
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceColumnar liquid-crystalline assemblies of X-shaped pyrene–oligothiophene conjugates: photoconductivities and mechanochromic functions†
Kian Ping
Gan
,
Masafumi
Yoshio
* and
Takashi
Kato
*
Department of Chemistry and Biotechnology, School of Engineering, The University of Tokyo, Hongo, Bunkyo-ku, Tokyo 113-8656, Japan. E-mail: yoshio@chembio.t-u-tokyo.ac.jp; kato@chiral.t.u-tokyo.ac.jp; Fax: +81-3-5841-8661; Tel: +81-3-5841-7440
First published on 11th April 2016
Abstract
π-Conjugated liquid crystals have great potential as organic semiconductors owing to their smooth thin-film forming and flexible properties, as well as charge carrier transport ability. In the present study, we have designed and synthesised X-shaped columnar liquid crystals with pyrene as the central core, which are fourfold conjugated with bithiophene moieties, and tethered with eight or twelve alkoxy chains at their extremities. These X-shaped molecules show hexagonal, tetragonal, and rectangular columnar liquid-crystalline (LC) phases over a wide range of temperature. The extended π-conjugation has led to the realisation of a moderately low bandgap. The hole mobilities of both molecules in their various LC states are revealed to be in the order of 10−4 to 10−5 cm2 V−1 s−1 using the time-of-flight (TOF) photoconductivity measurements. In addition, the mechano-responsive photoluminescent change has been observed for the columnar phase of the X-shaped molecule with eight alkoxy chains. The possible π-stacking configurations are discussed. The introduction of a gelator, N,N′-bis(lauroyl)hydrazine, into the X-shaped molecules leads to the formation of a LC gel that produces a change in the measured hole mobility. The presented strategy could lead to the design of new solution-processable, low bandgap semiconducting materials for optoelectronic devices.
Introduction
π-Conjugated liquid crystals have attracted considerable attention because of their ease of molecular alignment and anisotropic charge carrier transport ability.1–4 Since the early reports of carrier transport of triphenylenes in the columnar phases by Boden, Haarar, and Ringsdorf et al.,5,6 a variety of columnar liquid crystals that show charge carriers transport have been reported.7–30 Polycyclic aromatic hydrocarbon derivatives such as perylene, pyrene, and other heteroaromatic molecules including phthalocyanine and fused thiophene have come under intense scrutiny because of their potential to be employed as mesogenic cores. These columnar liquid-crystalline (LC) π-conjugated molecules have found applications in organic optoelectronic devices such as organic light emitting diodes (OLEDs),31,32 organic field-effect transistors (OFETs),33–35 and organic photovoltaics (OPVs).36–39 As such, π-conjugated columnar liquid crystals are emerging as promising semiconducting soft materials due to their abilities to form one-dimensional π–π columnar stacks that allow high charge carrier transport and their capabilities to form smooth thin films with a uniaxially oriented columnar domain, which are essential for optimal device performance.Oligothiophene derivatives are promising for applications in electronics because of their charge transport abilities and fluorescence properties. Smectic and nematic LC oligothiophenes are intensively studied,40–48 however the examples of the columnar LC assembly of oligothiophene are limited. We previously reported on the first example of columnar LC polycatenar oligothiophenes (Scheme 2) with electronic and photonic functions,49,50 and helical stacking of propeller-shaped oligothiophene-triazine conjugates in columnar LC phases that results in ambipolar charge transport.51 In addition, we developed mechanochromic liquid crystals containing π-conjugated chromophores such as pyrene and oligothiophene as their central cores.52–56 The luminescent colours vary by mechanical stimuli, which are attributed to the change in the LC molecular assembled structures between the columnar and cubic LC phases. However, LC molecules that exhibit both charge transport properties and mechanochromic functions have not been explored. If these properties could be realised, a new class of supramolecular optoelectronic materials could be created with possible applications to displays, memory devices, and sensors.57–60 We envisaged that the combination of rod-shaped oligothiophenes with a disc-like pyrene would produce a multi-functional neoteric columnar liquid crystal that could exhibit both charge carrier transport and stimuli-responsive photoluminescent properties.
Herein, we report X-shaped molecules 1a and 1b (Fig. 1a) consisting of intercrossing polycatenar oligothiophenes through a central pyrene core (see ESI,† Fig. S1 for molecular modelling). The X-shaped molecules can form hole-transporting columnar LC phases via one-dimensional stacks (Fig. 1b). The molecular stacks are also tunable by the application of mechanical shear force, inducing a change in photoluminescence colour.
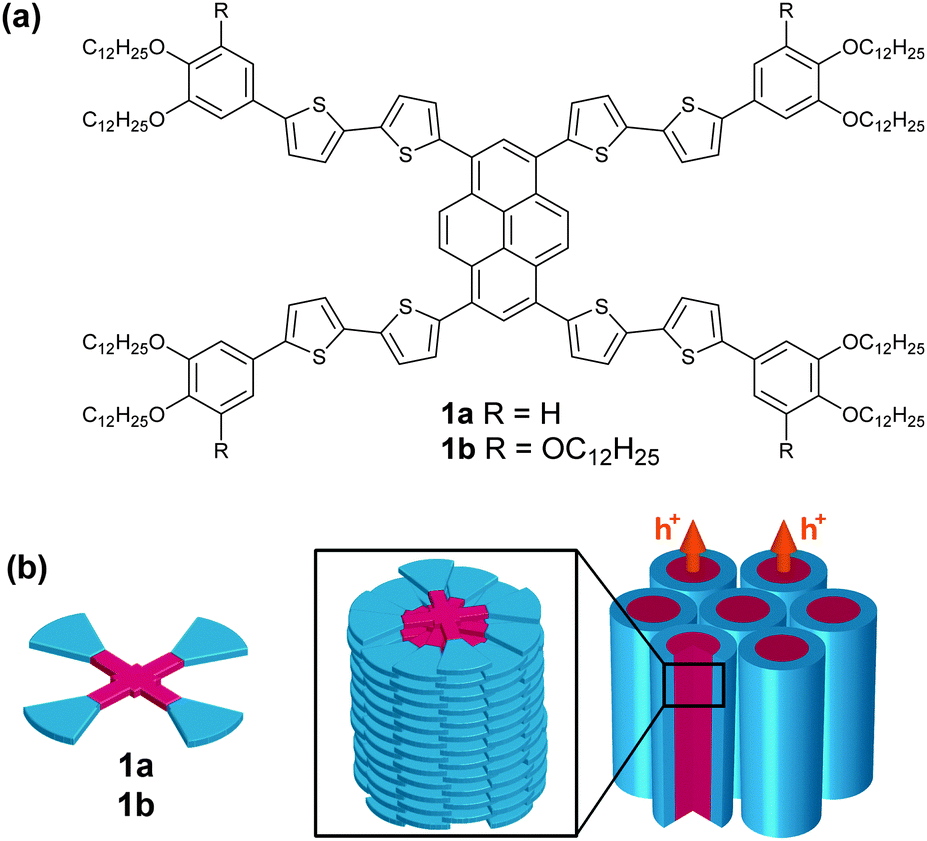 | ||
| Fig. 1 The molecular structure of the X-shaped molecules with a π-conjugated core (a), and the one-dimensional π–π columnar stacks that facilitate charge transport (b). | ||
Results and discussion
Synthesis and characterisation
The X-shaped pyrene–oligothiophene conjugates 1a and 1b were synthesised according to the synthetic route shown in Scheme 1 (see ESI,† Section S1 for detailed experimental procedures). The Suzuki–Miyaura cross-coupling reaction of alkoxy bromobenzene 2a with 2,2′-bithiophene-5-boronic acid pinacol ester yielded 3a, which is then followed by an iridium-catalysed borylation reaction61 with bis(pinacolato)diboron to afford the corresponding pinacol ester 4a. The fourfold Suzuki–Miyaura cross-coupling reaction of 4a with 1,3,6,8-tetrabromopyrene afforded the desired X-shaped molecule 1a. Compound 1b was obtained by the identical synthetic pathway. Both X-shaped molecules 1a and 1b were characterised and verified by 1H and 13C NMR spectroscopy, MALDI-TOF mass spectrometry, and elemental analyses.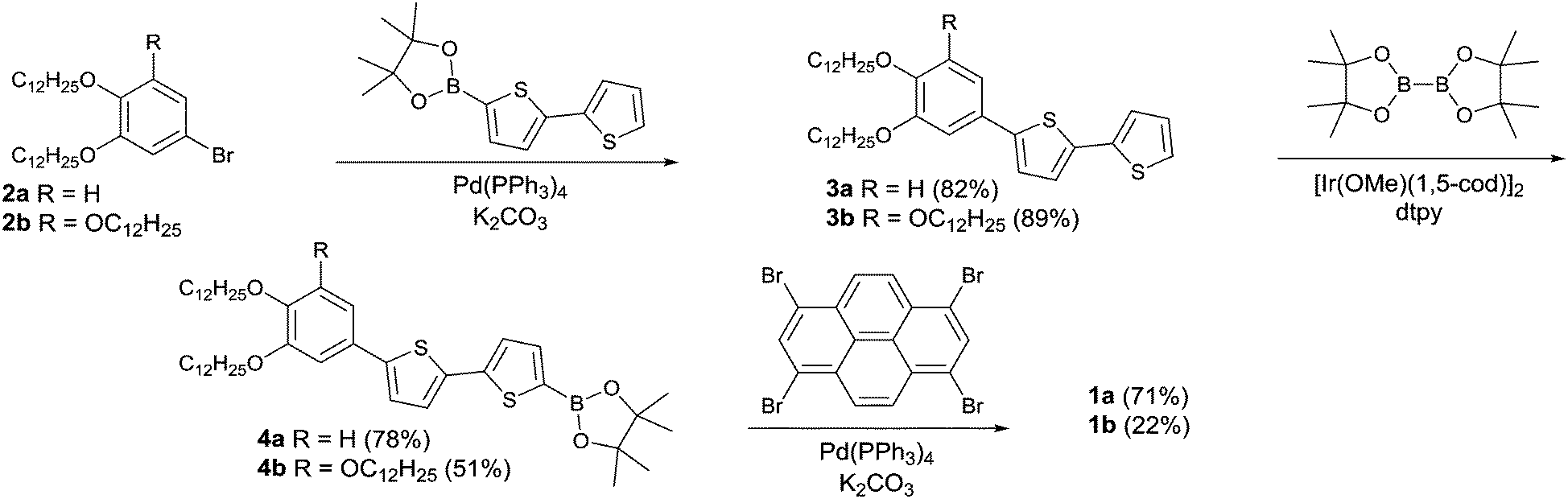 | ||
| Scheme 1 The synthetic scheme of the X-shaped pyrene-bithiophene conjugates 1a and 1b. | ||
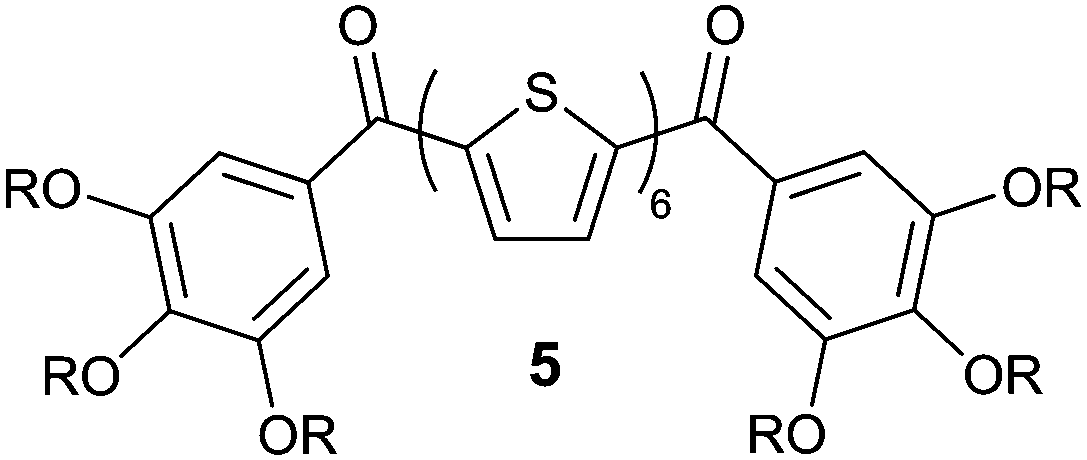 | ||
| Scheme 2 The polycatenar oligothiophene derivative 5 previously reported.49,50 | ||
Compounds 1a and 1b are highly soluble in organic solvents like hexane, toluene, dichloromethane, chloroform, and THF. Both compounds exhibit a high thermal stability of above 300 °C under air (see ESI,† Fig. S2). As such, these molecules could be processed and annealed under air without extensive oxidative decomposition.
Liquid-crystalline properties
The thermotropic LC properties and X-ray diffraction (XRD) data of the X-shaped molecules 1a and 1b are summarised in Table 1 (see ESI,† Fig. S3 for DSC thermograms). The polarising optical microscopic (POM) textures and XRD patterns are shown in Fig. 2 (see ESI,† Fig. S4 for additional POM images and XRD patterns). Compound 1a with 8 alkoxy chains at the peripheries exhibits enantiotropic hexagonal columnar phases (Colh). Observations under the POM show that 1a exhibits a fan-shaped texture with a large domain size when cooled from the isotropic melt at a rate of less than 1 °C min−1 (Fig. 2a). The wide angle X-ray diffraction (WAXD) pattern at 125 °C shows two peaks at 40.7 (100) and 20.0 Å (200) (Fig. 2b). In addition, the two- dimensional small angle X-ray diffraction (SAXD) pattern at the same temperature reveals diffraction spots with a six-fold symmetry at the (100) plane (Fig. 2c), indicating a Colh phase, with an intercolumnar distance of 47.0 Å (see ESI,† Fig. S5a and b). When 1a is cooled to below 117 °C, at which the DSC chart on a cooling process exhibits an exothermic peak, no visible change in the texture is seen (see ESI,† Fig. S4a). At 100 °C, the WAXD pattern of 1a shows three diffraction peaks at 43.2 (100), 21.2 (200), and 13.8 Å (300) (see ESI,† Fig. S4b) with a lattice parameter a of 49.9 Å. Furthermore, the two-dimensional SAXD pattern of 1a at 100 °C also exhibits a hexagonal symmetry at the (100) plane (see ESI,† Fig. S4c). These results suggest a transition from the Colh2 to a new Colh1 phase. The larger intercolumnar distance can be attributed to a change in the cofacial stacking configuration (vide infra), as well as more extended and rigid alkyl chains of the Colh1 phase as compared to that of the Colh2 phase. A transition to a glassy state is seen when 1a is cooled to below 20 °C.| Compound | Phase transition behavioura | XRD data | |||
|---|---|---|---|---|---|
| T/°C | Lattice parameters/Å | ||||
| a Transition temperatures (°C) and transition enthalpies (kJ mol−1) in parentheses, determined by DSC on cooling at a rate of 10 K min−1. G, glassy; Iso, isotropic; Colh, hexagonal columnar phase; Colt, tetragonal columnar phase; Colr, rectangular columnar phase. Subscript numbers 1 and 2 indicate first and second columnar phases, respectively. | |||||
| 1a | G 20 Colh1 | 117 Colh2 | 132 Iso | 125 | a = 47.0 |
| (26) | (1) | 100 | a = 49.9 | ||
| 1b | Colr1 10 Colr2 | 100 Colt | 135 Iso | 125 | a = 40.3 |
| (43) | (12) | (6) | 90 |
a = 75.8
b = 52.0 |
|
| −5 |
a = 68.8
b = 47.9 |
||||
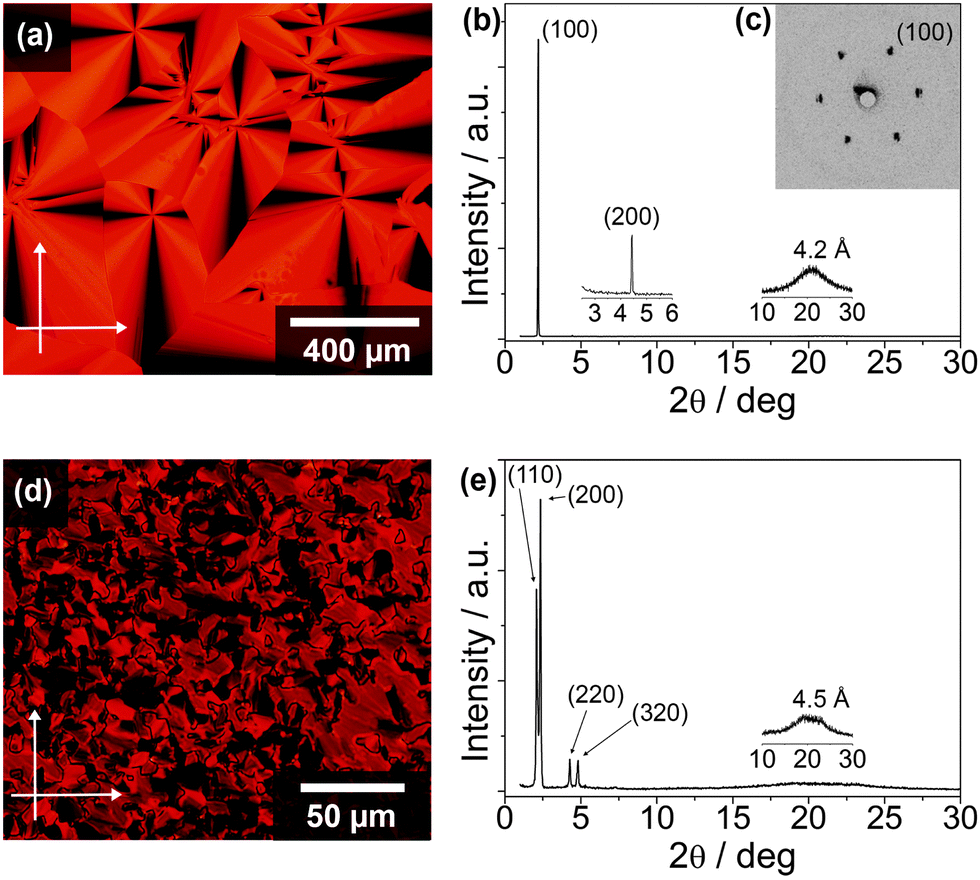 | ||
| Fig. 2 POM photographs (left panels) and WAXD patterns (right panels). Compound 1a in the Colh2 phase at 125 °C (a and b) with its SAXD pattern at the same temperature (c). Compound 1b in the Colr2 phase at 90 °C (d and e). | ||
Compound 1b with 12 alkoxy chains at the peripheries exhibits a similar isotropic transition temperature to 1a upon heating. Cooling from the isotropic melt reveals a polydomain texture of 1b at 125 °C (see ESI,† Fig. S4d). The WAXD and SAXD patterns of compound 1b at 125 °C show diffraction peaks at 40.3 (100), 29.3 (110), 19.6 (200), and 14.9 Å (300) (see ESI,† Fig. S4e and f), which indicate a tetragonal columnar (Colt) phase (see ESI,† Fig. S5c). The texture of 1b (Fig. 2d) remains relatively unchanged upon cooling to below 100 °C as compared to the texture observed at 125 °C. However, the WAXD pattern is significantly different with four distinct peaks at 42.9, 37.9, 20.7, and 18.4 Å (Fig. 2e) observed at 90 °C, which correspond to the diffractions from the (110), (200), (220), and (320) planes of a rectangular columnar phase (Colr) with a P21/a symmetry (see ESI,† Fig. S6a and S7). Using the Debye Scherer method, the lattice parameters a and b are evaluated to be 75.8 and 52.0 Å, respectively (see ESI,† Fig. S6a). Further cooling of 1b to below 10 °C induced a transition to a new Colr phase (Colr2 to Colr1 phase), with the lattice parameters a and b of 68.8 and 47.9 Å, respectively (see ESI,† Fig. S4g, h and S6b).
Hole carrier transport
The time-of-flight (TOF) photoconductivity method was used to examine the hole mobilities of compounds 1a and 1b. The samples form LC domains oriented parallel to the surface of the indium-tin-oxide (ITO) cells with a sample thickness of 9 μm by slow cooling from isotropic states to room temperature.Compounds 1a and 1b illuminated by a pulsed YAG laser at 355 nm under applied electric fields at room temperature show non-dispersive transient photocurrent curves of hole carriers (see ESI,† Fig. S8). The transit time is taken at the inflection point of the photocurrent curves. The hole mobilities measured are nearly independent of the applied voltage, which is the characteristic feature for charge carrier transport of semiconductors.40
Temperature dependence of the hole carrier mobilities of 1a and 1b is shown in Fig. 3. Compound 1a in the isotropic state shows hole mobilities in the order of 10−5 cm2 V−1 s−1. The transition from the isotropic state to the Colh2 phase induces an increase in the hole mobility. The maximum value in the Colh2 phase of 1a at 117 °C is 4.4 × 10−4 cm2 V−1 s−1. This increment in the hole mobility can be attributed to the facilitation of charge carrier transport due to the formation of the one-dimensional π–π columnar stacks. In the Colh1 phase of 1a, the hole mobilities slightly decrease with decreasing temperature.
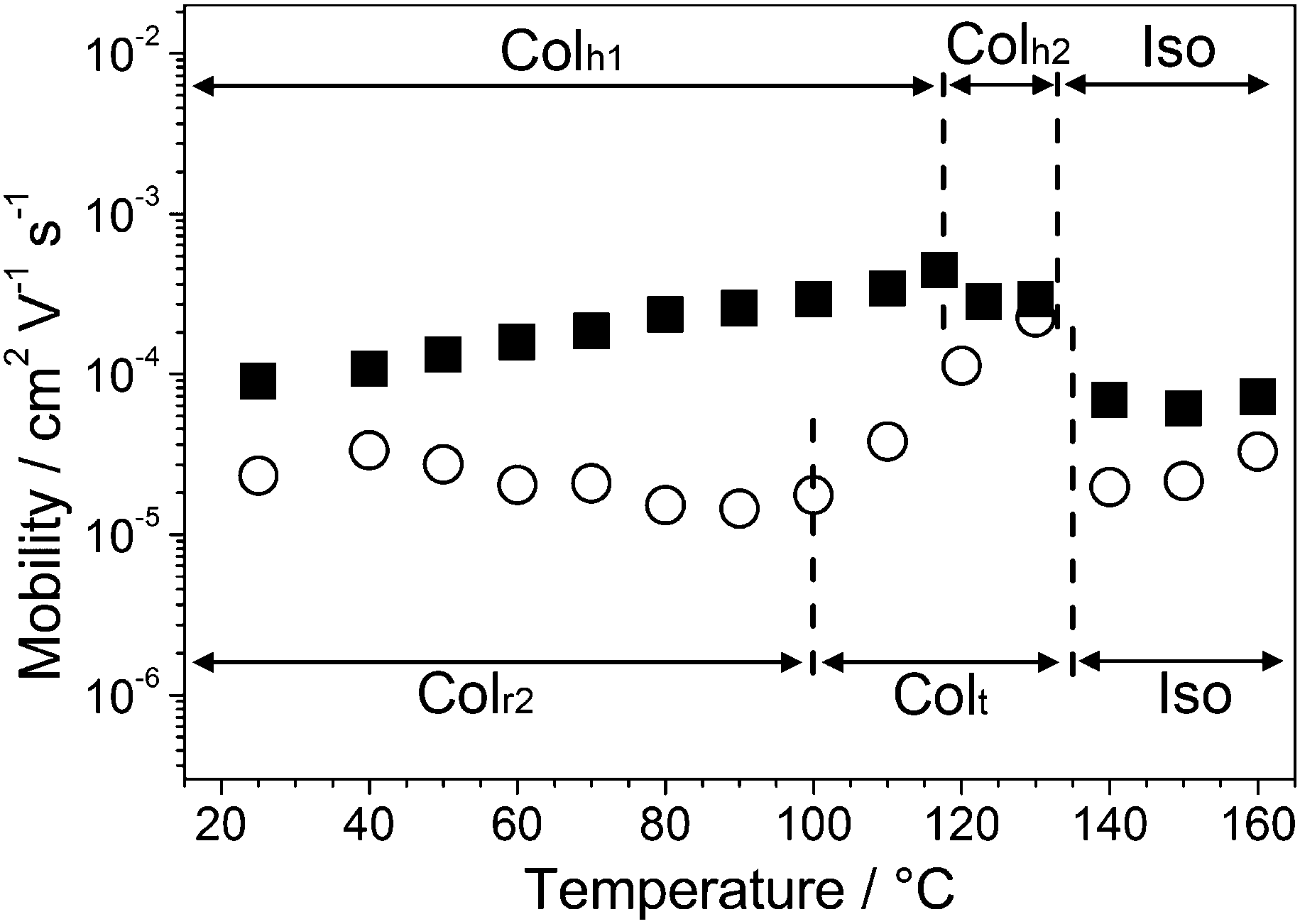 | ||
| Fig. 3 The logarithmic plot of hole mobilities of 1a (■) and 1b (○) as a function of temperature on cooling. Dotted lines denote phase transition temperatures. | ||
As for compound 1b, an abrupt increase of the hole mobilities is observed at the Iso to Colt transition. The maximum value of 2.3 × 10−4 cm2 V−1 s−1 is observed at 130 °C in the Colt phase. The hole mobilities of 1b in the Colt phase decrease by approximately one order of magnitude with decreasing temperature. In the Colr2 phase, no significant change in the hole mobilities is observed with varying temperature.
Mechanochromic properties
The stimuli-reponsive properties of the X-shaped molecules 1a and 1b were explored. Under the irradiation of UV light, the luminescence colour of compound 1a changes from red to orange when a mechanical shear force is applied on the sample in the Colh1 phase at room temperature (Fig. 4), forming a new disordered Col phase (vide infra). The red luminescence of the initial state (Colh1 phase) of 1a is recovered by heating the sheared sample to the isotropic state and then cooling to room temperature. In contrast, compound 1b shows no apparent difference in luminescence colour before or after shearing (see ESI,† Fig. S9). | ||
| Fig. 4 The photograph of 1a at room temperature on a quartz plate under illumination of UV light (365 nm) for the original Colh1 sample (left half) and the sheared sample (right half). | ||
To clarify the photophysical properties of 1a before and after mechanical shearing, UV-vis absorption (Fig. 5a, left) and photoluminescence spectra (Fig. 5a, right) of 1a have been measured on quartz substrates at ambient temperature. In addition, in the isotropic liquid phase at 150 °C and the Colh2 phase at 125 °C of 1a, the UV-vis absorption and emission spectra (Fig. 5b) have also been taken for comparison with those of the shear-induced phase.
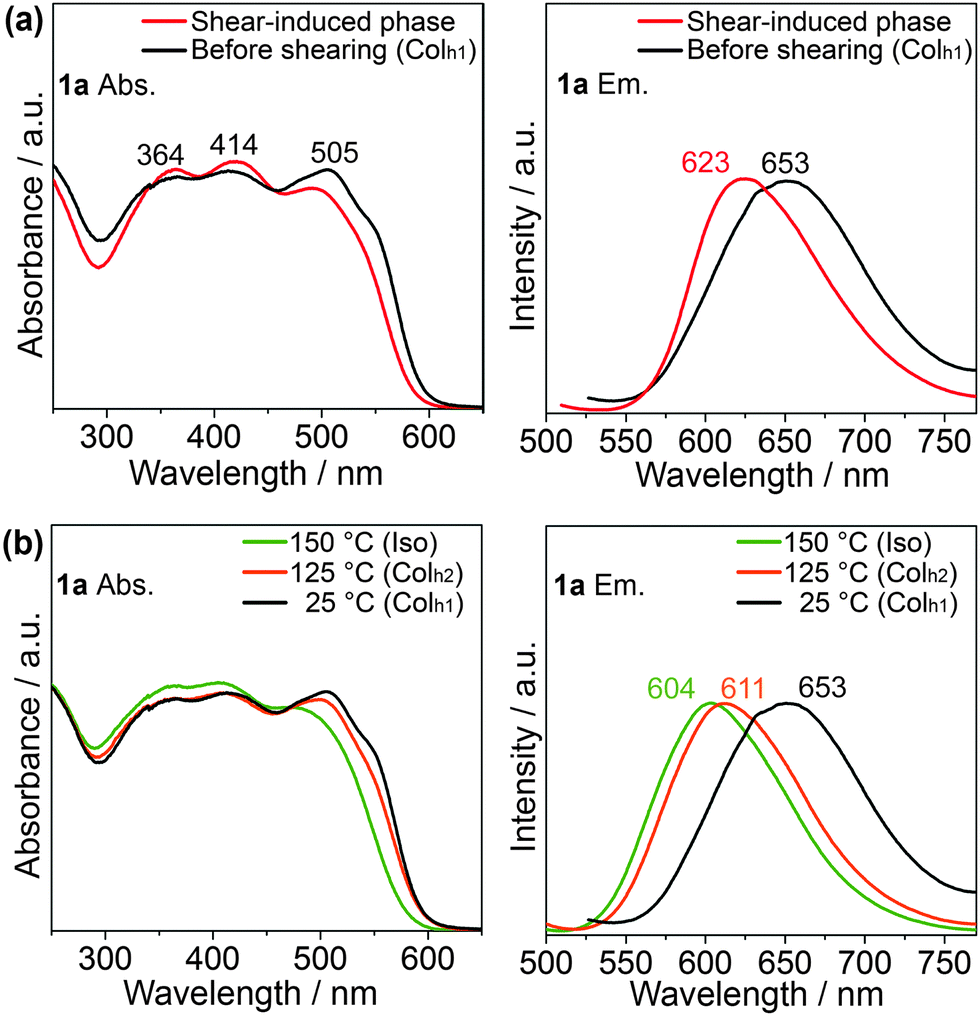 | ||
| Fig. 5 The normalized UV-vis absorbance (left panels) and emission (right panels) spectra (excitation wavelength of 420 nm) of the effect of shearing on 1a at room temperature (a), and at its various LC phases. | ||
The absorption spectra of 1a (Fig. 5a, left) show three absorption peaks (Fig. 5, left). The two absorption peaks with higher energy wavelengths at around 364 and 414 nm can be ascribed to the π → π* transition from the oligothiophene-conjugated pyrene core, while the absorption peak with lower energy wavelength at around 505 nm is probably due to n → π* from the sulfur heteroatom of the thiophene moiety.62 No significant differences in the absorption spectra before and after shearing are observed (Fig. 5a, left), although the absorption edge of the shear-induced phase is slightly blue-shifted to that of the Colh1 phase. In contrast, the maximum wavelength of the emission attributable to the S1 → So relaxation in the shear-induced phase (λmax = 623 nm) is blue-shifted by 30 nm relative to that in the Colh1 phase (Fig. 5a, right) while maintaining the absolute fluorescent quantum yield of 2% (see ESI,† Fig. S10). Moreover, the emission spectrum in the shear-induced phase at 25 °C (Fig. 5a, right) is found to be close to those in the Colh2 phase (λmax = 611 nm) at 125 °C and in the isotropic liquid phase (λmax = 604 nm) at 150 °C, as shown in Fig. 5b, right. From these results, we consider that the application of shear force to the Colh1 phase of 1a may induce disorder in the columnar stacking structure of the X-shaped molecule.
Further investigation of the shear-induced LC phase of 1a was performed. The thermal properties of the original LC sample and the sheared sample of 1a were characterised by DSC heating traces (Fig. 6a). It is observed that the sheared sample exhibits two distinct peaks at 132 and 136 °C, with transition enthalpies of 14 and 1 kJ mol−1, respectively. The total transition enthalpy from the shear-induced Colh phase to the isotropic state is considerably lower than the transition enthalpy from the Colh1 phase to the isotropic state of another sample of 1a that is not subject to shearing (30 kJ mol−1). This result suggests that shearing of the LC sample causes a disorder in the molecular arrangement. We suspect that the broad peak at 132 °C in the DSC trace of the shear-induced phase is due to the inherently incomplete shearing process.
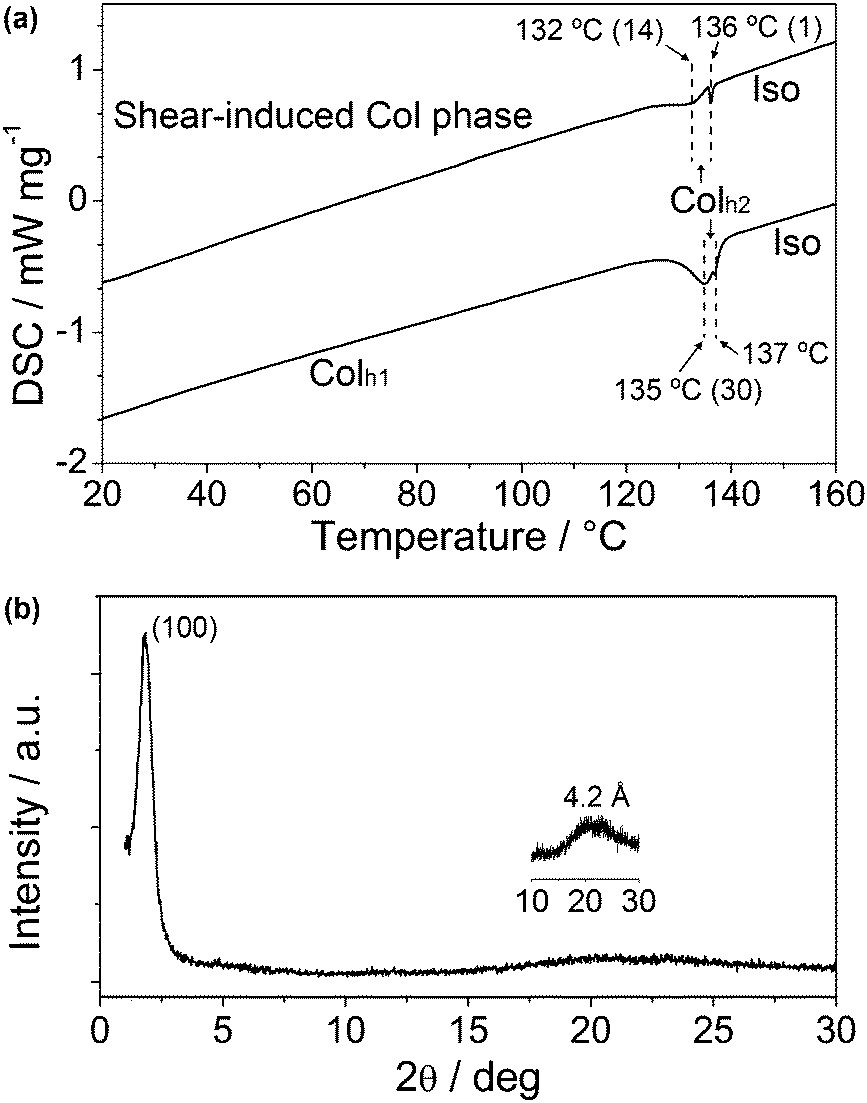 | ||
| Fig. 6 The DSC traces of a sample of 1a that is subject to shearing, and another sample that is cooled from an isotropic state to form the Colh2 phase (a). The heating rate is 10 K min−1 with transition enthalpies (kJ mol−1) shown in parentheses. The WAXD pattern of the sheared-induced Col phase of 1a (b) at room temperature. | ||
Furthermore, WAXD pattern of the sheared sample (Fig. 6b) shows only a broad peak at 47.7 Å (100), suggesting a new shear-induced Col phase. Assuming that the hexagonal lattice is maintained, the lattice parameter a of this disordered Colh phase is calculated to be 55.5 Å, which is noticeably larger than that of Colh1 and Colh2 phases as shown in Table 1.
From these observations, we propose that in the Colh1 phase, cofacial columnar stacks are formed with the individual X-shaped molecules of 1a rotated with an angle of about 15°, forming a helical packing structure (Fig. 7). This assembly was found to exhibit higher stabilisation energy.63 The blue-shift in the emission spectrum for the transition from the Colh1 phase to the Colh2 phase can be explained by the decreased interchromophoric interactions,64 which is evident by another blue-shift for the transition to the isotropic state. Based on these results, it can be deduced that 1a in the Colh2 phase presumably adopts a less cofacial stacking configuration with the X-shaped molecules rotated at a larger acute angle. (Fig. 7)
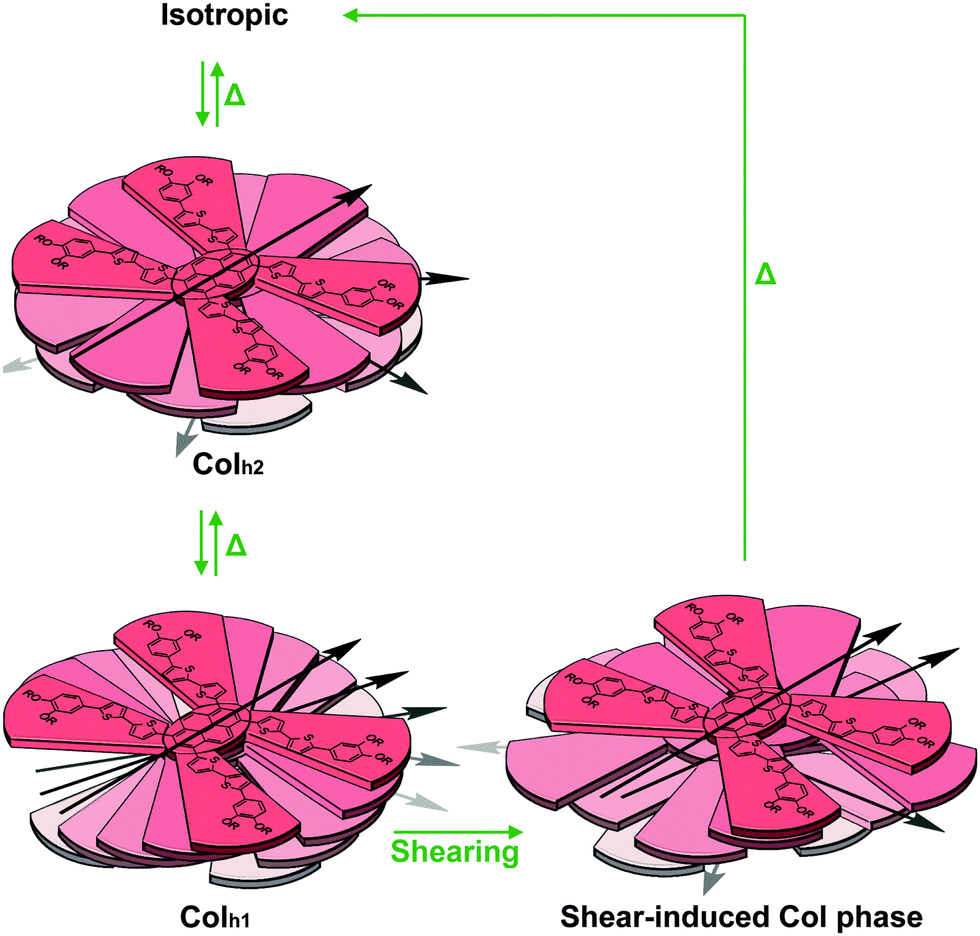 | ||
| Fig. 7 The proposed transition scheme and intermolecular stacking arrangement of 1a in the Colh1 phase, Colh2 phase and the shear-induced Col phase. The alkyl chains are omitted for clarity. | ||
It is proposed that the mechanochromic response exhibited for compound 1a can be explained in a similar way. The mechanical shearing force on 1a disrupts the ordered arrangement of the aromatic cores, causing the sheared sample to adopt a new disordered Col phase, and resulting in lesser intermolecular π–π interactions. Consequently, a visible blue-shift in the fluorescence spectra is observed.
As for compound 1b, shearing causes no observable change in the fluorescent properties. This can be attributed to the more rigid Colr phase as compared to the Colh phase.
LC gel properties
Incorporation of self-assembled fibres into the π-conjugated LC molecules is one approach to increase the hole mobility.65–71 We intend to investigate the influence on photoconductivities by the introduction of fibrous networks into the X-shaped liquid crystals. In our previous studies, triphenylene-based liquid crystals were found to form LC gels with gelator 671 (Scheme 3). However in the case of compound 1a, the introduction of gelator 6 forms a macroscopically phase-separated mixture under the POM and scanning electron microscope (SEM) observations (see ESI,† Fig. S11). Compound 7 was thus employed in our study as it was known to exhibit good gelation properties in common organic solvents.72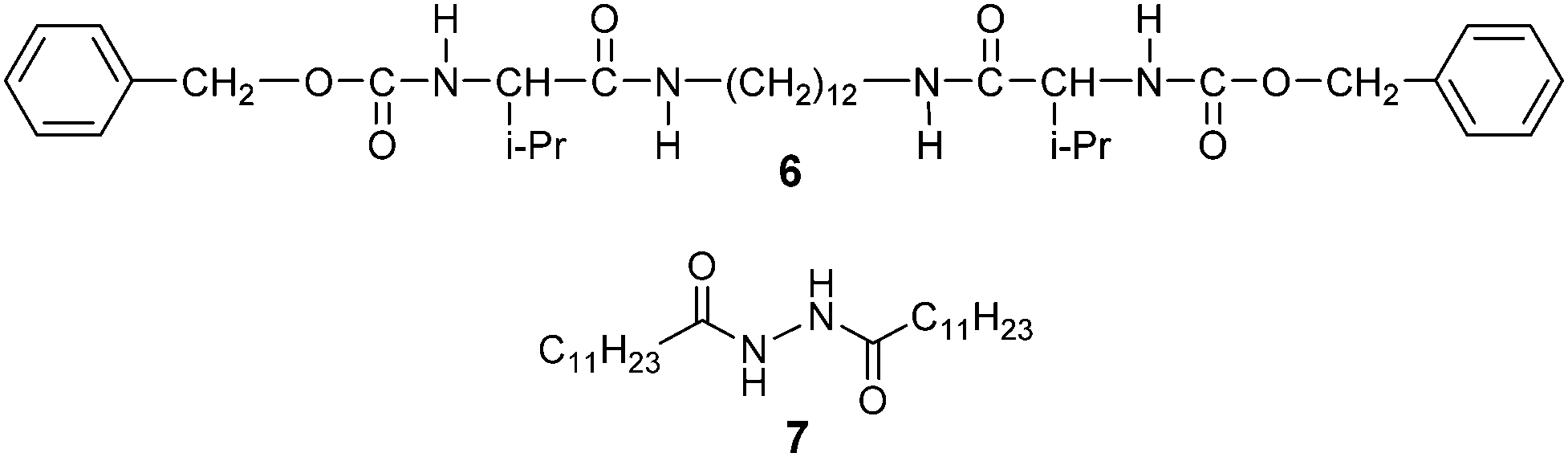 | ||
| Scheme 3 The molecular structures of the gelator candidates employed. | ||
The formation of a LC gel is achieved for compound 1a with 3 weight% of gelator 7. Upon cooling the mixture from the isotropic states, fibrous aggregates of 7 are first formed in the liquid of 1a. Upon subsequent cooling, the orientation of the LC domains of 1a along the fibrous network is observed under POM (Fig. 8a). The xerogel also reveals the fibrous network under SEM (Fig. 8b). The fibrous network is also observed by the introduction of gelator 7 into compound 1b (see ESI,† Fig. S12).
 | ||
| Fig. 8 The images of the LC gel of liquid crystal 1a and gelator 7 upon cooling from the isotropic state to 125 °C under POM (a), and the fibrous structure of the xerogel under SEM (b). | ||
The hole mobilities of LC gels of 1a and 1b with 3 weight% of gelator 7 are measured at varying temperatures and compared to their pure counterparts. For the LC gel of 1a, consistently higher hole mobility was observed as compared to pure 1a (see ESI,† Fig. S13). At 117 °C, the LC gel shows a maximum hole mobility of 8.4 × 10−4 cm2 V−1 s−1, almost double of that of pure 1a. In contrast, hole mobility of the LC gel of 1b shows no significant difference as compared to pure 1b (see ESI,† Fig. S14). In our previous studies, a significant increase in hole mobility was observed for the LC gels of triphenylene molecules, which could be attributed to the alignment of the liquid crystal on the fibrous structure that presumably suppresses the fluctuation of the molecular assembly of LC molecules.68,71 The observations in this study suggest that the gelation exerted a smaller effect on the fluctuation of the X-shaped molecule 1b as compared to 1a, suggesting a more rigid stacking structure in the Colt and Colr phases of 1b as compared to the Colh phases of 1a.
Conclusion
We have demonstrated that pyrene–oligothiophene based X-shaped molecules of 1a and 1b are capable of forming Colh and Colr phases over a wide range of temperature, including room temperature. TOF photoconductivity measurements reveal that the Colh and Colr phases of 1a and 1b show modest hole carrier transport in the order of 10−4 to 10−5 cm2 V−1 s−1. Compound 1a exhibits mechano-sensitive photoluminescence properties, and the possible intermolecular configurations in different columnar phases have been elucidated. The introduction of a fibrous network also causes a small increase in hole mobility for 1a. This simple strategy for the preparation of new and multi-functional liquid-crystalline pyrene–oligothiophene based X-shaped molecules would potentially contribute to the growing field of organic electronics.Acknowledgements
This study was partially supported by Grant-in-Aid for Scientific Research A (KAKENHI 23245030 for T. K.) and Scientific Research on Innovative Area (Stimuli-responsive Chemical Species, KAKENHI 15H00921 for M. Y.) from the Ministry of Education, Culture, Sports, Science and Technology of Japan (MEXT). K. P. G. is grateful for the support by MEXT on his research and pursue of graduate degrees in the University of Tokyo.Notes and references
- Chemistry of Discotic Liquid Crystals: From Monomers to Polymers, ed. S. Kumar, Taylor and Francis, CRC Press, Boca Raton, FL, 2011 Search PubMed.
- Handbook of Liquid Crystals, ed. J. W. Goodby, P. J. Collings, T. Kato, C. Tschierske, H. Gleeson and P. Raynes, Wiley-VCH, Weinheim, Germany, 2nd edn, 2014 Search PubMed.
- W. Pisula and K. Müllen, in Handbook of Liquid Crystals, ed. J. W. Goodby, P. J. Collings, T. Kato, C. Tschierske, H. Gleeson and P. Raynes, Wiley-VCH, Weinheim, Germany, 2nd edn, 2014, ch. 20, vol. 8, pp. 627–673 Search PubMed.
- M. Funahashi, T. Yasuda and T. Kato, in Handbook of Liquid Crystals, ed. J. W. Goodby, P. J. Collings, T. Kato, C. Tschierske, H. Gleeson and P. Raynes, Wiley-VCH, Weinheim, Germany, 2nd edn, 2014, ch. 21, vol. 8, pp. 675–708 Search PubMed.
- N. Boden, R. J. Bushby and J. Clements, J. Chem. Phys., 1993, 98, 5920–5931 CrossRef CAS.
- D. Adam, P. Schuhmacher, J. Simmerer, L. Haussling, K. Siemensmeyer, K. H. Etzbachi, H. Ringsdorf and D. Haarer, Nature, 1994, 371, 141–143 CrossRef CAS.
- F. J. M. Hoeben, P. Jonkheijm, E. W. Meijer and A. P. H. J. Schenning, Chem. Rev., 2005, 105, 1491–1546 CrossRef CAS PubMed.
- S. Kumar, Chem. Soc. Rev., 2006, 35, 83–109 RSC.
- J. Wu, W. Pisula and K. Müllen, Chem. Rev., 2007, 107, 718–747 CrossRef CAS PubMed.
- S. Sergeyev, W. Pisula and Y. H. Geerts, Chem. Soc. Rev., 2007, 36, 1902–1929 RSC.
- M. O’Neill and S. M. Kelly, Adv. Mater., 2011, 23, 566–584 CrossRef PubMed.
- R. J. Bushby and K. Kawata, Liq. Cryst., 2011, 38, 1415–1426 CrossRef CAS.
- S. Kumar, Isr. J. Chem., 2012, 52, 820–829 CrossRef CAS.
- S. Kumar, NPG Asia Mater., 2014, 6, e82 CrossRef CAS.
- T. Wohrle, I. Wurzbach, J. Kirres, A. Kostidou, N. Kapernaum, J. Litterscheidt, J. C. Haenle, P. Staffeld, A. Baro, F. Giesselmann and S. Laschat, Chem. Rev., 2016, 116, 1139–1241 CrossRef PubMed.
- V. Percec, M. Glodde, T. K. Bera, Y. Miura, I. Shiyanovskaya, K. D. Singer, V. S. K. Balagurusamy, P. A. Heiney, I. Schnell, A. Rapp, H. W. Spiess, S. D. Hudson and H. Duan, Nature, 2002, 417, 384–387 CrossRef PubMed.
- J. Wu, M. D. Watson, L. Zhang, Z. Wang and K. Müllen, J. Am. Chem. Soc., 2004, 126, 177–186 CrossRef CAS PubMed.
- M. G. Debije, J. Piris, M. P. de Haas, J. M. Warman, Ž. Tomović, C. D. Simpson, M. D. Watson and K. Müllen, J. Am. Chem. Soc., 2004, 126, 4641–4645 CrossRef CAS PubMed.
- H. Iino, J.-i. Hanna, R. J. Bushby, B. Movaghar, B. J. Whitaker and M. J. Cook, Appl. Phys. Lett., 2005, 87, 132102 CrossRef.
- M. J. Sienkowska, H. Monobe, P. Kaszynski and Y. Shimizu, J. Mater. Chem., 2007, 17, 1392–1398 RSC.
- X. Feng, V. Marcon, W. Pisula, M. R. Hansen, J. Kirkpatrick, F. Grozema, D. Andrienko, K. Kremer and K. Mullen, Nat. Mater., 2009, 8, 421–426 CrossRef CAS PubMed.
- S. Diring, F. Camerel, B. Donnio, T. Dintzer, S. Toffanin, R. Capelli, M. Muccini and R. Ziessel, J. Am. Chem. Soc., 2009, 131, 18177–18185 CrossRef CAS PubMed.
- B. Zhao, B. Liu, R. Q. Png, K. Zhang, K. A. Lim, J. Luo, J. Shao, P. K. H. Ho, C. Chi and J. Wu, Chem. Mater., 2010, 22, 435–449 CrossRef CAS.
- M.-C. Tzeng, S.-C. Liao, T.-H. Chang, S.-C. Yang, M.-W. Weng, H.-C. Yang, M. Y. Chiang, Z. Kai, J. Wu and C. W. Ong, J. Mater. Chem., 2011, 21, 1704–1712 RSC.
- T. Hirose, Y. Shibano, Y. Miyazaki, N. Sogoshi, S. Nakabayashi and M. Yasutake, Mol. Cryst. Liq. Cryst., 2011, 534, 81–92 CrossRef CAS.
- E. Venuti, R. G. Della Valle, I. Bilotti, A. Brillante, M. Cavallini, A. Calò and Y. H. Geerts, J. Phys. Chem. C, 2011, 115, 12150–12157 CAS.
- Q. Xiao, T. Sakurai, T. Fukino, K. Akaike, Y. Honsho, A. Saeki, S. Seki, K. Kato, M. Takata and T. Aida, J. Am. Chem. Soc., 2013, 135, 18268–18271 CrossRef CAS PubMed.
- T. Hirose, H. Takai, M. Watabe, H. Minamikawa, T. Tachikawa, K. Kodama and M. Yasutake, Tetrahedron, 2014, 70, 5100–5108 CrossRef CAS.
- S. R. McLaren, D. J. Tate, O. R. Lozman, G. H. Mehl and R. J. Bushby, J. Mater. Chem. C, 2015, 3, 5754–5763 RSC.
- T. Kushida, A. Shuto, M. Yoshio, T. Kato and S. Yamaguchi, Angew. Chem., Int. Ed., 2015, 54, 6922–6925 CrossRef CAS PubMed.
- A. Bacher, I. Bleyl, C. H. Erdelen, D. Haarer, W. Paulus and H.-W. Schmidt, Adv. Mater., 1997, 9, 1031–1035 CrossRef CAS.
- T. Hassheider, S. A. Benning, H.-S. Kitzerow, M.-F. Achard and H. Bock, Angew. Chem., Int. Ed., 2001, 40, 2060–2063 CrossRef CAS.
- A. M. van de Craats, N. Stutzmann, O. Bunk, M. M. Nielsen, M. Watson, K. Müllen, H. D. Chanzy, H. Sirringhaus and R. H. Friend, Adv. Mater., 2003, 15, 495–499 CrossRef CAS.
- I. O. Shklyarevskiy, P. Jonkheijm, N. Stutzmann, D. Wasserberg, H. J. Wondergem, P. C. M. Christianen, A. P. H. J. Schenning, D. M. de Leeuw, Ž. Tomović, J. Wu, K. Müllen and J. C. Maan, J. Am. Chem. Soc., 2005, 127, 16233–16237 CrossRef CAS PubMed.
- B. Mukherjee and M. Mukherjee, Org. Electron., 2009, 10, 1282–1287 CrossRef CAS.
- L. Schmidt-Mende, A. Fechtenkötter, K. Müllen, E. Moons, R. H. Friend and J. D. MacKenzie, Science, 2001, 293, 1119–1122 CrossRef CAS PubMed.
- J. L. Li, M. Kastler, W. Pisula, J. W. F. Robertson, D. Wasserfallen, A. C. Grimsdale, J. S. Wu and K. Müllen, Adv. Funct. Mater., 2007, 17, 2528–2533 CrossRef CAS.
- K. Takemoto, M. Karasawa and M. Kimura, ACS Appl. Mater. Interfaces, 2012, 4, 6289–6294 CAS.
- L. Li, S. W. Kang, J. Harden, Q. Sun, X. Zhou, L. Dai, A. Jakli, S. Kumar and Q. Li, Liq. Cryst., 2008, 35, 233–239 CrossRef CAS.
- M. Funahashi and J.-i. Hanna, Appl. Phys. Lett., 2000, 76, 2574–2576 CrossRef CAS.
- M. Funahashi and J. I. Hanna, Adv. Mater., 2005, 17, 594–598 CrossRef CAS.
- M. P. Aldred, A. E. A. Contoret, S. R. Farrar, S. M. Kelly, D. Mathieson, M. O'Neill, W. C. Tsoi and P. Vlachos, Adv. Mater., 2005, 17, 1368–1372 CrossRef CAS.
- A. J. J. M. van Breemen, P. T. Herwig, C. H. T. Chlon, J. Sweelssen, H. F. M. Schoo, S. Setayesh, W. M. Hardeman, C. A. Martin, D. M. de Leeuw, J. J. P. Valeton, C. W. M. Bastiaansen, D. J. Broer, A. R. Popa-Merticaru and S. C. J. Meskers, J. Am. Chem. Soc., 2006, 128, 2336–2345 CrossRef CAS PubMed.
- M. Funahashi, F. Zhang and N. Tamaoki, Adv. Mater., 2007, 19, 353–358 CrossRef CAS.
- F. Zhang, M. Funahashi and N. Tamaoki, Org. Electron., 2010, 11, 363–368 CrossRef CAS.
- X. Zhang, L. J. Richter, D. M. DeLongchamp, R. J. Kline, M. R. Hammond, I. McCulloch, M. Heeney, R. S. Ashraf, J. N. Smith, T. D. Anthopoulos, B. Schroeder, Y. H. Geerts, D. A. Fischer and M. F. Toney, J. Am. Chem. Soc., 2011, 133, 15073–15084 CrossRef CAS PubMed.
- M. Funahashi, F. Zhang, N. Tamaoki and J.-i. Hanna, ChemPhysChem, 2008, 9, 1465–1473 CrossRef CAS PubMed.
- K. Sun, Z. Xiao, S. Lu, W. Zajaczkowski, W. Pisula, E. Hanssen, J. M. White, R. M. Williamson, J. Subbiah, J. Ouyang, A. B. Holmes, W. W. H. Wong and D. J. Jones, Nat. Commun, 2015, 6, 6013 CrossRef CAS PubMed.
- T. Yasuda, K. Kishimoto and T. Kato, Chem. Commun., 2006, 3399–3401 RSC.
- T. Yasuda, H. Ooi, J. Morita, Y. Akama, K. Minoura, M. Funahashi, T. Shimomura and T. Kato, Adv. Funct. Mater., 2009, 19, 411–419 CrossRef CAS.
- T. Yasuda, T. Shimizu, F. Liu, G. Ungar and T. Kato, J. Am. Chem. Soc., 2011, 133, 13437–13444 CrossRef CAS PubMed.
- Y. Sagara and T. Kato, Angew. Chem., Int. Ed., 2008, 47, 5175–5178 CrossRef CAS PubMed.
- Y. Sagara and T. Kato, Nat. Chem., 2009, 1, 605–610 CrossRef CAS PubMed.
- Y. Sagara, S. Yamane, T. Mutai, K. Araki and T. Kato, Adv. Funct. Mater., 2009, 19, 1869–1875 CrossRef CAS.
- M. Mitani, S. Yamane, M. Yoshio, M. Funahashi and T. Kato, Mol. Cryst. Liq. Cryst., 2014, 594, 112–121 CrossRef CAS.
- M. Mitani, S. Ogata, S. Yamane, M. Yoshio, M. Hasegawa and T. Kato, J. Mater. Chem. C, 2016, 4, 2752–2760 RSC.
- Z. Chi, X. Zhang, B. Xu, X. Zhou, C. Ma, Y. Zhang, S. Liu and J. Xu, Chem. Soc. Rev., 2012, 41, 3878–3896 RSC.
- C. Löwe and C. Weder, Adv. Mater., 2002, 14, 1625–1629 CrossRef.
- J. Kunzelman, M. Kinami, B. R. Crenshaw, J. D. Protasiewicz and C. Weder, Adv. Mater., 2008, 20, 119–122 CrossRef CAS.
- G. Zhang, J. Lu, M. Sabat and C. L. Fraser, J. Am. Chem. Soc., 2010, 132, 2160–2162 CrossRef CAS PubMed.
- T. Ishiyama, J. Takagi, Y. Yonekawa, J. F. Hartwig and N. Miyaura, Adv. Synth. Catal., 2003, 345, 1103–1106 CrossRef CAS.
- K. R. Idzik, P. J. Cywinski, W. Kuznik, J. Frydel, T. Licha and T. Ratajczyk, Phys. Chem. Chem. Phys., 2015, 17, 22758–22769 RSC.
- V. de Halleux, J. P. Calbert, P. Brocorens, J. Cornil, J. P. Declercq, J. L. Brédas and Y. Geerts, Adv. Funct. Mater., 2004, 14, 649–659 CrossRef CAS.
- A. Hayer, V. de Halleux, A. Köhler, A. El-Garoughy, E. W. Meijer, J. Barberá, J. Tant, J. Levin, M. Lehmann, J. Gierschner, J. Cornil and Y. H. Geerts, J. Phys. Chem. B, 2006, 110, 7653–7659 CrossRef CAS PubMed.
- T. Kato, N. Mizoshita and K. Kishimoto, Angew. Chem., Int. Ed., 2006, 45, 38–68 CrossRef CAS PubMed.
- T. Kato, Y. Hirai, S. Nakaso and M. Moriyama, Chem. Soc. Rev., 2007, 36, 1857–1867 RSC.
- T. Kato, T. Yasuda, Y. Kamikawa and M. Yoshio, Chem. Commun., 2009, 729–739 RSC.
- T. Kato and K. Tanabe, Chem. Lett., 2009, 38, 634–639 CrossRef CAS.
- N. Mizoshita, H. Monobe, M. Inoue, M. Ukon, T. Watanabe, Y. Shimizu, K. Hanabusa and T. Kato, Chem. Commun., 2002, 428–429 RSC.
- M. Moriyama, N. Mizoshita and T. Kato, Polym. J., 2004, 36, 661–664 CrossRef CAS.
- Y. Hirai, H. Monobe, N. Mizoshita, M. Moriyama, K. Hanabusa, Y. Shimizu and T. Kato, Adv. Funct. Mater., 2008, 18, 1668–1675 CrossRef CAS.
- K. Tomioka, T. Sumiyoshi, S. Narui, Y. Nagaoka, A. Iida, Y. Miwa, T. Taga, M. Nakano and T. Handa, J. Am. Chem. Soc., 2001, 123, 11817–11818 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental, molecular modelling, thermogravimetric curves, DSC traces, POM images and XRD patterns, lattice analyses, transient photocurrent curves, UV-vis and fluorescence spectra, POM/SEM images and hole mobilities of LC gels. See DOI: 10.1039/c6tc00808a |
| This journal is © The Royal Society of Chemistry 2016 |