes. Coercivity fields increase from 1060 Oe (0.1CFO–0.9BTO) to a maximum of 1230 Oe for xCFO = 0.2 and decreases with higher CoFe2O4 contents down to a minimum of 970 Oe for xCFO = 0.5. It is commonly known that coercivity values for CoFe2O4 are dependent on its grain shape and structure. Therefore, the change in coercivity is probably due to the formation of CoFe2O4 clusters and interconnected structures with increasing xCFO values (see SEM part).
 | ||
| Fig. 12 (a) Magnetic coercive fields and (b) saturation and remanent magnetization at 300 K. | ||
3.7 Magnetoelectric coupling
An example of the magnetic DC field dependence of the magnetoelectric coefficient αME is shown in Fig. 13 for the composite with xCFO = 0.4. For all samples, the αME values show maxima/minima around ±2.5 kOe and clear hysteretic behavior with coercive fields between ±400 and ±600 Oe as well as distinct remanent αME values of ±7 to ±150 μV Oe−1 cm−1. Thus, the sign of αME is switchable with the direction of the applied magnetic DC field. | ||
| Fig. 13 Magnetic DC field dependent magnetoelectric coefficient of 0.4CFO–0.6BTO. | ||
It is remarkable that the maximum values of αME at ±2.5 kOe are dependent on the history of the magnetic DC field. Upon increasing the DC field strength (both in positive and negative direction), the αME values are higher than for decreasing field strengths. For example, in the case of xCFO = 0.4 the maximum αME value is 420 μV Oe−1 cm−1 when the magnetic field is increased up to 10 kOe (red circles, Fig. 13), whereas αME(max) amounts to only 337 μV Oe−1 cm−1 when the field strength is lowered down from 10 kOe (blue diamonds, Fig. 13). To the best of our knowledge, such an effect has not yet been described for αME and its origin needs to be investigated in further experiments.
A comparison between the magnetic and ME hystereses (Fig. 14) reveals that the ME coercivities are about half the size of the magnetic coercivites, i.e. the ME voltage diminishes although magnetization still exists. It is noteworthy that the DC field at which the maximum αME is ovserved (±2.5 kOe) shows no obvious relation to the characteristic points of the magnetic hysteresis. In particular, it is much smaller than the saturation field (roughly ±20 kOe) and also significantly different from the inflection points of magnetization which occur at about ±1 kOe. It seems that the ME effect is more affected by magnetostriction. For example, van Run et al. found the highest ME values at the maximum of the piezomagnetic strain per Oe.37 In laminated PZT–Terfenol-D systems, ME maxima were found at the saturation point of the magnetostrictive strain38 and at the maximum of the piezomagnetic coefficient.39
 | ||
| Fig. 14 Comparison of magnetic and magnetoelectric hystereses of 0.3CFO–0.7BTO. | ||
Our paper is one of the few examples in which a hysteretic behavior of the ME coefficient is reported for CoFe2O4–BaTiO3 composites.37,40–43 In the majority of articles (e.g.ref. 13 and 44–47), αME values were found to be zero when the magnetic DC field was switched off, or only initial curves, i.e. without cycling of Hdc, were published.
Fig. 15 shows the maximum αME values for the investigated samples depending on xCFO and the frequency of Hac. The αME values increase with higher CoFe2O4 contents up to a maximum at xCFO = 0.4 with 420 μV Oe−1 cm−1 at 500 Hz and decrease again for xCFO = 0.5 at all investigated frequencies. The magnetoelectric remanences follow this trend and the sample 0.4CFO–0.6BTO shows the highest remanent αME value with 150 μV Oe−1 cm−1. The maximum αME values are within the typical range of a few μV Oe−1 cm−1 to some mV Oe−1 cm−1 reported for CoFe2O4–BaTiO3 composites with 0–3 connectivity measured under similar conditions.13–15,41,43
 | ||
| Fig. 15 Frequency dependence (Hac) of the maximal ME coefficients. | ||
ME measurements at five different Hac frequencies showed that αME is significantly lower for f = 100 Hz than for higher frequencies, while for frequencies from 300 to 900 Hz the obtained αME values show only little deviations. This is in contrast to previously reported frequency dependencies where αME values increase more or less linearly up to 1 kHz.44,45 Other articles show a giant increase of αME (up to 2.5 V Oe−1 cm−1) under resonance conditions in the range between 15 and 430 kHz.46–48 Measurements at such higher frequencies are therefore planned for the future.
4. Conclusions
In multiferroic composites, the strength of the magnetoelectric coupling depends on the quality of the interface and therefore an intimate contact between the two components is mandatory. Because of the low sintering activity of BaTiO3, such high-quality interfaces require high sintering temperatures, which usually lead to the formation of secondary phases like BaFe12O19 and/or ionic exchanges like the incorporation of iron or cobalt ions in BaTiO3. To avoid these problems we describe a new approach starting with the polyol-assisted soft-chemistry preparation of a CoFe2O4–BaTiO3 composite precursor consisting of submicrometer-sized particles. This composite powder is sintered under a reducing atmosphere, resulting in CoFe2–BaTiO3 composites. In a final reaction step these ceramics are reoxidized under mild conditions leading to the final dense CoFe2O4–BaTiO3 composites (≥80% of the crystallographic density), which according to XRD are completely free of secondary phases. The presence of tetragonal BaTiO3 with lattice parameters independent of the ferrite fraction further indicates that no considerable amounts of Co2+ or Fe3+ are incorporated in BaTiO3. This finding is additionally corroborated by dielectric measurements showing the occurrence of the ferroelectric–paraelectric phase transition, which is shifted to 150 °C probably due to defect-related dielectric relaxations. EDX investigations gave no hints for secondary phases or detectable Ti/Fe/Co exchange between the two phases. All composites show ferrimagnetic behavior. Their (normalized) saturation magnetizations slightly increase with the CoFe2O4 content, reflecting the increasing grain sizes of the ferrite. Field-dependent measurements of the magnetoelectric coupling show a hysteresis of αME with maxima at DC magnetic fields of ±2.5 kOe and remanent αME values in the order of 10–150 μV Oe−1 cm−1. We observed distinct deviations between the coercivities of the magnetic moment and the ME effect, i.e. the magnetoelectric coupling diminishes already at roughly ±500 Oe while the required field to extinct the magnetization is about twice as high. Concerning the effect of the Hac frequency, we found almost constant αME values from 300 Hz to 900 Hz while the magnetoelectric coupling is much smaller at 100 Hz. In addition, an increase of the ME effect with increasing CoFe2O4 content was observed leading to a maximum in the order of 400 μV Oe−1 cm−1 for x = 0.4. With a higher ferrite content, αME decreases again. Most interestingly, for all samples we observed strong differences for the maximum αME values depending on the direction of the magnetic dc-field sweep. When the field strength is increased (both in positive and negative directions) the maximum ME values are up to 20% larger than upon decreasing Hdc. Thus, the measured ME voltages of the CoFe2O4–BaTiO3 composites not only depend on the Hdc field strength but also on its history. This ‘memory effect’ may give rise to additional future applications.Acknowledgements
The authors thank F. Syrowatka for the SEM investigations and Th. Müller for TGA experiments. Financial support by the German Research Foundation within the Collaborative Research Centre SFB 762 Functionality of Oxide Interfaces is gratefully acknowledged.References
- N. A. Spaldin and M. Fiebig, Science, 2005, 309, 391–392 CrossRef CAS PubMed.
- M. Bibes and A. Barthélémy, Nat. Mater., 2008, 7, 425–426 CrossRef CAS PubMed.
- J. Ma, J. Hu, Z. Li and C.-W. Nan, Adv. Mater., 2011, 23, 1062–1087 CrossRef CAS PubMed.
- N. Ortega, A. Kumar, J. F. Scott and R. S. Katiyar, J. Phys.: Condens. Matter, 2015, 27, 504002 CrossRef CAS PubMed.
- W. Eerenstein, N. D. Mathur and J. F. Scott, Nature, 2006, 442, 759–765 CrossRef CAS PubMed.
- J. van den Boomgaard, D. R. Terrell, R. A. J. Born and H. F. J. I. Giller, J. Mater. Sci., 1974, 9, 1705–1709 CrossRef CAS.
- C.-W. Nan, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 6082–6088 CrossRef CAS.
- M. Avellaneda and G. Harshé, J. Intell. Mater. Syst. Struct., 1994, 5, 501–513 CrossRef CAS.
- M. I. Bichurin, V. M. Petrov and G. Srinivasan, J. Appl. Phys., 2002, 92, 7681–7683 CrossRef CAS.
- J. Ryu, S. Priya, K. Uchino and H.-E. Kim, J. Electroceram., 2002, 8, 107–119 CrossRef CAS.
- R. E. Newnham, D. P. Skinner and L. E. Cross, Mater. Res. Bull., 1978, 13, 525–536 CrossRef CAS.
- C.-W. Nan, M. I. Bichurin, S. Dong, D. Viehland and G. Srinivasan, J. Appl. Phys., 2008, 103, 031101 CrossRef.
- R. P. Mahajan, K. K. Patankar, M. B. Kothale, S. C. Chaudhari, V. L. Mathe and S. A. Patil, Pramana, 2002, 58, 1115–1124 CrossRef CAS.
- G. Schileo, Prog. Solid State Chem., 2013, 41, 87–98 CrossRef CAS.
- A. Baji, Y.-W. Mai, R. Yimnirun and S. Unruan, RSC Adv., 2014, 4, 55217–55223 RSC.
- J. S. Andrew, J. D. Starr and M. A. K. Budi, Scr. Mater., 2014, 74, 38–43 CrossRef CAS.
- X. Chen and G. Tan, 2012, arXiv:1201.3963 [cond-mat.mtrl-sci].
- T. Kimura, Annu. Rev. Condens. Matter Phys., 2012, 3, 93–110 CrossRef CAS.
- Z. Dong, Y. Pu, Z. Gao, P. Wang, X. Liu and Z. Sun, J. Eur. Ceram. Soc., 2015, 35, 3513–3520 CrossRef CAS.
- J. D. S. Guerra, S. Betal, M. Pal, J. E. Garcia, A. J. A. Oliveira, J.-C. M’Peko, A. C. Hernandes, R. Guo and A. S. Bhalla, J. Am. Ceram. Soc., 2015, 98, 1542–1547 CrossRef CAS.
- N. Koga and T. Tsutaoka, J. Magn. Magn. Mater., 2007, 313, 168–175 CrossRef CAS.
- T. Tsutaoka and N. Koga, J. Magn. Magn. Mater., 2013, 325, 36–41 CrossRef CAS.
- D. Caruntu, Y. Remond, N. H. Chou, M.-J. Jun, G. Caruntu, J. He, G. Goloverda, C. O’Connor and V. Kolesnichenko, Inorg. Chem., 2002, 41, 6137–6146 CrossRef CAS PubMed.
- M. Siemons, T. Weirich, J. Mayer and U. Simon, Z. Anorg. Allg. Chem., 2004, 630, 2083–2089 CrossRef CAS.
- S. Gablenz, H.-P. Abicht, E. Pippel, O. Lichtenberger and J. Woltersdorf, J. Eur. Ceram. Soc., 2000, 20, 1053–1060 CrossRef CAS.
- V. Ischenko, E. Pippel, R. Köferstein, H.-P. Abicht and J. Woltersdorf, Solid State Sci., 2007, 9, 21–26 CrossRef CAS.
- V. Ischenko, J. Woltersdorf, E. Pippel, R. Köferstein and H.-P. Abicht, Solid State Sci., 2007, 9, 303–309 CrossRef CAS.
- G. W. Marks and L. A. Monson, Ind. Eng. Chem., 1955, 47, 1611–1620 CrossRef CAS.
- A. Hanumaiah, T. Bhimasankaram, S. V. Suryanarayana and G. S. Kumar, Bull. Mater. Sci., 1994, 17, 405–409 CrossRef CAS.
- K. Raidongia, A. Nag, A. Sundaresan and C. N. R. Rao, Appl. Phys. Lett., 2010, 97, 062904 CrossRef.
- A. Gupta and R. Chatterjee, J. Eur. Ceram. Soc., 2013, 33, 1017–1022 CrossRef CAS.
- R. Köferstein and S. G. Ebbinghaus, RSC Adv., 2015, 5, 71491–71499 RSC.
- L. Mitoseriu and V. Buscaglia, Phase Transitions, 2006, 79, 1095–1121 CrossRef CAS.
- K. W. Wagner, Arch. Elektrotech., 1914, 2, 371–387 CrossRef.
- J. Nie, G. Xu, Y. Yang and C. Cheng, Mater. Chem. Phys., 2009, 115, 400–403 CrossRef CAS.
- L. M. Hrib and O. F. Caltun, J. Alloys Compd., 2011, 509, 6644–6648 CrossRef CAS.
- A. M. J. G. van Run, D. R. Terrell and J. H. Scholing, J. Mater. Sci., 1974, 9, 1710–1714 CrossRef CAS.
- S. Dong, J.-F. Li and D. Viehland, J. Appl. Phys., 2004, 95, 2625–2630 CrossRef CAS.
- C. P. Zhao, F. Fang and W. Yang, Smart Mater. Struct., 2010, 19, 125004 CrossRef.
- J. van den Boomgaard, A. M. J. G. Van Run and J. Van Suchtelen, Ferroelectrics, 1976, 10, 295–298 CrossRef CAS.
- M. E. Botello-Zubiate, D. Bueno-Baqués, J. de Frutos Vaquerizo, L. E. Fuentes Cobas and J. A. Matutes-Aquino, Ferroelectrics, 2006, 338, 247–253 CrossRef CAS.
- G. V. Duong, R. Groessinger and R. Sato Turtelli, J. Magn. Magn. Mater., 2007, 310, 1157–1159 CrossRef CAS.
- G. V. Duong, R. S. Turtelli and R. Groessinger, J. Magn. Magn. Mater., 2010, 322, 1581–1584 CrossRef CAS.
- A. Khamkongkaeo, P. Jantaratana, C. Sirisathitkul, T. Yamwong and S. Maensiri, Trans. Nonferrous Met. Soc. China, 2011, 21, 2438–2442 CrossRef CAS.
- H. Yang, G. Zhang and Y. Lin, J. Alloys Compd., 2015, 644, 390–397 CrossRef CAS.
- W. P. Wang, H. Yang, T. Xian and R. C. Yu, Chem. Phys. Lett., 2015, 618, 72–77 CrossRef CAS.
- R. A. Islam, M. Bichurin and S. Priya, in Ferroelectrics – Characterization and Modeling, ed. M. Lallart, InTech, 2011, pp. 61–78 Search PubMed.
- S. Q. Ren, L. Q. Weng, S.-H. Song, F. Li, J. G. Wan and M. Zeng, J. Mater. Sci., 2005, 40, 4375–4378 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2016 |