
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe development of aryl-substituted 2-phenylimidazo[1,2-a]pyridines (PIP) with various colors of excited-state intramolecular proton transfer (ESIPT) luminescence in the solid state†
Toshiki
Mutai
*,
Tatsuya
Ohkawa
,
Hideaki
Shono
and
Koji
Araki
*
Department of Materials and Environmental Science, the University of Tokyo, 4-6-1, Komaba, Meguro-ku, Tokyo 153-8505, Japan. E-mail: mutai@iis.u-tokyo.ac.jp; araki@iis.u-tokyo.ac.jp
First published on 30th March 2016
Abstract
A series of solid-state luminescent dyes based on 2-phenylimidazo[1,2-a]pyridine (PIP) displaying a wide range of emitting colors from blue to red have been developed. Whereas 2′-methoxy PIP (2′MeOPIP, 10) shows blue luminescence, 2′-hydroxy PIP (HPIP, 1) exhibits emission with large Stokes shift at around 500 nm that is known as the excited-state intramolecular proton transfer (ESIPT) luminescence, which can be tuned from blue-green to red by simply introducing aryl group(s) into HPIP through Pd-catalyzed cross coupling reactions. It is shown that the energy of ESIPT luminescence decreases as the electron-withdrawing nature of the para-substituent on the aryl group increases. Varying the substitution position is also an effective tuning method, because the ESIPT luminescence wavelength is in the order 6-aryl < 8-aryl < 6,8-diaryl. Although the quantum yields of these compounds are quite low in organic solutions (Φ ∼ 0.01), they generally display a much stronger ESIPT luminescence in the solid state. For all compounds except for 9 having long C6-alkyl chains, the similar emission properties in the dilute frozen matrix and the solid state indicated that ESIPT emission in the solid state is from the monomeric species, even though π-stacked motifs of the HPIP cores and the aryl groups introduced are confirmed by crystallographic analysis. Time-dependent DFT calculations reasonably explained the effect of substitution on ESIPT luminescence in the solid state. The results show that aryl-substitution is a convenient approach to tuning the radiation energy of the ESIPT luminescence of HPIPs without suffering the quenching effect due to intermolecular interactions, and thus a series of PIP compounds that exhibit a wide range of luminescence colors can be realized.
Introduction
The development of organic solid-state luminescent materials is a fundamental and important issue in various applications.1 However, fluorescence is often quenched in the solid state because of unfavorable intermolecular interactions that promote the non-radiative deactivation of the excited state.2 A common strategy to overcome this problem is the introduction of a bulky group3 or the design of non-planar structures4 to suppress inter-fluorophore electronic interactions. In recent years, compounds that are non-emissive in solution but show efficient luminescence in the solid state, i.e., the phenomena of aggregation- or crystallization-induced emission (AIE or CIE, respectively),5 have attracted increasing interest, and their photophysical properties are being actively studied in view of their conformational confinement and/or intermolecular interactions in the solid state.6Photo-induced proton transfer through an intramolecular hydrogen bond is termed the excited-state intramolecular proton transfer (ESIPT).7 Upon excitation of an intramolecularly hydrogen-bonded species (closed-enol, Scheme 1), ESIPT takes place immediately and subsequent conformational relaxation leads to the formation of the ESIPT state. The emission from the ESIPT state is characterized by a large Stokes shift (∼10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cm−1), which enables long-wavelength luminescence upon UV-excitation.8 Luminescence with a large Stokes shift is especially beneficial in the solid state because of the absence of absorption and emission spectral overlap, which indicates the suppression of intermolecular electronic interference. Hence, colorless luminescent materials,7b,9 white-light emitting materials,10 and multi-component luminophore systems that can realize a variety of emission colors11 are actively being studied. To this end, ESIPT emitters exhibiting bright luminescence in the solid state are required, but are limited to date.12 Color tuning by chemical modification of a parent luminophore is a convenient and practical approach for developing solid ESIPT luminophores showing a variety of emission colors.13,14
000 cm−1), which enables long-wavelength luminescence upon UV-excitation.8 Luminescence with a large Stokes shift is especially beneficial in the solid state because of the absence of absorption and emission spectral overlap, which indicates the suppression of intermolecular electronic interference. Hence, colorless luminescent materials,7b,9 white-light emitting materials,10 and multi-component luminophore systems that can realize a variety of emission colors11 are actively being studied. To this end, ESIPT emitters exhibiting bright luminescence in the solid state are required, but are limited to date.12 Color tuning by chemical modification of a parent luminophore is a convenient and practical approach for developing solid ESIPT luminophores showing a variety of emission colors.13,14
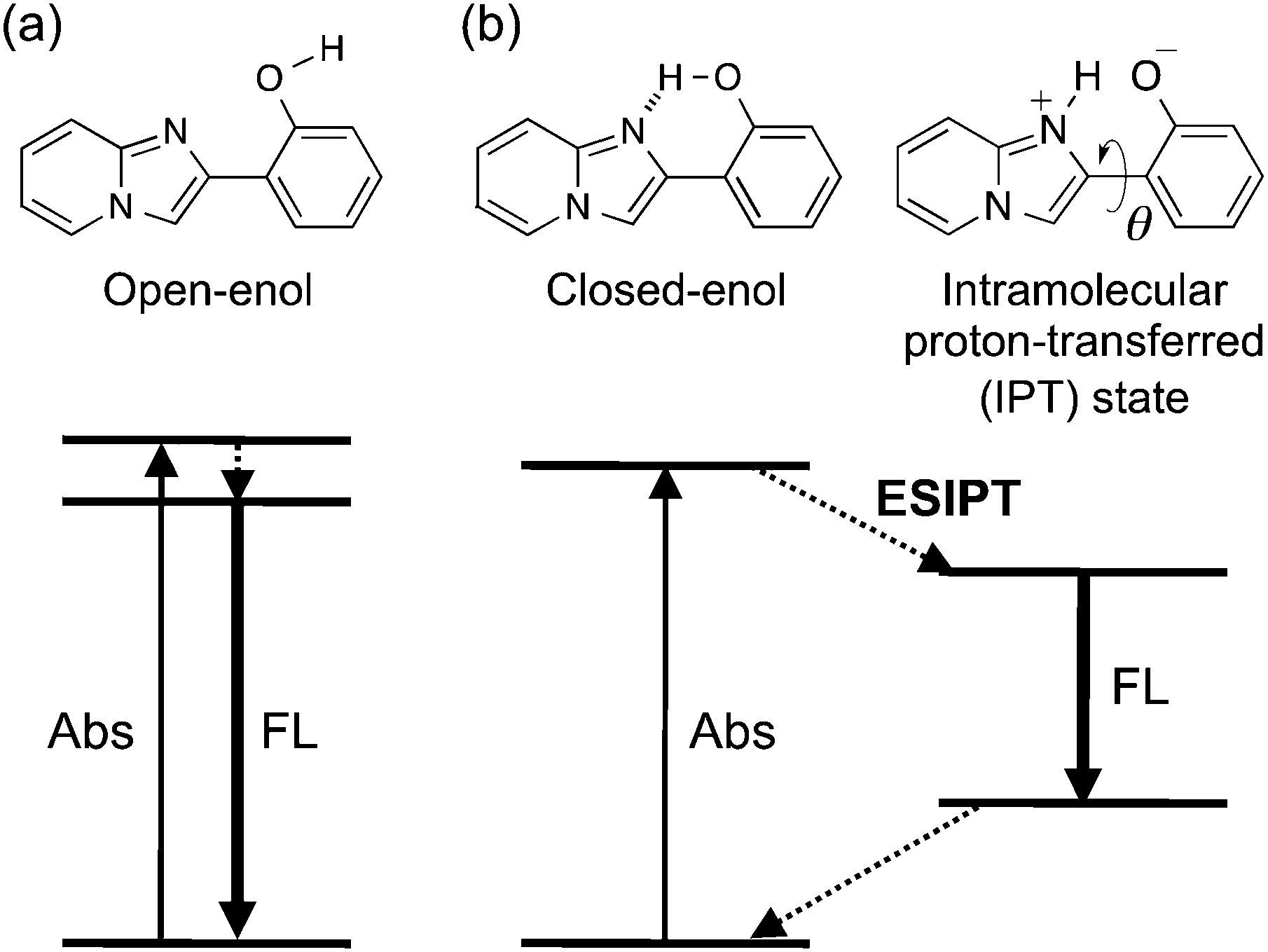 | ||
| Scheme 1 Energy diagrams of normal (a) and ESIPT (b) fluorescence. | ||
2-(2′-Hydroxyphenyl)imidazo[1,2-a]pyridine (HPIP, 1) in the closed-enol form shows weak ESIPT fluorescence (Φ ∼ 0.05) at around 600 nm in organic solutions.15 We found16 that 1 also exhibits bright ESIPT luminescence (Φ ∼ 0.5) in the solid state, and the mechanism of luminescence enhancement in the solid state was examined by quantum chemical calculations.17 The effects of electron donating/withdrawing substituents on the luminescence properties were also studied.14
In this report, a series of HPIP derivatives 2–9 having aryl groups at the 6 and/or 8 positions were synthesized (Scheme 2) and their ESIPT luminescence properties in the solid state were examined. Aryl substitution is an effective method for modifying the π-electronic state, and could also be effective in controlling the luminescence properties. Although aromatic units tend to stack in densely packed solids, which causes unfavorable luminescence quenching, the π-stacking was found to be negligible in the case of aryl-substituted HPIPs. Thus, novel solid-state luminescent materials showing a variety of bright emission colors can be realized by aryl substitution.
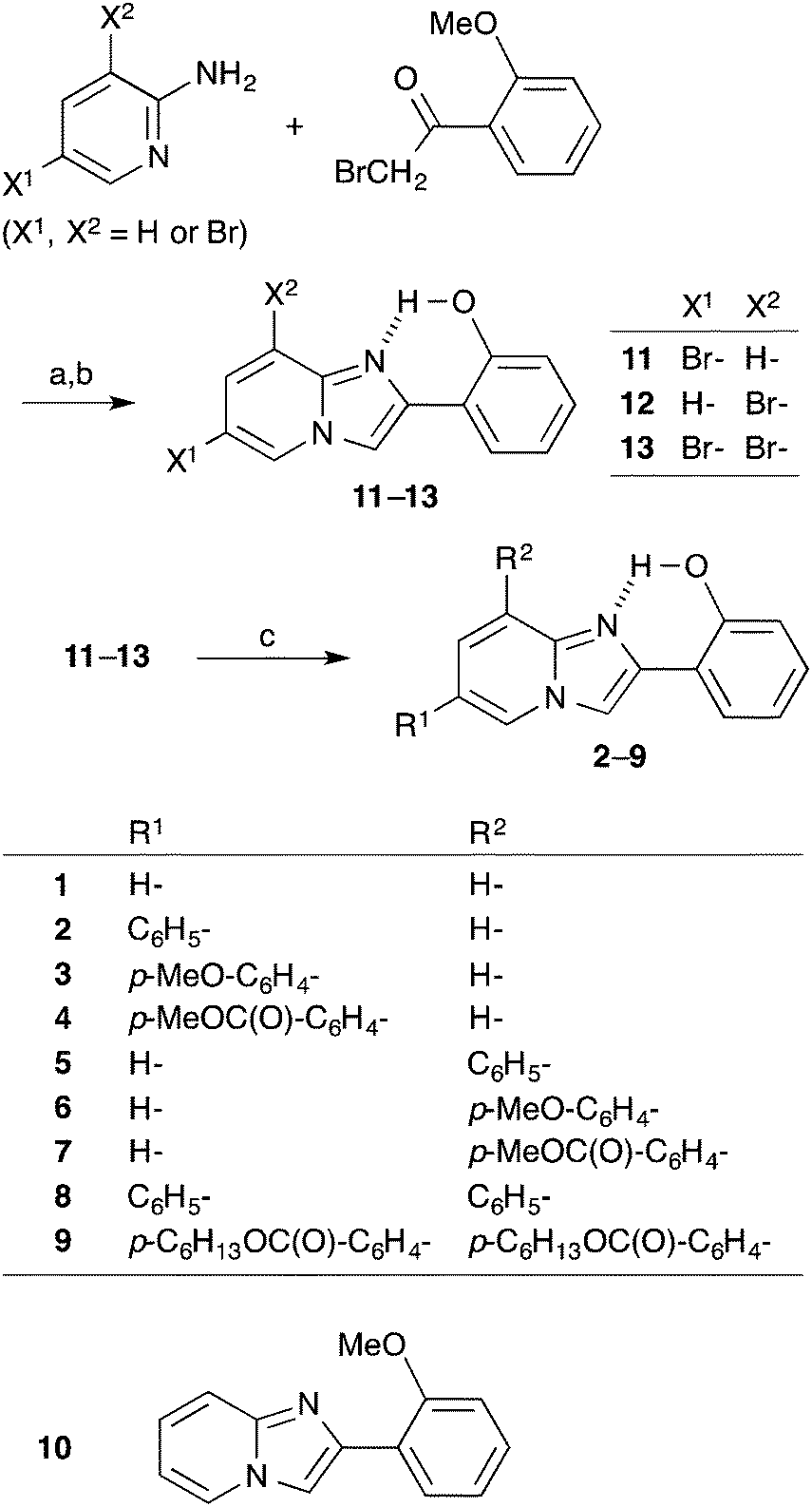 | ||
| Scheme 2 Synthetic scheme of 1–13. (a) NaHCO3, CH3CN, and reflux; (b) BBr3 and CH2Cl2; (c) Ar–B(OH)2, Pd(PPh3)4, K2CO3, 1,4-dioxane–H2O, and reflux. | ||
In order to understand the photophysical properties of aryl-substituted HPIPs in the solid state, the electronic effects of aryl substituents at the single molecular level, the role of conformational confinement, and the effects of intermolecular interactions were analyzed by studying their luminescence in dilute fluid solutions, dilute frozen matrices, and densely packed solids. Based on crystallographic analysis and quantum chemical calculations, the photophysical properties of aryl-substituted HPIPs in the solid state were discussed in further detail.
Results and discussion
Synthesis
The synthetic scheme and molecular structures of aryl-substituted HPIPs 2–9 are shown in Scheme 2. The parent compounds 1 and 10 were synthesized according to the method described in the literature.16 The coupling of bromo-substituted 2-aminopyridine and α-bromoacetophenone yielded the corresponding bromo 2′-methoxy PIPs, which were further demethylated with boron tribromide to produce 11–13. Subsequent Suzuki–Miyaura cross-coupling gave 6-aryl (2–4), 8-aryl (5–7), and 6,8-diaryl (8, 9) HPIPs. In the infrared spectra of the solid samples, the O–H stretching band of 1–9 appeared at around 3150 cm−1, indicating the formation of an intramolecular hydrogen bond between O–H and nitrogen (N1) in the imidazopyridine ring (“closed-enol” in Scheme 1).Absorption and fluorescence properties in dilute fluid solution
In fluid tetrahydrofuran (THF, 1.0 × 10−5 mol dm−3) solution, the absorption bands of HPIP (1) and its 6-phenyl (2) and 8-methoxyphenyl (6) derivatives were observed at around 330 nm (Fig. 1). As shown in Table 1, the absorption spectra of the aryl HPIPs (2–9) appeared in the near-UV region with the maximum wavelength in the range of 320–339 nm. Detailed examination indicated that the introduction of 6-aryl groups (2–4) caused a slight red shift (∼5 nm) regardless of the para-substituent (–COOR, –H, –OCH3), whereas 8-aryl derivatives 5–7 showed a blue shift (∼10 nm). Nevertheless, such small shifts indicate that aryl substitution had little effect on the absorption properties of the HPIP core in dilute fluid solutions.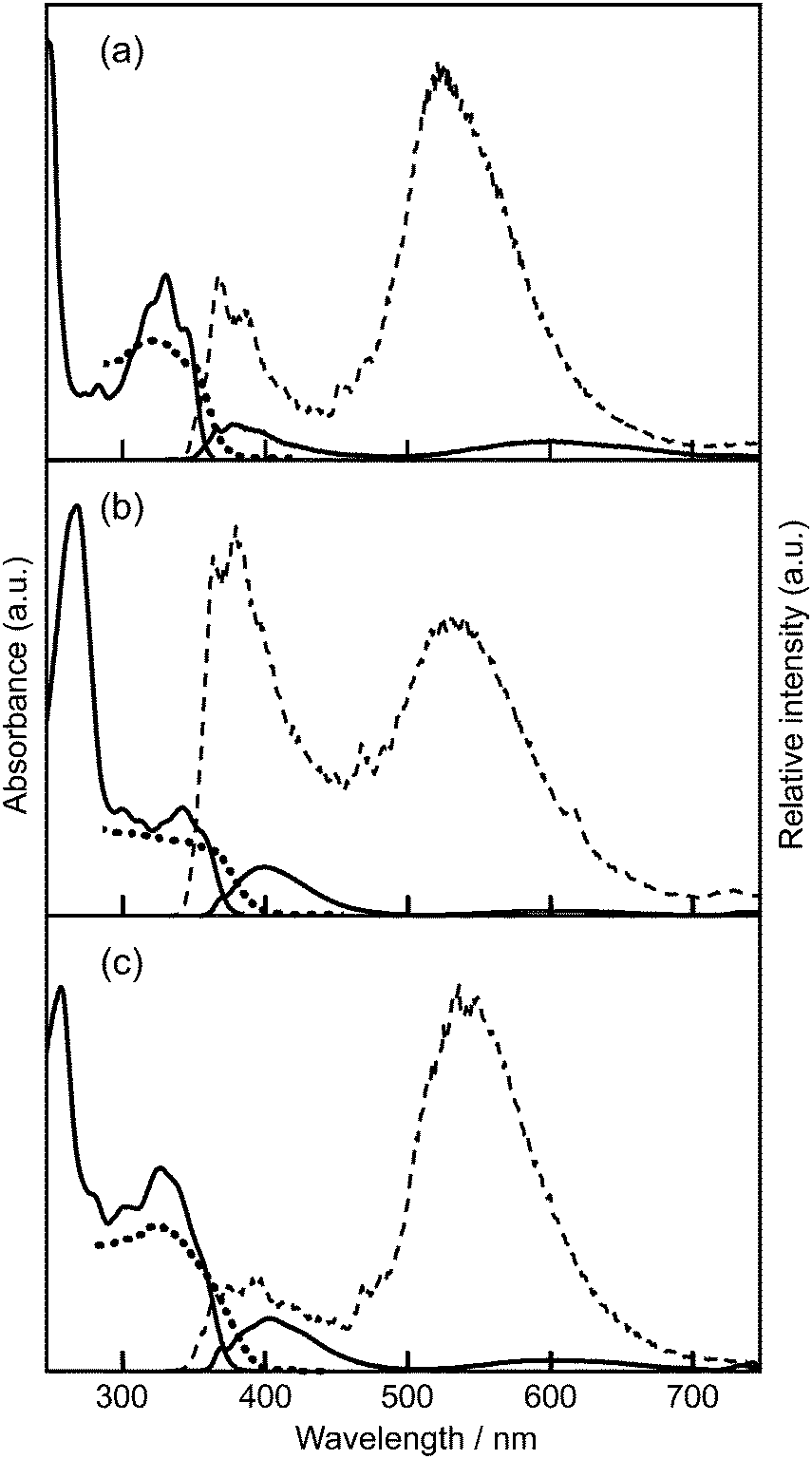 | ||
| Fig. 1 Absorption and luminescence spectra of 1 (a), 2 (b), and 6 (c). Absorption and fluorescence in a fluid THF solution (solid line), fluorescence in a frozen THF solution at 77 K (broken line), and the Kubelka–Munk spectra of the solid (dotted line). | ||
| Compd | Cyclohexane (R.T.) | (77 K) | THF (R.T.) | (77 K) | Solid | τ M/ns (χ2) | CIE coordinate | ||
|---|---|---|---|---|---|---|---|---|---|
| λ abs/nm | λ em/nm (Φ) | λ em/nm | λ abs/nm | λ em/nm (Φ) | λ em/nm | λ em/nm (Φ) | |||
| a Measured in benzene due to low solubility in cyclohexane. b s: shoulder peak. | |||||||||
| 1 | 337 | 578 (0.04) | 525 | 333 | 377, 602 (0.08) | 370, 521 | 1BG 496 (0.50) | 5.91 (2.26) | (0.24, 0.51) |
| 1Y 529 (0.39) | 5.84 (1.69) | (0.36, 0.55) | |||||||
| 2 | 340 | 581 (0.01) | 536 | 336 | 399, 605 (0.01) | 377, 536 | 527 (0.34) | 4.77 (2.55) | (0.38, 0.55) |
| 3 | 343 | 575 (<0.01) | 533 | 339 | 397, 598 (0.01) | 379, 528 | 527 (0.31) | 5.83 (3.13) | (0.33, 0.50) |
| 4 | 345 | 599 (0.01) | 542 | 338 | 427, 629 (<0.01) | 397, 543 | 536 (0.34) | 5.17 (1.29) | (0.42, 0.51) |
| 5 | 322 | 602 (0.01) | 549 | 323 | 412, 620 (<0.01) | 399, 548 | 537 (0.31) | 7.15 (1.22) | (0.41, 0.50) |
| 6 | 329 | 584 (<0.01) | 538 | 323 | 402, 610 (0.01) | 394, 536 | 546 (0.31) | 5.23 (1.88) | (0.40, 0.45) |
| 7 | 326 | 639 (<0.01) | 561 | 324 | 438, −(0.01) | 420, 586 | 579 (0.28) | 4.94 (2.35) | (0.54, 0.43) |
| 8 | 320a | 402, 614 (<0.01)a | 540a | 332 | 420, 629 (<0.01) | 405, 572 | 569 (0.25) | 4.16 (1.41) | (0.48, 0.45) |
| 9 | 351sb | 403, 653 (<0.01) | 394, 589 | 358sb | 442, −(0.01) | 432, 596 | 630 (0.05) | 1.62 (0.97) | (0.42, 0.31) |
| 10 | 336 | 366, 384 (0.10) | 377 | 332 | 378 (0.11) | 348, 366, 383 | 382 (0.24) | 2.75 (1.86) | (0.23, 0.22) |
Upon excitation at 330 nm (1.0 × 10−6 mol dm−3), most compounds in THF showed weak dual fluorescence (quantum yield Φ ∼ 0.01) in the visible region, i.e., blue fluorescence at 380–440 nm and orange/red fluorescence at 600–630 nm (Table 1). On the other hand, 2′MeOPIP 10 having no intramolecular hydrogen bonds showed a normal blue fluorescence at around 380 nm.15 Based on a previous report,15,16 the blue fluorescence is ascribed to a normal excitation/emission process from a THF-mediated “open-enol” species, while the longer-wavelength emission with a large Stokes shift is the fluorescence from the closed-enol species in the ESIPT state (Scheme 1). In cyclohexane solution, where the formation of open-enol species is not expected, the ESIPT fluorescence was observed dominantly. The very large Stokes shift and low quantum yield are attributed to the conformational relaxation of the ESIPT species through an increase in the torsion angle (θ) from the co-planar to the twisted form and rearrangement of solvent molecules around the excited species.17,18 The dual fluorescence in THF remained weak at lower temperatures as long as the solution was kept in the fluid state. The fluorescence of the selected compounds (1, 2, 5, and 8) in viscous solvents such as dimethylsulfoxide (viscosity at 25 °C η = 1.99 mPa s19) and 1,2-dimethoxyethane (η = 0.42 mPa s20) was similarly weak.
Fluorescence properties in a dilute frozen matrix
Although the dilute THF solution was cloudy in the frozen state (77 K), reliable emission spectra were recorded. In the frozen THF matrix, dual emission intensified more than 50-fold compared to the fluid solution (Fig. 1). The ESIPT fluorescence was shifted to a significantly shorter wavelength (70–90 nm) regardless of the position of aryl group(s) and the para-substituent (Table 1). Such a marked blue shift and intensified emission in a frozen rigid matrix is explained by the suppression of the conformational relaxation of the ESIPT emitting species16 because this species is restricted to a co-planar conformation. Similar spectral behaviors were also observed in cyclohexane solution. Our recent quantum chemical study on the potential energy surfaces of HPIP also supports this discussion:17 the energy gap between the HOMO and the LUMO of the proton-transferred species, i.e., the ESIPT fluorescence energy, decreases as the torsion angle (θ) between the phenyl and imidazo[1,2-a]pyridine rings increases.The ESIPT luminescence was found to be sensitive to the introduced aryl substituents. The emission band showed a successively increasing red shift as the electron-withdrawing nature of the para-substituent increased (–OCH3 < –H < –COOR; that is, compound 3 < 2 < 4 and 6 < 5 < 7) (Table 1). The ESIPT luminescence of diaryl HPIPs 8 and 9 was red-shifted more than that of the corresponding mono-aryl HPIPs, i.e.2 and 5 for 8, and 4 and 7 for 9, indicating that varying the number of aryl groups is also an effective tool for tuning the luminescence color.
These results demonstrate that conformational confinement in the rigid matrix greatly enhances the ESIPT fluorescence with a noticeable blue shift, and the color of the ESIPT fluorescence could be finely tuned by changing the p-substituent on the phenyl group and the number of aryl substituents.
Absorption and fluorescence properties in the solid state
We then examined the absorption and luminescence properties in the solid state at ambient temperature. Solid-state absorption spectra were obtained via Kubelka–Munk conversion of the diffuse reflectance spectra. As shown in Fig. 1, the spectra of 1, 2, and 6 appeared close to those observed in fluid THF solutions, indicating that there were no obvious molecular interactions affecting the ground state (S0) in the solid state.Upon excitation at 330 nm, 1–9 showed a single emission band attributed to the ESIPT luminescence. Compounds 1–8 exhibited blue-green (496 nm) to orange (579 nm) ESIPT luminescence with high quantum yields (Φ: 0.25–0.50), which were around 40 times higher than those in fluid solutions (Table 1). The observed luminescence of the solid samples of 2–8 was virtually the same as that observed in the dilute frozen matrix, wherein the molecules mainly exist in a solvent-separated monomeric state with fixed conformation. Therefore, the ESIPT luminescence in the solid state is an emission from the monomeric species and is not affected by the surrounding molecules. From the luminescence decay profiles, which fit reasonably with mono- or bi-exponential curves, the weighted average lifetimes (τM) of all the compounds were found to be 4.16–7.15 ns, indicating that the luminescence was emitted from the singlet species. This implies that intermolecular interactions did not have a prominent effect on the solid-state luminescence of 2–8 even though the aryl substituents were introduced into the parent PIP unit. However, the ESIPT luminescence of 9 having long C6-alkyl chains was further red-shifted to 630 nm (the red region) with a decreased quantum yield (Φ = 0.05, τM = 1.62 ns). Even when the solid sample was cooled to 77 K, the emission maximum was not altered much and remained in the red region. This suggests that the observed red shift of the solid sample of 9 is not because of the loose conformational confinement at ambient temperature, but because of intermolecular interactions in the densely packed state.
Upon including blue-emitting 2′-methoxy PIP (10), a series of PIP derivatives showing emission bands from 382 nm to 630 nm are realized by the aryl-substitution of HPIP (Fig. 2a). Fig. 2b shows a CIE chromaticity diagram of the solid-state luminescence of 1–10, including the ESIPT luminescence from two polymorphic crystals of 1 [blue-green (1BG) and yellow (1Y)].16 The luminescence was spread over the blue (10), green-to-yellow (6-aryl HPIP 2–4), yellow-to-orange (8-aryl HPIP 5–7), and orange-to-red (6,8-diaryl HPIP 8–9) regions. These results indicate that the aryl-substitution of HPIP successfully imparted solid-state luminescence with a wide range of colors. Since aryl-substituted HPIPs can be easily prepared by metal-catalyzed cross-coupling reactions, aryl-substitution is a simple and effective method for fine-tuning the nature of luminescence.
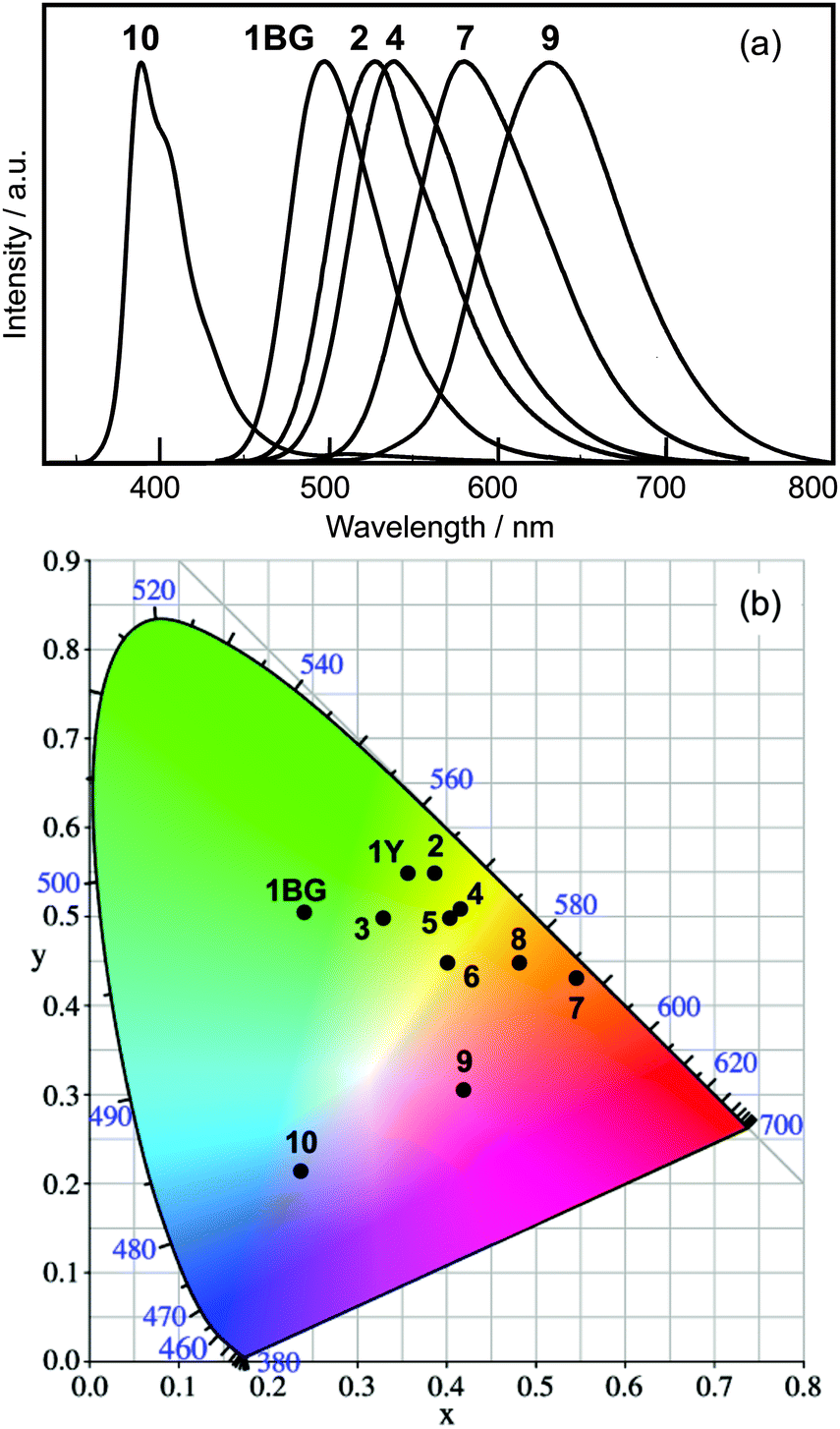 | ||
| Fig. 2 Normalized luminescence spectra (lex = 330 nm) (a) and a CIE chromaticity diagram (b) in the solid state. | ||
Crystallographic analysis
To further clarify the photophysical properties of the compounds in the solid state, the molecular conformation and packing of compounds that yielded suitable crystals, i.e., 3, 4, 7, and 8, were examined by X-ray crystallographic analysis. Fig. 3 shows the ORTEP drawings of the asymmetric unit of crystals of 3 (P21, Z = 4), 4 (P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , Z = 4), 7 (P21/c, Z = 4), and 8 (P21/c, Z = 4).21 The values of the torsion angles between hydroxyphenyl and imidazo[1,2-a]pyridine rings (θ) and between aryl and imidazo[1,2-a]pyridine rings (φ) are also shown. The torsion angle θ is below 10° and the distance between 2′-O and 1-N is less than 2.6 Å in all asymmetric units, indicating the nearly co-planar conformation of the PIP core because of the formation of an intramolecular hydrogen bond (O–H⋯N1). On the other hand, the aryl rings were largely twisted with respect to the PIP core (φ: 25–45°) in all crystals.
, Z = 4), 7 (P21/c, Z = 4), and 8 (P21/c, Z = 4).21 The values of the torsion angles between hydroxyphenyl and imidazo[1,2-a]pyridine rings (θ) and between aryl and imidazo[1,2-a]pyridine rings (φ) are also shown. The torsion angle θ is below 10° and the distance between 2′-O and 1-N is less than 2.6 Å in all asymmetric units, indicating the nearly co-planar conformation of the PIP core because of the formation of an intramolecular hydrogen bond (O–H⋯N1). On the other hand, the aryl rings were largely twisted with respect to the PIP core (φ: 25–45°) in all crystals.
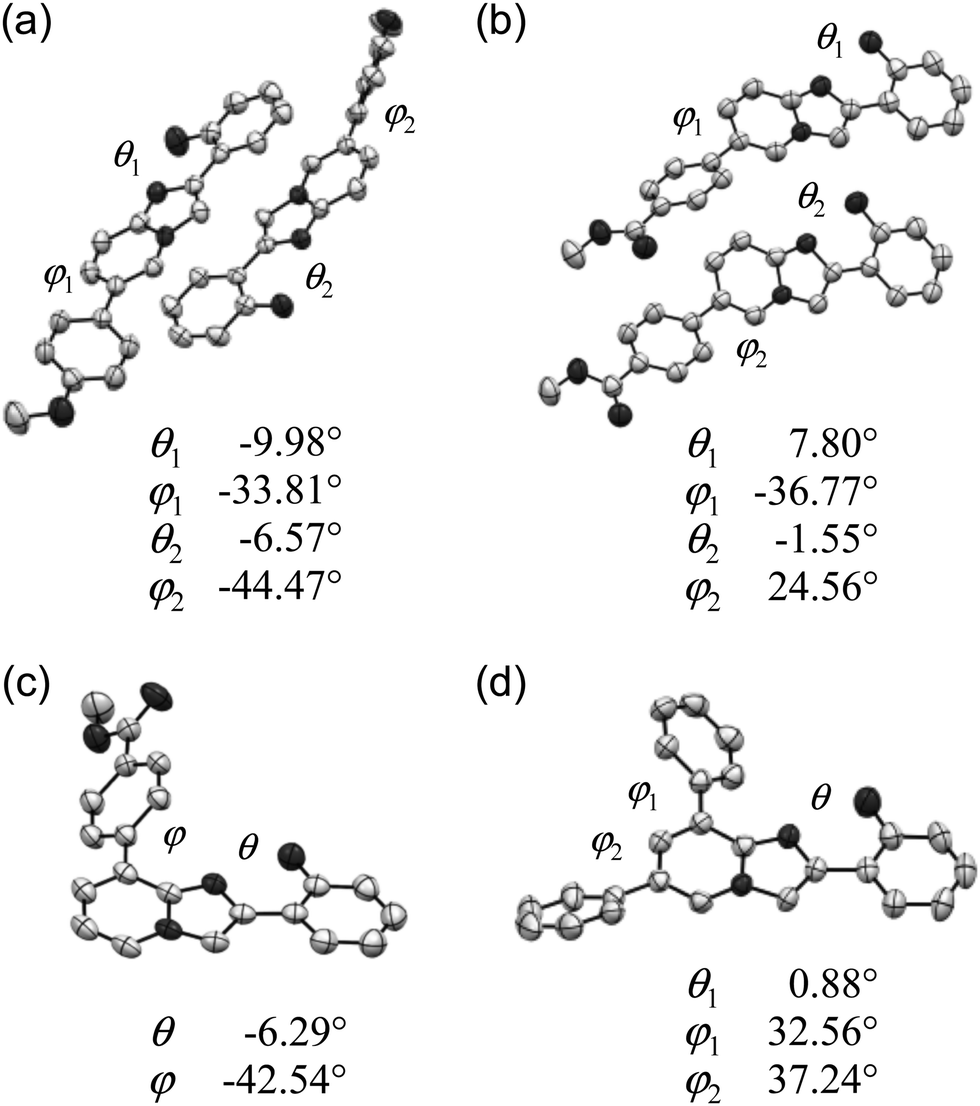 | ||
| Fig. 3 ORTEP drawings of 3 (a), 4 (b), 7 (c), and 8 (d). | ||
The molecular packing of each type of crystal is shown in Fig. S4–S7 in the ESI.† In the crystals of 3, the molecules formed an anti-parallel π-stacked dimer (d = 3.39 Å), and the dimers were further arranged perpendicular to each other (Fig. S4b, ESI†). As shown in Fig. S4c (ESI†), the methoxyphenyl (MeOPh) and HPIP parts were separated. The MeOPh parts were arranged perpendicular to each other, and no π–π interaction with other aromatic units was observed.
In the crystals of 4, there were two modes of anti-parallel π-stacking of the PIP core: partial stacking over imidazo[1,2-a]pyridines (Mode-A, d = 3.79 Å) and full stacking of PIP (Mode-B, d = 3.43 Å) (Fig. S5b, ESI†). In Mode-A, the methoxycarbonylphenyl (MeOCOPh) part was further stacked with the MeOCOPh part of an adjacent molecule (d = 3.41 Å), thereby forming an alternate π-stacking motif along the c-axis between the PIP and MeOCOPh parts (Fig. S5c, ESI†). In Mode-B, on the other hand, another anti-parallel stacking was observed between the MeOCOPh-imidazo[1,2-a]pyridine parts, even though they were not fully stacked because of the twisted conformation (φ2 = 24.5(4)°). Similar to Mode-A, an alternate stacking motif of Mode-B was also observed along the c-axis. The two motifs were arranged in the A–B–A–B manner in the bc-plane (Fig. S5d, ESI†).
Compound 7 also formed anti-parallel π-stacked dimers (d = 3.42 Å), which were further arranged in a slip-stacked column (d = 3.26 Å) (Fig. S6b, ESI†). The MeOCOPh parts were arranged in a columnar structure along the c-axis, but no π–π stacking with PIP was observed (Fig. S6c and d, ESI†).
In the crystals of diphenyl HPIP 8, the PIP parts of two molecules were stacked anti-parallel (d = 3.57 Å) (Fig. S7b, ESI†). The π-stacked PIP dimers did not further show apparent π–π interaction with other molecules. The phenyl groups were arranged at a tilt angle of about 60° to each other (Fig. S7c and d, ESI†).
The formation of intramolecular hydrogen bonds supports the conclusion that emission in the solid state is exclusively ESIPT luminescence. As discussed above, the solid-state emission of 2–8 was monomeric ESIPT luminescence, even though the π-stacked PIP dimers that potentially cause excimer formation or energy migration were found in all the crystals examined. Since simultaneous excitation of the stacked pair of molecules is unlikely to occur under conventional photo-irradiation conditions, the observed monomeric ESIPT luminescence indicates that the excited-state IPT molecule has no remarkable electronic interactions with the paired enol molecule in the ground state. These results show the advantageous point of ESIPT luminescence in solid-state emissive materials. Though further study is required, the observed absence of electronic interactions can be attributed to the significantly different electronic properties of the ESIPT species and the ground-state molecule.17
The aryl groups are either π-stacked (4 and 7) or not (3 and 8). Partial aryl-PIP stacking was also observed in the crystals of 4. Despite having a greater variety of stacking motifs compared to the PIP core, aryl-related interactions had no prominent effect on ESIPT luminescence in the crystalline state. Although the number of examples is limited, the results suggest that the aryl substitution does not cause unfavorable quenching of the solid-state HPIPs through π-stacking.
Molecular orbital calculations
Subsequently, the ESIPT luminescence properties were examined using quantum chemical methods. The geometry of the excited-state species in the IPT state was optimized by the time-dependent density functional theory (TD-DFT) using the CAM-B3LYP/6-31G(d) basis set.22 To estimate the ESIPT luminescence properties in fluid solutions, the geometries of 3 and 4 extracted from the crystal structure were optimized without geometrical restriction. The result shows that the initially co-planar PIP core (θ < 10°) formed a largely twisted geometry (θ ∼ 90°), which is consistent with a previous report.15 The twisted species had a very small energy gap (Table 2) that was consistent with the observed weak emission in a fluid medium.| Observed λem/nm | Calculated energy gap/eV (λem/nm) | |
|---|---|---|
| Fluid solution | ||
| 3 | 598 | 2.1515 (576.27), 1.0344 (1198.60) |
| 4 | 629 | 2.0051 (618.34), 0.9505 (1304.41) |
| Solid state | ||
| 3 | 527 | 2.7467 (451.39) |
| 4 | 536 | 2.6959 (459.89) |
| 7 | 579 | 2.5336 (489.35) |
| 8 | 569 | 2.6508 (467.73) |
We further performed TD-DFT calculations to assess the ESIPT luminescence in the solid state. The initial geometry of the molecules was extracted from the crystal structures of 3, 4, 7, and 8, and the position of heavy atoms except that of 2′-oxygen was locked during geometry optimization. The order of the computed energy gap (3 > 4 > 8 > 7) was consistent with the order of observed ESIPT luminescence (Table 2), although the estimated transition energy was consistently larger (ca. 0.4 eV) than the observed energy.
Fig. 4 shows the calculated electron densities and energy levels of the HOMO and LUMO of 3, 4, 7, and 8. In all cases, the HOMO showed similar localization on the phenyl ring of the PIP core with very similar energy levels. On the other hand, the electronic configuration and energy levels of the LUMO were obviously dependent on the aryl group and substituent position, indicating that the major factor determining the luminescence properties was the LUMO level.
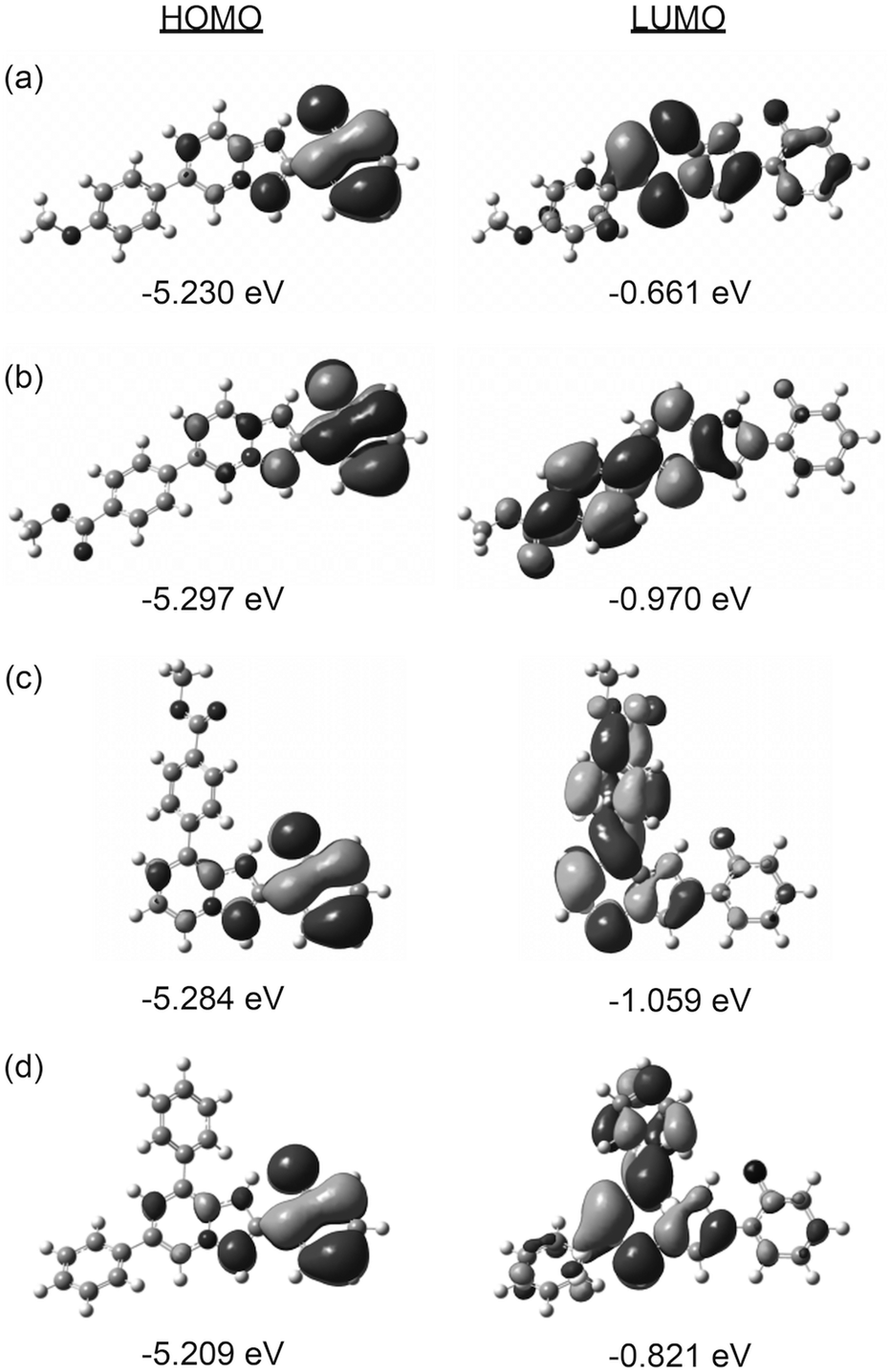 | ||
| Fig. 4 Calculated HOMO and LUMO of the IPT form of 3 (a), 4 (b), 7(c), and 8 (d). | ||
Thus, the ESIPT luminescence of the aryl substituted PIPs with a variety of emission colors in the solid state is reasonably explained by the TD-DFT calculations.
Conclusion
In this report, aryl-substituted HPIPs 2–9 were synthesized and their ESIPT luminescence properties were studied. Although these compounds showed weak dual (normal and ESIPT) fluorescence in solution, they exhibited intensified ESIPT emission with a wide range of emitting colors in the solid state. π-Stacked anti-parallel dimer motifs of the HPIP cores were formed in the crystals of 3, 4, 7 and 8, and π-stacking of the introduced aryl groups was also observed in the crystals of 4 and 7. However, except for 9 having long C6-alkyl chains, the similar emission properties for all compounds in the dilute frozen matrix and the solid state indicated that there is ESIPT emission from the monomeric species even in the presence of a π-stacked packing motif. Thus, the observed emission enhancement is ascribed to conformational confinement in the densely packed solid state.The introduction of methoxy (3, 6) or alkoxycarbonyl (4, 7, 9) groups into the para-position of the aryl groups caused blue or red shifts of the ESIPT fluorescence, respectively. In comparison with those having the same aryl group(s), the ESIPT luminescence wavelength became longer in the order of 6-, 8-, and then 6,8-substituted derivatives. TD-DFT calculations of the IPT species indicated that the observed emission color change is ascribed to the alteration of the LUMO levels of this species because of aryl substitution.
Through these studies, we have shown that the aryl-substituted HPIPs are promising organic solid-state luminescent materials because of their intensified and stacking-insensitive ESIPT luminescence. Upon including the non-substituted HPIP 1 and 2′-methoxy PIP 10, a series of PIP derivatives showing a wide range of luminescence colors from blue (382 nm) to reddish-orange (630 nm) was realized. Facile and convenient cross-coupling reactions of different para-substituted aryl groups into the luminescent core led to the fine-tuning of the luminescence color over a wide range of colors, offering a promising way to tune the ESIPT luminescence properties of HPIPs in the solid state without deterioration caused by intermolecular interaction.
Experimental
Methods
The UV-vis absorption and fluorescence spectra in the organic solutions were recorded using a Shimadzu UV-2500PC spectrophotometer and a JASCO FP-6600 spectrofluorometer, respectively. The fluorescence quantum yield in a fluid THF solution was calculated using 2-aminopyridine (Φ = 0.37; ethanol; excitation 285 nm) as the standard. A time-resolved emission decay was measured via excitation of the sample solutions using a nitrogen laser pulse (337 nm). The emission was dispersed using a Hamamatsu Photonics C-2830 disperser and monitored on a Hamamatsu Photonics M-2548 streak camera.Solid-state absorption spectra were obtained via the Kubelka–Munk conversion of a diffractive reflectance spectrum measured on a JASCO FP-6600 spectrofluorophotometer equipped with a JASCO ILF-533 integral sphere. The samples were prepared from finely ground compounds (1 mg) and powdered sodium chloride (1 g), which were mixed in a glass tube and then placed in a 5 mm quartz cell. The luminescence spectra in the solid state were obtained by spreading a finely powdered solid on a quartz plate that was placed in an ILF-533 integral sphere and measured on a JASCO FP-6600 spectrofluorophotometer. The quantum yield in the solid state was obtained using software installed in the spectrofluorophotometer. The x, y coordinates on a Commission Internationale de l'Eclairage (CIE) 1931 chromaticity diagram were calculated from an emission spectrum measured on a HAMAMATSU PMA-11 multi-channel analyzer equipped with an integral sphere via excitation at 365 nm.
The quantum chemical calculations were performed on a Gaussian 09W, Gaussian Inc. (Revision D.01), package,23 and the results were processed on GaussView 5.0.9.
Materials
2-Bromo-1-(2-methoxyphenyl)ethanone was purchased from Wako Chem. Co. 2-Amino-3-bromopyridine, 2-amino-5-bromopyridine, 2-amino-3,5-dibromopyridine, aryl boronic acids, dichloromethane solution of boron tribromide (1.0 M) and other chemicals were also obtained commercially and used as received. The syntheses of 2-(2′-hydroxyphenyl)imidazo[1,2-a]pyridine (HPIP, 1) and 2-(2′-methoxyphenyl)imidazo[1,2-a]pyridine (2′MeOPIP, 10) are described elsewhere.16 The synthetic procedures of the aryl HPIPs 2–9 and their bromo precursors 11–13 are described in the ESI.†Acknowledgements
This work was supported by Grant-in-Aid for Scientific Research (B) (21350109) and (C) (24550222) from the Japan Society for the Promotion of Science (JSPS).References
- (a) H. Sasabe and J. Kido, Chem. Mater., 2011, 23, 621–630 CrossRef CAS; (b) A. Mishra and P. Bauerle, Angew. Chem., Int. Ed., 2012, 51, 2020–2067 CrossRef CAS PubMed; (c) H. Uoyama, K. Goushi, K. Shizu, H. Numra and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed; (d) N. Yanai, K. Kitayama, Y. Hijikata, H. Sato, R. Matsuda, Y. Kubota, M. Takata, M. Mizuno, T. Uemura and S. Kitagawa, Nat. Mater., 2011, 10, 787–793 CrossRef CAS PubMed; (e) Y. S. Zhao, H. Fu, A. Peng, Y. Ma, Q. Liao and J. Yao, Acc. Chem. Res., 2010, 43, 409–418 CrossRef CAS PubMed; (f) G. Qian and Z. Y. Wang, Chem. – Asian J., 2010, 5, 1006–1029 CrossRef CAS PubMed; (g) K. T. Kamtekar, A. P. Monkman and M. R. Bryce, Adv. Mater., 2010, 22, 572–582 CrossRef CAS PubMed.
- (a) J. B. Birks, Photophysics of Aromatic Molecules, Wiley, New York, 1970 Search PubMed; (b) M. Shimizu and T. Hiyama, Chem. – Asian J., 2010, 5, 1516–1531 CrossRef CAS PubMed.
- (a) J. N. Moorthy, P. Natarajan, P. Venkatakrishnan, D.-F. Huang and T. J. Chow, Org. Lett., 2007, 9, 5215–5218 CrossRef CAS PubMed; (b) T. Qin, G. Zhou, H. Scheiber, R. E. Bauer, M. Baumgarten, C. E. Anson, E. J. W. List and K. Müllen, Angew. Chem., Int. Ed., 2008, 47, 8292–8296 CrossRef CAS PubMed; (c) B. Li, J. Li, Y. Fu and Z. Bo, J. Am. Chem. Soc., 2004, 126, 3430–3431 CrossRef CAS PubMed.
- (a) M. Li, Y. Niu, X. Zhu, Q. Peng, H. Lu, A. Xia and C. Chen, Chem. Commun., 2014, 50, 2993–2995 RSC; (b) M. Shimizu, R. Kaki, Y. Takeda, T. Hiyama, N. Nagai, H. Yamagishi and H. Furutani, Angew. Chem., Int. Ed., 2012, 51, 4095–4099 CrossRef CAS PubMed; (c) M. Shimizu, Y. Takeda, M. Higashi and T. Hiyama, Angew. Chem., Int. Ed., 2009, 48, 3653–3656 CrossRef CAS PubMed.
- (a) J. Mei, Y. Hong, J. W. Y. Lam, A. Qin, Y. Tang and B. Z. Tang, Adv. Mater., 2014, 26, 5429–5479 CrossRef CAS PubMed; (b) Aggregation-Induced Emission: Fundamentals, ed. A. Qin and B. Z. Tang, Wiely, Chichester, UK, 2014 Search PubMed; (c) Y. Hong, J. W. Y. Lama and B.-Z. Tang, Chem. Soc. Rev., 2011, 40, 5361–5388 RSC; (d) J. Luo, Z. Xie, J. W. Y. Lam, L. Cheng, H. Chen, C. Qiu, H.-S. Kwok, X. Zhan, Y. Liu, D. Zhu and B.-Z. Tang, Chem. Commun., 2001, 1740–1741 RSC.
- (a) K. Araki and T. Mutai, in Specialist Periodical Reports, Photochemistry, ed. E. Fasani, The Royal Society of Chemistry, London, 2015, vol. 43, pp. 191–225 Search PubMed; (b) D. Ding, K. Li, B. Liu and B. Z. Tang, Acc. Chem. Res., 2013, 46, 2441–2453 CrossRef CAS PubMed.
- (a) J. Zhao, S. Ji, Y. Chen, H. Guo and P. Yang, Phys. Chem. Chem. Phys., 2012, 14, 8803–8817 RSC; (b) J. E. Kwon and S. Y. Park, Adv. Mater., 2011, 23, 3615–3642 CrossRef CAS PubMed; (c) J. Wu, W. Liu, J. Ge, H. Zhang and P. Wang, Chem. Soc. Rev., 2011, 40, 3483–3495 RSC.
- (a) M. Barbatti, A. J. A. Aquino, H. Lischka, C. Schriever, S. Lochbrunner and E. Riedle, Phys. Chem. Chem. Phys., 2009, 11, 1406–1415 RSC; (b) S. Lochbrunner, T. Schultz, M. Schmitt, J. P. Shaffer, M. Z. Zgierski and A. J. Stolow, Chem. Phys., 2001, 114, 2519–2522 CAS.
- S. M. Ormson and R. G. Brown, Prog. React. Kinet., 1994, 19, 45–91 CAS.
- (a) S. Park, J. E. Kwon, S. H. Kim, J. Seo, K. Chung, S.-Y. Park, D.-J. Jang, B. Milián Medina, J. Gierschner and S. Y. Park, J. Am. Chem. Soc., 2009, 131, 14043–14049 CrossRef CAS PubMed; (b) K. C. Tang, M. J. Chang, T. Y. Lin, H. A. Pan, T. C. Fang, K. Y. Chen, W. Y. Hung, Y. H. Hsu and P. T. Chou, J. Am. Chem. Soc., 2011, 133, 17738–17745 CrossRef CAS PubMed; (c) K. Benelhadj, J. Massue, P. Retailleau, G. Ulrich and R. Ziessel, Org. Lett., 2013, 15, 2918–2921 CrossRef CAS PubMed; (d) H. Shono, T. Ohkawa, H. Tomoda, T. Mutai and K. Araki, ACS Appl. Mater. Interfaces, 2011, 3, 654–657 CrossRef CAS PubMed.
- J. E. Kwon, S. Park and S. Y. Park, J. Am. Chem. Soc., 2013, 135, 11239–11246 CrossRef CAS PubMed.
- For review, see V. S. Padalkar and S. Seki, Chem. Soc. Rev., 2016, 45, 169–202 RSC.
- (a) D. Yao, S. Zhao, J. Guo, Z. Zhang, H. Zhang, Y. Liu and Y. Wang, J. Mater. Chem., 2011, 21, 3568–3570 RSC; (b) S. Park, J. E. Kwon and S. Y. Park, Phys. Chem. Chem. Phys., 2012, 14, 8878–8884 RSC; (c) W.-T. Chuang, C.-C. Hsieh, C.-H. Lai, C.-H. Lai, C.-W. Shih, K.-Y. Chen, W.-Y. Hung, Y.-H. Hsu and P.-T. Chou, J. Org. Chem., 2011, 76, 8189–8202 CrossRef CAS PubMed.
- T. Mutai, H. Sawatani, T. Shida, H. Shono and K. Araki, J. Org. Chem., 2013, 78, 2482–2489 CrossRef CAS PubMed.
- (a) A. Douhal, F. Amat-Guerri and A. U. Acuña, J. Phys. Chem., 1995, 99, 76–80 CrossRef CAS; (b) A. J. Stasyuk, M. Banasiewicz, M. K. Cyrański and D. T. Gryko, J. Org. Chem., 2012, 77, 5552–5558 CrossRef CAS PubMed.
- T. Mutai, H. Tomoda, T. Ohkawa, Y. Yabe and K. Araki, Angew. Chem., Int. Ed., 2008, 47, 9522–9524 CrossRef CAS PubMed.
- Y. Shigemitsu, T. Mutai, H. Houjou and K. Araki, J. Phys. Chem. A, 2012, 116, 12041–12048 CrossRef CAS PubMed.
- (a) A. Douhal, F. Amat-Guerri and A. U. Acuña, Angew. Chem., Int. Ed., 1997, 36, 1514–1516 CrossRef CAS; (b) A. Douhal, Ber. Bunsenges. Phys. Chem., 1998, 102, 448–451 CrossRef CAS.
- CRC Handbook of Chemistry and Physics, ed. D. R. Lide, CRC Press, Boca Raton, FL, 85th edn, 2004 Search PubMed.
- P. K. Muhuri and D. K. Hazra, J. Chem. Eng. Data, 1994, 39, 375–377 CrossRef CAS.
- Details of the crystallographic analysis and CCDC numbers of 3, 4, 7, and 8 are described in the ESI.†.
- T. Yanai, D. Tew and N. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian, Inc., Wallingford CT, 2009.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1447749–1447752. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6tc00376a |
| This journal is © The Royal Society of Chemistry 2016 |