Colour-tunable asymmetric cyclometalated Pt(II) complexes and STM-assisted stability assessment of ancillary ligands for OLEDs†
Received
8th January 2016
, Accepted 3rd March 2016
First published on 4th March 2016
Abstract
We combined an asymmetric cyclometalated C^N^N Pt(II) complex with three different classes of monodentate ancillary ligands, namely a bulky phosphine, a tilted pyridine, and either a tilted or a coplanar isocyanide. The controlled planarization of the coordination environment yields green, orange or red phosphors. Electronic structure calculations using DFT reveal the excimeric intermolecular interactions in the excited triplet state, resulting in tunable photophysical properties. Finally, we assess the suitability of the ancillary ligands for optoelectronic applications by investigating the molecular integrity upon sublimation with the aid of scanning tunneling microscopy (STM). The ancillary ligand controls the surface-induced chiral self-assembly of the molecules on metallic substrates, as well as the availability of loose coordination sites. The concordance between the photoluminescence spectra and electroluminescence profiles for solution-processed and vapour-deposited devices confirm the predictions attained by STM regarding molecular integrity and processability.
Introduction
Triplet emitters have found application as dopants in optoelectronic devices, such as organic light-emitting diodes1 (OLEDs) and light-emitting electrochemical cells (LEECs).2 Besides Ir(III),3 Rh(III),4 Re(I)5 and Cu(I),6 coordination compounds featuring Pt(II)7 and Au(III)8 cations have been described displaying strong phosphorescence, fluorescence or thermally-activated delayed fluorescence. The latter possess a d8 electronic configuration and a mostly square-planar coordination environment, and have found applications in photocatalysis, bioimaging and therapy.9 Both bidentate10 as well as tridentate11 luminophores have been employed, yielding highly (electro)luminescent coordination compounds. We have also recently shown that neutral Pt(II) complexes bearing dianionic tridentate N^N^N ligands can be used as triplet emitters, particularly in electroluminescent devices. Depending on the substitution pattern, they can be found either as monomeric12 or as aggregated phosphorescent species.13 For the monomeric species, the emission occurs from metal-perturbed ligand-centered triplet states (3MP-LC), whereas the aggregates emit from excimeric or metal–metal-to-ligand charge-transfer states (3MMLCT).11a Planar species can be also coupled with metallic substrates by hybridization of the protruding dz2 orbitals with the surface. Such behavior can be accurately unraveled with the aid of scanning tunneling microscopy (STM) and spectroscopy (STS), as we have recently shown.14 In supramolecular chemistry, intermolecular interactions give rise to new properties.15 For instance, M. Kato et al.16 have investigated the capabilities of supramolecular arrays consisting of interacting Pt(II) complexes. De Cola et al. have elegantly shown that biomimetic self-assembly of Pt complexes can be monitored in real time and correlated with kinetic and thermodynamic parameters.17 We have previously shown that tridentate bis-diazole, bis-triazole- or bis-tetrazole-substituted pyridine luminophores (or asymmetric combinations thereof) with pyridine ancillary ligands give rise to fully planar coordination environments, whereas the bulky triphenylphosphine completely suppresses the intermolecular interactions, due to their out-of-plane phenyl units.12,14,18
We have recently also described phosphorescent organo- and hydrogelators,13a,b,e as well as crystalline arrays in which π-accepting ancillary ligands can switch-on Pt⋯Pt interactions, regardless of the presence of bulky adamantyl substituents.19a Moreover, 2D-confinement on insulating or semiconducting substrates can also be used to force the interaction between adjacent metallic centers.19b However, a deeper understanding and a defined control are still required, as stacking most frequently causes lowered solubility, undesired triplet–triplet annihilation, lowered quantum yields and unpredictable red-shifts.15
In order to achieve high photoluminescence quantum yields, good σ-donors as well as strong π-acceptors with large ligand field splitting are required to destabilize the otherwise thermally accessible, dark metal-centred states.11a Yersin and co-workers have recently published an interesting concept for bright Pt(II) complexes bearing doubly cyclometallating carborane-substituted luminophores and isocyanide ancillary ligands,20 whereas combinations of C^N^N luminophores and isocyanides have been used previously by Che et al.21
In a previous work, we have shown that it is possible to find submolecular electronic set-screws with the aid of scanning tunnelling microscopy (STM) and scanning tunnelling spectroscopy (STS), enabling the realization of deep-blue phosphorescent Pt(II) complexes.14c Herein we present a notably straightforward strategy towards fully tunable intermolecular interactions on the basis of controllable planarization of the coordination environment of Pt(II) complexes. We demonstrate that the introduction of a cyclometallating phenyl ring on a strongly σ-donating 2-(1,2,4-triazol-5-yl)-pyridine chelate yields an outstanding dianionic C^N^N ligand for luminescent Pt(II) complexes. Moreover, the introduction of a C–H bond adjacent to the ancillary ligand (Lanc) forces the out-of-plane tilt of the monodentate unit. Depending on the bulkiness of the fourth ligand, variable tilt angles are achievable, which in turn hinder intermolecular interactions to different extents, yielding green, orange and red phosphors. The excimeric nature of the emissive states of monomeric and dimeric species was assessed by density functional theory (DFT). Finally, we elucidated the integrity of the complexes upon sublimation with the aid of STM, in order to consider the suitability of each of the ancillary ligands, and corroborated the concordance between the photoluminescence spectra and electroluminescence profiles for solution-processed and vacuum-deposited devices.
Results and discussion
Synthesis and structural characterization
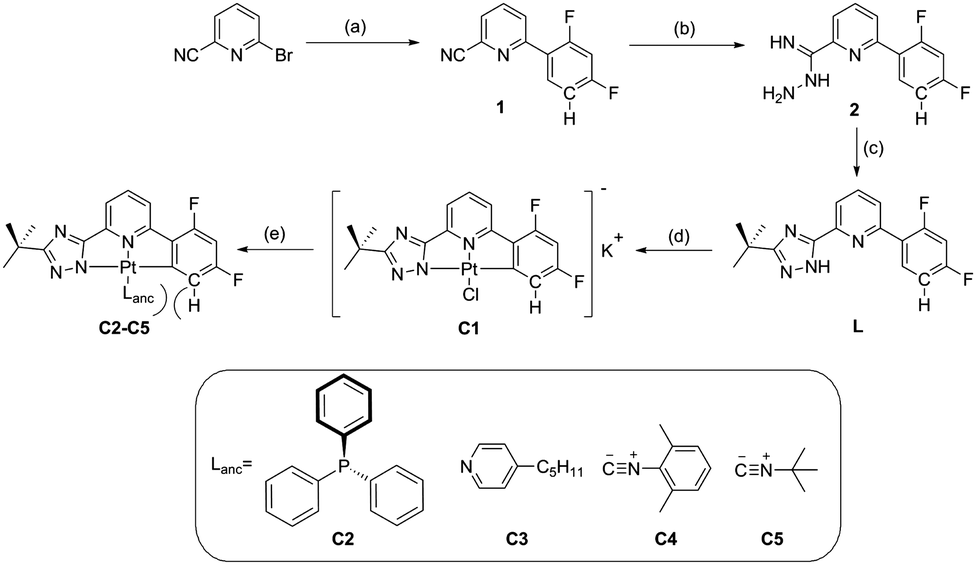
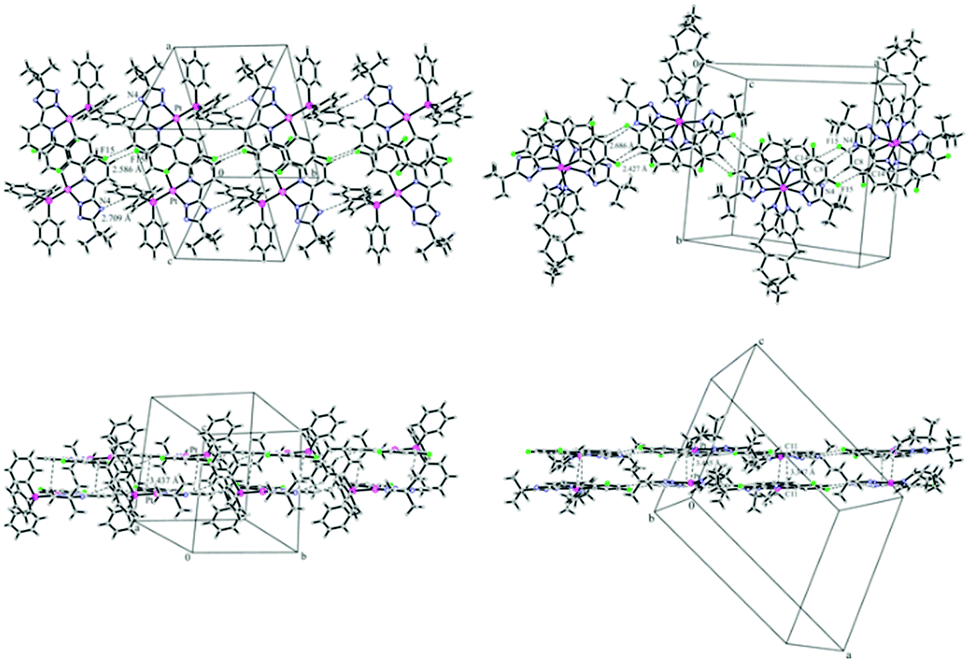
The synthetic route (Scheme 1) starts with a Suzuki coupling, leading to a 2-cyanopyridine substituted with a difluorophenyl moiety. The triazole ring was introduced by reaction with hydrazine monohydrate followed by an acylation with trimethylacetyl chloride and a heat-aided ring closure. Cyclometallation with K2PtCl4 in acetic acid yielded the anionic complex C1, which upon reaction with a neutral monodentate ligand precursor led to complexes C2–C5. A detailed description of the conditions, purification methods as well as 1H, 13C, 19F, 31P and 195Pt NMR characterization can be found in the ESI† (Sections S1.1 and S1.2). C2 and C3 yielded crystals suitable for X-ray diffractometric analysis (Section S1.3, ESI†). Complex C2 presents the formation of dimers through π⋯π-interactions involving only the phenylpyridine moieties of the luminophore (the shortest distance between aromatic rings is 3.437 Å). No interaction between adjacent metallic centers of the dimeric structures was observed (the Pt⋯Pt distance is 7.33 Å). These dimers are further stabilized by C–H⋯F and C–H⋯N interactions in linear chains along the ac-plane (2.586 Å and 2.709 Å, respectively, Fig. 1 (left)). In contrast, the packing diagram of C3 shows the formation of dimers with clear Pt⋯Pt contacts and stronger π⋯π-interactions involving the entire luminophore (3.485 Å and 3.347 Å, respectively). Also for C3, C–H⋯F and C–H⋯N interactions were observed in zig-zag chains along the ac-plane (2.427 Å and 2.686 Å, respectively, Fig. 1 (right)).
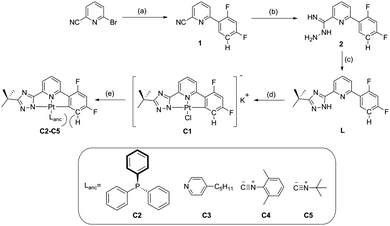 |
| | Scheme 1 Synthetic route towards the C^N^N ligand and the corresponding Pt(II) complex. (a) 2,4-Difluorophenylboronic acid, Pd(PPh3)4, K2CO3, THF, N2, reflux, 24 h. (b) N2H4·H2O, EtOH, rt. (c) (1) t-BuC(O)Cl, K2CO3, DMF, 0 °C to rt, (2) ethylene glycol, 160 °C. (d) K2PtCl4, acetic acid, reflux. (e) Lanc, THF, 12 h. | |
 |
| | Fig. 1 Left: Packing diagram presenting the C–H⋯F and C–H⋯N interactions along the ac-plane (top) and π–π stacking involving only the phenylpyridine moiety (bottom) in complex C2. Right: Packing diagram presenting the C–H⋯F and C–H⋯N interactions along the ac-plane (top) and Pt–Pt interactions and π–π stacking (bottom) in complex C3. | |
Photophysical characterization
All the complexes were characterized in terms of UV-Vis absorption and emission spectroscopy in the solid state (powders, neat films and doped into PMMA), as well as in solution at room temperature and in frozen glassy matrices at 77 K, including absolute photoluminescence quantum yields (ΦL), time-resolved luminescence decays as well as radiative and non-radiative rate constants. The detailed results are shown in the (Section S4, ESI†). A summary of the most relevant photophysical data is presented in Table 1. The complexes show absorption bands around 220–260 nm, which can be assigned to π–π* transitions, as well as bands around 320 nm, which can be ascribed to a transition with mixed MLCT/LC character. Furthermore, the long excited state lifetimes (>10 μs) and vibrational progressions observed in frozen matrices at 77 K indicate that the emission occurs mainly from 3MP-LC excited states. All the complexes (C2–C5) show green phosphorescence in diluted solutions (10−5 M) with high ΦL for deaerated samples (up to 73%, C5). Their emission maxima shift only slightly from 504 nm for C3 to 511 nm for C4, indicating a minor contribution of the ancillary ligand to the ground- and the excited states of the chromophore. These experimental results also correlate with the DFT calculations (ESI,† Section S3), showing that the highest occupied molecular orbital (HOMO) as well as the lowest unoccupied molecular orbital (LUMO) are mostly located on the chelating luminophore, with marginal participation of the metallic center (see ESI,† Section S3). In the solid state, complex C2 shows a blue-shifted, monomeric (3MP-LC) emission with a slightly blue-shifted maximum at 503 nm, due to the lack of stabilizing solvent molecules (Fig. 1). The bulky ancillary P(Ph)3 ligand suppresses aggregation, avoiding bimolecular quenching processes, and yielding a high ΦL (84%). On the other hand, complexes C3–C5 show a characteristic broad, red-shifted 3MMLCT emission band that is attributed to stacked species. The emission wavelength red-shifts from 599 nm (C3) and 622 nm (C4) down to 698 nm (C5) in powders and neat films, depending on the bulkiness of the ancillary ligand (C2vs.C5) and its tilt angle with respect to the coordination plane minimizing the sterical hindrance with the tridentate moiety (C3vs.C4). Interestingly, the highest ΦL for the aggregated species was achieved for the deepest red-emitting compound (C5, 31%), whereas C3 and C4 have rather low ΦL (10% and 14%, respectively). Pt⋯Pt interactions occur mainly in the excited triplet state (3MMLCT excimer), as indicated by DFT calculations (ESI,† Section S3). Consequently, the binding energies and intermetallic distances of singlet and triplet dimers show that the Pt⋯Pt distance significantly shrinks in the excited triplet state with respect to the ground state (Table 1). This shortening also nicely correlates with the observed bathochromic shift, and the calculated emission spectra are in good agreement with the experimental observations. The lowest triplet states of the dimers actually show the electron transfer from an intermetallic antibonding (dz2–dz2) to a bonding interligand (π*–π*) orbital (3MMLCT). As a consequence of the bulky P(Ph)3, however, the C2 dimer is less stable by 0.51 eV than two monomers (i.e., one triplet plus one ground state), whereas all the other dimers are stable with respect to dissociation. For all the complexes, the binding of the dimers in the ground state can be attributed to van der Waals interactions, as the Pt⋯Pt distances lie above 3.5 Å (Fig. 2).
Table 1 Representative photophysical properties, binding energies and intermetallic distances for compounds C2–C5
| Complex |
C2
|
C3
|
C4
|
C5
|
|
In tetrahydrofuran.
In dichloromethane.
Powder, measured at room temperature.
Measured at room temperature in an integrating sphere.
Obtained from the bluest emission maximum. Amplitude-averaged lifetimes are given when multiexponential decays are observed.
In frozen glassy matrices (2-MeTHF).
Dimer binding energies calculated as the difference between the S0 dimer energy and the energy of two S0 monomers, or as the difference between the T1 dimer energy and the sum of the T1 energy and the S0 energy of the monomer.
Calculated for a S0 dimer or for a T1 dimer.
|
|
λ
abs/nm |
217, 324a |
253, 323b |
262, 327b |
256, 313, 338b |
|
λ
em,solid
/nm |
503 |
599 |
622 |
698 |
|
λ
em,deaer
/nm |
508a |
504b |
511b |
507b |
|
Φ
L,solid
(±0.02) |
0.84 |
0.10 |
0.14 |
0.31 |
|
Φ
L,deaer
(±0.02) |
0.70a |
0.08b |
0.71b |
0.73b |
|
τ
deaer
/μs (±0.1 μs) |
15.2a |
2.0b |
13.6b |
16.5b |
|
τ
77K
,
/μs (±0.1 μs) |
15.2 |
12.2 |
10.6 |
13.5 |
| ΔEg (eV) S0/T1 |
−0.107/0.513 |
−0.358/−0.353 |
−0.255/−0.212 |
−0.236/−0.167 |
| Pt⋯Pt distanceh (Å) S0/T1 |
5.952/2.867 |
3.927/3.849 |
3.625/2.822 |
4.492/2.818 |
 |
| | Fig. 2 Left: Normalized emission spectra of complexes C2–C5 in the solid state (red: experimental; black: calculated). Right: DFT-calculated geometries of monomeric species and Pt⋯Pt distances of triplet dimers. | |
STM analysis upon sublimation
The STM images in Fig. 3 show the molecular arrangement of complexes C2–C5 vapour-deposited onto Au(111) under ultrahigh vacuum conditions (p < 10−10 mbar). C2 (a and b) was evaporated at 436 K and self-assembles at low coverages into 2D-twisted chains with random length and direction. The submolecular details could not be resolved most likely due to the bulkiness of the P(Ph)3 and therefore, the orientation of the complex remains unclear. However, we assume these complexes to be intact and the ancillary ligand to appear brightest in the images as the measured structures match in size with C2. Schneider and others have shown that planar asymmetric aromatic systems without a stereogenic centre become prochiral upon adsorption to a surface.22 This adsorption-induced chirality results in two possible stereoisomers for complex C2, which influence the orientation of the growing chain either clock- or anticlockwise (Fig. S5b, ESI†). As seen in the close-up image (b), also a closed ring can be formed, consisting of only one type of stereoisomer. The STM images of C3 (c and d) show densely packed islands as well as irregularly arranged stripes on the substrate. A close-up image (d) reveals that the monolayer islands consist of paired elongated structures each containing a bright protrusion at one end. Based on the comparison of the features with molecular models and our finding with a similar Pt complex,14a we conclude that the images show only the ancillary ligand (4-amylpyridine), which indicates a decomposition of C3. In this case, the self-assembly is probably favoured by van der Waals interactions. One explanation for the decomposition is the lower dissociation energy of the Pt–N bond due to sterical hindrance (vide supra), but also the enhanced planarity maybe relevant, as it favours aggregation and hinders sublimation. However, the evaporation temperature is only slightly increased (443 K) as compared to C2. C4 does not assemble in long-range regular structures at low coverages but seems to prefer pairing of a few complexes along the fcc regions of the Au(111) surface (e). Although the sublimation temperature is again slightly higher (453 K) due to further planarization, no decomposition seems to occur as we only found intact complexes (f). The increased stability is based on the strong isonitrile ligand (both a σ-donor and a π-acceptor). Complex C5 (g and h) sublimes with the highest temperature (513 K) and assembles analogously to C2 in rings or twisted chains with prochiral behavior. From the close-up images we deduce that the structures consist only of the tridentate ligand and the Pt-core. As we could not find any indications of the ancillary ligand adsorbed onto the surface, the decomposition in the evaporator during the sublimation process seems the most probable scenario. These results suggest that a free coordination site is available at the Pt centre, which probably loosely interacts with the triazole moiety of a neighbouring complex (Fig. 3).
 |
| | Fig. 3 Overview and close-up STM images of self-assembled monolayers of C2 (a and b), C3 (c and d), C4 (e and f) and C5 (g and h). While C2 and C4 remain stable upon sublimation, C3 and C5 have not been found intact on the surface. Larger images are found in the ESI† (Section S2). | |
OLED fabrication
Our findings indicate that only C2 and C4 would make adequate dopants for vapour-deposited devices. Since C2 shows the highest color purity and ΦL, it was chosen for a comparative test regarding its ability to perform in electroluminescent devices. In order to find a suitable host material for spin coated devices, cyclic voltammetry, UPS, UV-Vis absorption and photoluminescence emission spectra of neat films from the host material and complex C2 were measured as well as the host materials doped with 5, 10 and 15 wt% of complex C2. The final device architecture and the optimization procedure are found in the ESI† (Section S5). The integrity of the complex itself was confirmed by comparing vapour-deposited and solution-processed OLEDs (Fig. 4), which clearly show an invariable electroluminescence that nicely overlaps with the photoluminescence in the solid state and in solution (see Fig. 1).
 |
| | Fig. 4 Normalized electroluminescence spectra of complex C2 in optimized solution processed (solid) and sublimation processed (dashed) OLEDs. | |
Conclusions
In summary, we demonstrated that the controllable intermolecular interaction gives rise to tunable Pt(II) complexes displaying green, orange or red phosphorescence. DFT calculations of monomeric and dimeric species showed the excimeric character of the excited state of red-emitting aggregates, and STM was used to verify the molecular integrity upon thermal sublimation. Combination of steady-state spectroscopic and STM measurements allowed us to assess the best candidate with the highest stability and color purity for the realization of both vapour-deposited and solution-processed electroluminescent devices. DFT calculations helped us to understand the nature of the intermolecular interactions within aggregated species, which is stronger in the excited state than in the ground state, and showed that bulky substituents suppress aggregation. These results regarding green and red emitters complement our previous STM/STS-guided development of deep-blue triplet emitters, as required for RGB-based electroluminescent technologies. They also show that the synergetic interaction between synthesis, spectroscopy, theory and microscopy constitute a virtuous approach towards the design and optimization of improved optoelectronic materials. The implementation of phosphorescent stacks in electroluminescent devices by direct coupling with metallic substrates lies at hand, as we have recently observed the hybridization and charge injection at metallic substrates for Pt(II) complexes. The ligand-dependent prochiral assembly with a loose coordination site could be interesting for surface catalysis. A clear understanding of molecular orientation in electroluminescent films might also play a crucial role in the design and realization of new optoelectronic devices, as the outcoupling efficiency is influenced by the relative direction of the transition dipole moments, a phenomenon that has been recently shown by Brütting and Thompson.23 Further studies regarding different substitution patterns on the luminophoric chelate are underway, as well as the synthesis of multinuclear complexes with enhanced intermolecular interactions. One goal to be achieved is an enhanced radiative rate constant and a reduced excited state lifetime, which would further enhance the efficiency of the devices by avoiding triplet–triplet annihilation especially at high current densities and luminances. Based on the tunable red-shifted wavelengths, applications in bioimaging can be envisaged as well.
Acknowledgements
J. S. wrote the manuscript, synthesized and characterized the ligand precursors and complexes, fabricated and characterized the OLED devices with the help of D. L., N. L. and D. Z.; L. S. carried out the photophysical characterization of the Pt-complexes; P. R. E. and D. W. rendered the STM measurements; C. D. performed the X-ray diffractometric analysis; C. S. and N. L. D. carried out the DFT calculations; C. A. S. conceived the experiments, discussed the results and wrote the manuscript. Financial support from the DFG (SFB-TRR 61, projects B13 and C07) is gratefully acknowledged.
References
-
(a) M. A. Baldo, D. O’Brien, Y. You, A. Shoustikov, S. Sibley, M. E Thompson and S. R. Forrest, Nature, 1998, 395, 151 CrossRef CAS;
(b)
Organic Light-Emitting Devices, ed. K. Müllen and U. Scherf, Wiley-VCH, Weinheim, 2006 Search PubMed;
(c)
Highly Efficient OLEDs with Phosphorescent Materials, ed. H. Yersin, Wiley-VCH, Weinheim, 2008 Search PubMed;
(d) W.-Y. Wong and C.-L. Ho, J. Mater. Chem., 2009, 19, 4457 RSC;
(e) W.-Y. Wong and C.-L. Ho, Coord. Chem. Rev., 2009, 253, 1709 CrossRef CAS;
(f) G. Zhou, W.-Y. Wong and S. Suo, J. Photochem. Photobiol., C, 2010, 11, 133 CrossRef CAS;
(g) G. Zhou, W.-Y. Wong and X. Yang, Chem. – Asian J., 2011, 6, 1706 CrossRef CAS PubMed;
(h) L. Ying, C.-L. Ho, H. Wu, Y. Cao and W.-Y. Wong, Adv. Mater., 2014, 26, 2459 CrossRef CAS PubMed;
(i) X. Yang, G. Zhou and W.-Y. Wong, J. Mater. Chem. C, 2014, 2, 1760 RSC;
(j) C.-L. Ho, H. Li and W.-Y. Wong, J. Organomet. Chem., 2014, 751, 261 CrossRef CAS;
(k) C.-L. Ho and W.-Y. Wong, New J. Chem., 2013, 37, 1665 RSC.
-
(a) Q. Pei, G. Yu, C. Zhang, Y. Yang and A. J. Heeger, Science, 1995, 269, 1086 CrossRef CAS PubMed;
(b) J.-K. Lee, D. S. Yoo, E. S. Handy and M. F. Rubner, Appl. Phys. Lett., 1996, 69, 1686 CrossRef CAS;
(c) J. C. de Mello, N. Tessler, S. C. Graham, X. Li, A. B. Holmes and R. H. Friend, Synth. Met., 1997, 85, 1277 CrossRef CAS;
(d) C. H. Lyons, E. D. Abbas, J. K. Lee and M. F. Rubner, J. Am. Chem. Soc., 1998, 20, 12100 CrossRef;
(e) S. Welter, K. Brunner, J. W. Hofstraat and L. De Cola, Nature, 2003, 421, 54 CrossRef CAS PubMed;
(f) J. D. Slinker, J. A. De Franco, M. J. Jaquith, W. R. Silveira, Y.-W. Zhong, J. M. Moran-Mirabal, H. G. Craighead, H. D. Abruna, J. A. Marohn and G. G. Malliaras, Nat. Mater., 2007, 6, 894 CrossRef CAS PubMed;
(g) M. Mydlak, C. Bizzarri, D. Hartmann, W. Sarfert, G. Schmid and L. De Cola, Adv. Funct. Mater., 2010, 20, 1812 CrossRef CAS.
-
(a) C. Adachi, M. A. Baldo, S. R. Forrest and M. E. Thompson, Appl. Phys. Lett., 2000, 77, 904 CrossRef CAS;
(b) L. Flamigni, A. Barbieri, C. Sabatini, B. Ventura and F. Barigelletti, Top. Curr. Chem., 2007, 281, 143 CrossRef CAS;
(c) C. Ulbricht, B. Beyer, C. Friebe, A. Winter and U. S. Schubert, Adv. Mater., 2009, 21, 4418 CrossRef CAS.
- A. Steffen, M. G. Tay, A. S. Batsanov, J. A. K. Howard, A. Beeby, K. Q. Vuong, X.-Z. Sun, M. W. George and T. B. Marder, Angew. Chem., Int. Ed., 2010, 49, 2349 CrossRef CAS PubMed.
- M. Mauro, E. Q. Procopio, Y. Sun, C.-H. Chien, D. Donghi, M. Panigati, P. Mercandelli, P. Mussini, G. D’Alfonso and L. De Cola, Adv. Funct. Mater., 2009, 19, 2607 CrossRef CAS.
-
(a) V. A. Krylova, P. I. Djurovich, M. T. Whited and M. E. Thompson, Chem. Commun., 2010, 46, 6696 RSC;
(b) V. A. Krylova, P. I. Djurovich, J. W. Aronson, R. Haiges, M. T. Whited and M. E. Thompson, Organometallics, 2012, 31, 7983 CrossRef CAS;
(c) H. Yersin, R. Czerwieniec and A. Hupfer, Proc. SPIE, 2012, 8435, 843508 CrossRef;
(d) D. M. Zink, M. Bächle, T. Bauman, M. Nieger, M. Kühn, C. Wang, W. Klopper, U. Monkowius, T. Hofbeck, H. Yersin and S. Bräse, Inorg. Chem., 2013, 52, 2292 CrossRef CAS PubMed;
(e) J. Nitsch, C. Kleeberg, R. Fröhlich and A. Steffen, Dalton Trans., 2015, 44, 6944 RSC.
-
(a) V. Adamovich, J. Brooks, A. Tamayo, A. M. Alexander, P. I. Djurovich, B. W. D’Andrade, C. Adachi, S. R. Forrest and M. E. Thompson, New J. Chem., 2002, 26, 1171 RSC;
(b) J. A. G. Williams, A. Beeby, E. S. Davies, J. A. Weinstein and C. Wilson, Inorg. Chem., 2003, 42, 8609 CrossRef CAS PubMed;
(c) W. Lu, B.-X. Mi, M. C. W. Chan, Z. Hui, C.-M. Che, N. Zhu and S.-T. Lee, J. Am. Chem. Soc., 2004, 126, 4958 CrossRef CAS PubMed;
(d) S.-W. Lai and C.-M. Che, Top. Curr. Chem., 2004, 24, 127 Search PubMed;
(e) H.-F. Xiang, S.-C. Chan, K. K.-Y. Wu, C.-M. Che and P. T. Lai, Chem. Commun., 2005, 1408 RSC;
(f) S. C. F. Kui, I. H. T. Sham, C. C. C. Cheung, C.-W. Ma, B. Yan, N. Zhu, C.-M. Che and W.-F. Fu, Chem. – Eur. J., 2007, 13, 417 CrossRef CAS PubMed;
(g) A. F. Rausch, H. H. H. Homeier and H. Yersin, Top. Organomet. Chem., 2010, 193 CrossRef CAS;
(h) C.-M. Che, C.-C. Kwok, S.-W. Lai, A. F. Rausch, W. J. Finkenzeller, N. Zhu and H. Yersin, Chem. – Eur. J., 2010, 16, 233 CrossRef CAS PubMed;
(i) K. M.-C. Wong and V. W.-W. Yam, Acc. Chem. Res., 2011, 44, 424 CrossRef CAS PubMed;
(j) A. Y.-Y. Tam, D. P.-K. Tsang, M.-Y. Chan, N. Zhu and V. W.-W. Yam, Chem. Commun., 2011, 47, 3383 RSC;
(k) M. C.-L. Yeung and V. W.-W. Yam, Chem. – Eur. J., 2011, 17, 11987 CrossRef CAS PubMed;
(l) S. C. F. Kui, P. K. Chow, G. Cheng, C.-C. Kwok, C. L. Kwong, K.-H. Low and C.-M. Che, Chem. Commun., 2013, 49, 1497 RSC;
(m) A. Tronnier and T. Strassner, Dalton Trans., 2013, 42, 9847 RSC;
(n) A. Jackel, M. Linseis, C. Häge and R. F. Winter, Inorganics, 2015, 3, 55 CrossRef CAS.
-
(a) V. K.-M. Au, K. M.-C. Wong, N. Zhu and V. W.-W. Yam, Chem. – Eur. J., 2011, 17, 130 CrossRef CAS PubMed;
(b) V. K.-M. Au, W. H. Lam, W.-T. Wong and V. W.-W. Yam, Inorg. Chem., 2012, 51, 7537 CrossRef CAS PubMed;
(c) W.-P. To, G. S.-M. Tong, W. Lu, C. Ma, J. Liu, A. L.-F. Chow and C.-M. Che, Angew. Chem., Int. Ed., 2012, 51, 2654 CrossRef CAS PubMed;
(d) W. Lu, K. T. Chan, S.-X. Wu, Y. Chen and C.-M. Che, Chem. Sci., 2012, 3, 752 RSC;
(e) M.-C. Tang, D. P.-K. Tsang, M. M.-Y. Chan, K. M.-C. Wong and V. W.-W. Yam, Angew. Chem., Int. Ed., 2013, 52, 446 CrossRef CAS PubMed;
(f) W.-P. To, K. T. Chan, G. S. M. Tong, C. Ma, W.-M. Kwok, X. Guan, K.-H. Low and C.-M. Che, Angew. Chem., Int. Ed., 2013, 52, 6648 CrossRef CAS PubMed.
-
(a) T. Zou, J. Liu, C. T. Lum, C. Ma, R. C.-T. Chan, C.-N. Lok, W.-M. Kwok and C.-M. Che, Angew. Chem., Int. Ed., 2014, 53, 10119 CrossRef CAS PubMed;
(b) P. Wu, E. L.-M. Wong, D.-L. Ma, G. S.-M. Tong, K.-M. Ng and C.-M. Che, Chem. – Eur. J., 2009, 15, 3652 CrossRef CAS PubMed;
(c) C. Y.-S. Chung, S. P.-Y. Li, M.-W. Louie, K. K. W. Lo and V. W.-W. Yam, Chem. Sci., 2013, 4, 2453 RSC;
(d) D.-L. Ma, T. Y.-T. Shum, F. Zhang, C.-M. Che and M. Yang, Chem. Commun., 2005, 4675 RSC;
(e) M. T. Proetto, A. M. Rush, M.-P. Chien, P. A. Baeza, J. P. Patterson, M. P. Thompson, N. H. Olson, C. E. Moore, A. L. Rheingold, C. Andolina, J. Millstone, S. B. Howell, N. D. Browning, J. E. Evans and N. C. Gianneschi, J. Am. Chem. Soc., 2014, 136, 1162 CrossRef CAS PubMed;
(f) W. A. Tarran, G. R. Freeman, L. Murphy, A. M. Benham, R. Kataky and J. A. G. Williams, Inorg. Chem., 2014, 53, 5738 CrossRef CAS PubMed;
(g) E. Baggaley, J. A. Weinstein and J. A. G. Williams, Coord. Chem. Rev., 2012, 256, 1762 CrossRef CAS.
-
(a) M. Hissler, W. B. Connick, D. K. Geiger, J. E. McGarrah, D. Lipa, R. J. Lachicotte and R. Eisenberg, Inorg. Chem., 2000, 39, 447 CrossRef CAS PubMed;
(b) C. E. Whittle, J. A. Weinstein, M. W. George and K. S. Schanze, Inorg. Chem., 2001, 40, 4053 CrossRef CAS PubMed;
(c) F. Hua, S. Kinayyigit, J. R. Cable and F. N. Castellano, Inorg. Chem., 2005, 44, 471 CrossRef CAS PubMed;
(d) F. Castellano, I. E. Pomestchenko, E. Shikhova, F. Hua, M. L. Muro and N. Rajapakse, Coord. Chem. Rev., 2006, 250, 1819 CrossRef CAS;
(e) F. Hua, S. Kinayyigit, J. R. Cable and F. N. Castellano, Inorg. Chem., 2006, 45, 4304 CrossRef CAS PubMed;
(f) F. Hua, S. Kinayyigit, A. A. Rachford, E. R. Shikhova, S. Goeb, J. R. Cable, C. J. Adams, K. Kirschbaum, A. A. Pinkerton and F. N. Castellano, Inorg. Chem., 2007, 46, 8771 CrossRef CAS PubMed;
(g) C. Bronner, S. Baudron, M. W. Hosseini, C. A. Strassert, A. Guenet and L. De Cola, Dalton Trans., 2010, 39, 180 RSC;
(h) I. Stengel, C. A. Strassert, E. A. Plummer, C.-H. Chien, L. De Cola and P. Bäuerle, Eur. J. Inorg. Chem., 2012, 1795 CrossRef CAS;
(i) I. Stengel, C. A. Strassert, L. De Cola and P. Bäuerle, Organometallics, 2014, 33, 1345 CrossRef CAS;
(j) Y. Unger, D. Meyer, O. Molt, C. Schildknecht, I. Münster, G. Wagenblast and T. Strassner, Angew. Chem., 2010, 49, 10214 CrossRef CAS PubMed;
(k) M. Mydlak, C.-H. Yang, F. Polo, A. Galstyan, C. G. Daniliuc, M. Felicetti, J. Leonhardt, C. A. Strassert and L. De Cola, Chem. – Eur. J., 2015, 21, 5161 CrossRef CAS PubMed;
(l) A. R. Naziruddin, A. Galstyan, A. Iordache, C. G. Daniliuc, C. A. Strassert and L. De Cola, Dalton Trans., 2015, 44, 8467 RSC.
-
(a) J. A. G. Williams, Top. Curr. Chem., 2007, 281, 205 CrossRef CAS;
(b) L. Murphy and J. A. G. Williams, Top. Organomet. Chem., 2010, 28, 75 CrossRef CAS;
(c) C. Bronner, M. Veiga, A. Guenet, L. De Cola, M. W. Hosseini, C. A. Strassert and S. A. Baudron, Chem. – Eur. J., 2012, 18, 4041 CrossRef CAS PubMed.
-
(a) M. Mydlak, M. Mauro, F. Polo, M. Felicetti, J. Leonhardt, G. Diener, L. De Cola and C. A. Strassert, Chem. Mater., 2011, 23, 3659 CrossRef CAS;
(b) C. Cebrian, M. Mauro, D. Kourkoulos, P. Mercandelli, D. Hertel, K. Meerholz, C. A. Strassert and L. De Cola, Adv. Mater., 2013, 25, 437 CrossRef CAS PubMed.
-
(a) C. A. Strassert, C.-H. Chien, M. D. Galvez-Lopez, D. Kourkoulos, D. Hertel, K. Meerholz and L. De Cola, Angew. Chem., Int. Ed., 2011, 50, 946 CrossRef CAS PubMed;
(b) N. K. Allampally, C. A. Strassert and L. De Cola, Dalton Trans., 2012, 41, 13132 RSC;
(c) M. Mauro, A. Aliprandi, C. Cebrian, D. Wang, C. Kübel and L. De Cola, Chem. Commun., 2014, 50, 7269 RSC;
(d) M. E. Robinson, D. J. Lunn, A. Nazemi, G. R. Whittell, L. De Cola and I. Manners, Chem. Commun., 2015, 51, 15921 RSC;
(e) N. K. Allampally, M. Bredol, C. A. Strassert and L. De Cola, Chem. – Eur. J., 2014, 20, 16863 CrossRef CAS PubMed.
-
(a) P. R. Ewen, J. Sanning, N. L. Doltsinis, M. Mauro, C. A. Strassert and D. Wegner, Phys. Rev. Lett., 2013, 111, 267401 CrossRef PubMed;
(b) P. R. Ewen, J. Sanning, T. Koch, N. L. Doltsinis, C. A. Strassert and D. Wegner, Beilstein J. Nanotechnol., 2014, 5, 2248 CrossRef PubMed;
(c) J. Sanning, P. R. Ewen, L. Stegemann, J. Schmidt, C. G. Daniliuc, T. Koch, N. L. Doltsinis, D. Wegner and C. A. Strassert, Angew. Chem., Int. Ed., 2015, 54, 786 CrossRef CAS PubMed.
-
C. A. Strassert, M. Mauro and L. De Cola, Adv. Inorg. Chem., ed. R. van Eldik and G. Stochel, Elsevier, 2011, vol. 63, p. 47 Search PubMed.
-
(a) K. Kobayashi, H. Sato, S. Kishi, M. Kato, S. Ishizaka, N. Kitamura and A. Yamagishi, J. Phys. Chem. B, 2004, 108, 18665 CrossRef CAS;
(b) M. Kato, C. Kosuge, K. Morii, J. S. Ahn, H. Kitagawa, T. Mitani, M. Matsushita, T. Kato, S. Yano and M. Kimura, Inorg. Chem., 1999, 38, 1638 CrossRef CAS;
(c) A. Kobayashi and M. Kato, Eur. J. Inorg. Chem., 2014, 4469 CrossRef CAS;
(d) M. Kato, Bull. Chem. Soc. Jpn., 2007, 80, 287 CrossRef CAS.
- A. Aliprandi, M. Mauro and L. De Cola, Nat. Chem., 2016, 8, 10–15 CrossRef CAS PubMed.
- A. Galstyan, A. R. Naziruddin, C. Cebrian, A. Iordache, C. G. Daniliuc, L. De Cola and C. A. Strassert, Eur. J. Inorg. Chem., 2015, 5822–5831 CrossRef CAS.
-
(a) N. K. Allampally, C. G. Daniliuc, C. A. Strassert and L. De Cola, Inorg. Chem., 2015, 54, 1588 CrossRef CAS PubMed;
(b) D. K. Bhowmick, L. Stegemann, M. Bartsch, N. K. Allampally, C. A. Strassert and H. Zacharias, J. Phys. Chem. C, 2015, 119, 5551 CrossRef CAS.
- A. M. Prokhorov, T. Hofbeck, R. Czerwienic, A. F. Suleymanova, D. N. Kozhevnikov and H. Yersin, J. Am. Chem. Soc., 2014, 136, 9637 CrossRef CAS PubMed.
- Y. Chen, W. Lu and C.-M. Che, Organometallics, 2013, 32, 350 CrossRef CAS.
-
(a) M. Böhringer, K. Morgenstern, W.-D. Schneider, R. Berndt, F. Mauri, A. De Vita and R. Car, Phys. Rev. Lett., 1999, 83, 324 CrossRef;
(b) B.-I. Kim, C. C. X. Deng and S. S. Perry, Surf. Sci., 2003, 538, 45 CrossRef CAS.
-
(a) M. J. Jurow, C. Mayr, T. D. Schmidt, T. Lampe, P. I. Djurovich, W. Brütting and M. E. Thompson, Nat. Mater., 2016, 15, 85–91 CrossRef CAS PubMed;
(b) C. Mayr and W. Brütting, Chem. Mater., 2015, 27, 2759 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, NMR and x-Ray data, STM and DFT procedures and data, photophysics and OLED fabrication. CCDC 1416852 (C2), 1416853 (C3) and 1416854. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6tc00093b |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence