
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A model for carbon incorporation from trimethyl gallium in chemical vapor deposition of gallium nitride†
Örjan
Danielsson
 *,
Xun
Li
,
Lars
Ojamäe
,
Erik
Janzén
,
Henrik
Pedersen
and
Urban
Forsberg
*,
Xun
Li
,
Lars
Ojamäe
,
Erik
Janzén
,
Henrik
Pedersen
and
Urban
Forsberg
Department of Physics, Chemistry and Biology, Linköping University, SE-581 83 Linköping, Sweden. E-mail: orjan.danielsson@liu.se
First published on 22nd December 2015
Abstract
Semi-insulating buffer layers are utilized to prevent leakage currents in Gallium Nitride (GaN) high power semiconductor devices. To make the GaN material semi-insulating, it can be doped with carbon. Carbon is inherently present in the process for producing GaN thin films by chemical vapor deposition (CVD), through the use of trimethyl gallium (TMGa) as a precursor. TMGa decomposes in the gas phase, releasing its methyl groups, which act as a carbon source for doping. It is previously known that carbon doping levels can be controlled by tuning CVD process parameters, such as temperature, pressure and precursor flow rates. However, the mechanism for carbon incorporation from TMGa is not yet understood. In this paper, a reactor independent model for predicting carbon incorporation from TMGa in GaN layers grown by CVD is proposed. The model is based on ab initio quantum chemical calculations of molecular adsorption and reaction energies. Computational Fluid Dynamics, including a chemical kinetic model for the decomposition of the precursors and reactions in the gas phase, is used to calculate gas mixture composition under realistic process conditions. These results are used together with the proposed model to obtain carbon doping concentrations as well as growth rates, varying inlet NH3/TMGa ratios (157–625) and temperature (800–1100 °C). The model predictions are then correlated with measurements with good agreement. It is concluded that the contribution of gallium to the GaN layer shifts from GaCH3 at low temperatures to atomic Ga at higher temperatures. In the same way there is a shift in carbon doping contribution, from CH3 at low temperatures to C2Hx at higher temperatures.
1. Introduction
Gallium nitride (GaN) is a stable, direct bandgap semiconductor that has been used in bright, blue LED devices for several decades, which was highlighted with the Nobel Prize in Physics in 2014. GaN is also a principal material for the fabrication of high-power and high-frequency devices. One of its unique and important characteristics is the fact that GaN can operate at elevated temperatures, making it an ideal material for power amplifiers. GaN has the possibility to form alloys with the other group 13 nitrides (also known as III-nitrides), particularly aluminum nitride (AlN) and indium nitride (InN) which also have very favorable electronic properties positioning the group 13 nitrides as key materials for future electronic devices for applications ranging from solid state lighting to high frequency telecommunication. Regardless of the application, the performance of GaN based devices is highly affected by impurities incorporated into the material during processing. Sometimes impurities are wanted, for example as dopants to control the material’s electrical conductivity, and sometimes impurities can have severe negative effects on device performance.A crucial aspect in the realization of GaN based devices is the possibility to synthesize thin layers of these materials uniformly over large areas. The most common approach to achieve this with controlled thicknesses is to use chemical vapor deposition (CVD), which is a process where the layers are formed through surface chemical reactions between molecules containing the atoms needed for the target film, in this case nitrogen and aluminum, gallium or indium. The standard chemical route for CVD of the group 13 nitrides is to use trimethyl-metal complexes (M(CH3)3, where M = Al, Ga or In) in combination with ammonia (NH3). Even though CVD of GaN can be regarded as a mature synthesis route for GaN films, the mechanisms for incorporation of impurities into the material is still very much unknown. Understanding and controlling impurity incorporation is of highest importance in order to further advance the development of semiconductor technology based on the group 13 nitrides.
One of the most important and common impurities in GaN is carbon, which can be utilized to obtain a semi-insulating material1,2 but also acts as a p-type dopant.3 Carbon is inherently present in CVD of GaN from the use of trimethyl gallium (TMGa) as a precursor.4 It can be noted that the main alternative to TMGa is triethyl gallium (TEGa) which will also form hydrocarbon species in the gas phase.5 The carbon free precursors GaCl and GaCl3 are generally avoided in CVD of GaN as their use may lead to the formation of solid particles of NH4Cl, which will destroy the GaN layers. Also, high amounts of solid NH4Cl downstream from the reaction cell will badly affect the vacuum system and pumps.
At the temperatures used for GaN CVD, 600–1100 °C, TMGa decomposes in the gas phase releasing its methyl groups; however the mechanism for the incorporation of carbon from TMGa is not yet understood. It is tempting to think that a released CH3 will be very reactive towards the GaN surface, but it must then be remembered that GaN CVD is done in a mixture of hydrogen and nitrogen in which the TMGa is diluted on the order of 1000 times and that the methyl groups released in the gas phase therefore likely will react with hydrogen to form methane, which is very unreactive due to its high symmetry. It is known experimentally that the carbon content in the GaN layers can be controlled to some degree by varying process conditions such as temperature, NH3/TMGa ratio and pressure.4,6,7 However, changing process parameters to control impurity incorporation may lead to non-optimal conditions for the actual GaN growth, which in turn may lead to layers with rough morphology or bad thickness and doping uniformity. Separate carbon precursors have therefore been suggested for applications where a controlled amount of carbon is desired in the layers.8 For applications where essentially carbon free GaN is desired an alternative Ga precursor is most likely needed. From a better understanding of how carbon from the Ga precursor is incorporated into GaN, new precursors can be designed that may still contain carbon atoms in the ligands but with ligands designed to give a precursor decomposition that may lead to significantly lower carbon incorporation.
In this paper we use thermochemical and quantum chemical modelling of the gas phase and surface chemistry, respectively, to form a better understanding of the CVD chemistry of GaN. Our modelling is then combined with the experimental results of carbon incorporation into GaN CVD to propose an empirical model for the carbon incorporation chemistry in GaN CVD layers.
2. Methods
Gas-phase species concentrations are calculated by chemical kinetic simulations using computational fluid dynamics, while surface reaction energies are calculated by quantum chemistry methods. Experimental data, obtained in a hot-wall CVD system, are used for validation of the model. The details of the computational and experimental methods are given below.2.1 Chemical kinetic simulations
The development of the CVD gas mixture composition with time under relevant process conditions was studied by Computational Fluid Dynamics (CFD) using a finite-rate chemical kinetic model for the decomposition of precursors, and for reactions between the resulting products. The chemical kinetic model consists of 220 elementary reaction steps for hydrocarbon decomposition and reactions with hydrocarbon molecules containing up to four carbon atoms.9–15 It is likely that the number of relevant reaction steps are less, but the focus of the present study is to investigate the doping mechanism of GaN and not to optimize a gas-phase reaction modeling scheme, and therefore the gas-phase reaction scheme is kept large not to limit the number of possible species or to produce “short cuts” to more abundant species through a simplified scheme. For decomposition of TMGa, the model from Sengupta et al. was used.16 The formation of a Lewis-adduct between TMGa and NH3, and subsequent decomposition to a three membered ring compound, [Ga(CH3)2NH2]3, has in the past been discussed in the GaN literature, and it has been suggested as a main pathway to GaN growth.17–19 While this might be the case at low temperatures (<1000 K), experimental studies have shown no evidence of adduct formation or its decomposition products at the high temperatures typically used to grow high quality GaN.20,21 Sengupta22 used DFT and QRRK theories to calculate reaction rate constants for the adduct related reactions and found negligible concentrations of the monomer Ga(CH3)2NH2, indicating an almost zero possibility to form the trimer ring compound, further supporting the experimental findings. Thus, in the gas-phase model used here, reactions leading to the trimer ring compound are excluded, while reactions leading to the formation of the adduct, as well as its other decomposition products are included.16 It was further assumed in the chemical model that N2 and NH3 do not react with hydrocarbons to form N–C species. Reaction pathways to form such species were e.g. studied by Zhu et al.,23 who showed that the probability to form N–C bonds, for example by reactions between CH3 and NH3, is reduced due to the much more probable reaction CH4 + NH2 → CH3 + NH3. All reaction steps used in the gas-phase kinetic model and their respective rate constants are provided in the ESI.†Since we are using CFD, the whole CVD reactor could, in principle, be modeled and used in the simulations. However, the complex design of the real CVD reactor would lead to very time consuming simulations only for solving the flow inside the chamber. Adding heat and chemical models increase the complexity even further. Alternatively, several simplifications would be needed which inevitably introduce large uncertainties in the results. Also, process parameters and conditions differ between different reactor designs and the purpose here is not to develop a model adapted to a specific reactor setup, but rather to study the fundamentals of carbon incorporation into GaN. Therefore a more general reactor geometry is chosen here. A so-called plug flow reactor24 was used as the model geometry for the simulations. In this type of reactor the flow profile is completely flat (a ‘plug’), which together with imposed isothermal conditions makes the axial distance of the reactor equal to a time coordinate of the reacting system. The results from the CFD simulations could then be analyzed at a time corresponding to the residence time of the gas in the real CVD reactor. In the study this time was set to 0.6 s, estimated from a more detailed CFD simulation of the CVD system used for the experimental part.
2.2 Quantum chemistry
Molecular quantum-chemical calculations were performed for models of the GaN surface with different adsorbates. From these computations, data for the adsorption and activation energies were obtained, which were subsequently used as input for kinetic simulations. The program Gaussian0925 was utilized for the calculations, in which the hybrid density functional B3LYP26 in combination with the 6-31G(d,p) basis set27 was used. This methodology is the same as that used in our earlier studies of GaN.28,29The GaN (0001) surface was simulated by a cluster model, cut out from the crystal structure,30 that consisted of 24 Ga and 24 N atoms forming two GaN layers perpendicular to the [0001] direction (a cluster size that was also studied in ref. 28). Dangling bonds at all surface planes were saturated by capping H atoms, which is a reasonable assumption under H-rich atmosphere CVD conditions. When studying a chemisorbed adsorbate one or two capping H at the center part of the (0001) surface were replaced by the adsorbate in an appropriate adsorption mode. The energy-minimum structures of the clusters were obtained by geometry optimizations31 and the enthalpies and Gibbs free energies at 1300 K for the optimized geometries were obtained from vibrational normal-coordinate calculations32 as described in ref. 33 using the Gaussian09 program. All atoms were allowed to relax in the geometry optimizations. When one H vacancy was present at the otherwise H-saturated GaN cluster the spin state was a doublet state, whereas for two H vacancies the triplet state was the state lowest in energy. In case the adsorbate was in a doublet spin state (e.g. CH3) the dangling bond on the GaN cluster would be saturated so that a singlet state was formed, etc. The lowest-energy state of an isolated Ga atom was a doublet and that of the GaCH3 molecule a singlet.
For computational convenience, in the identification of transition states and computation of activation energies involving H, Ga and GaCH3 a smaller GaN cluster was used, which consisted of merely 4 GaN units. The numerical values of the activation energies obtained from these small cluster studies will be very approximate, but we consider them as initial guesses that can be further refined by comparison with experiment. The molecular geometries of the large and the small clusters can be seen in Fig. 1. For the molecules containing two C atoms the activation energies were obtained from the energy profiles when the molecule in a symmetric fashion approached two surface H vacancies at the larger GaN cluster.
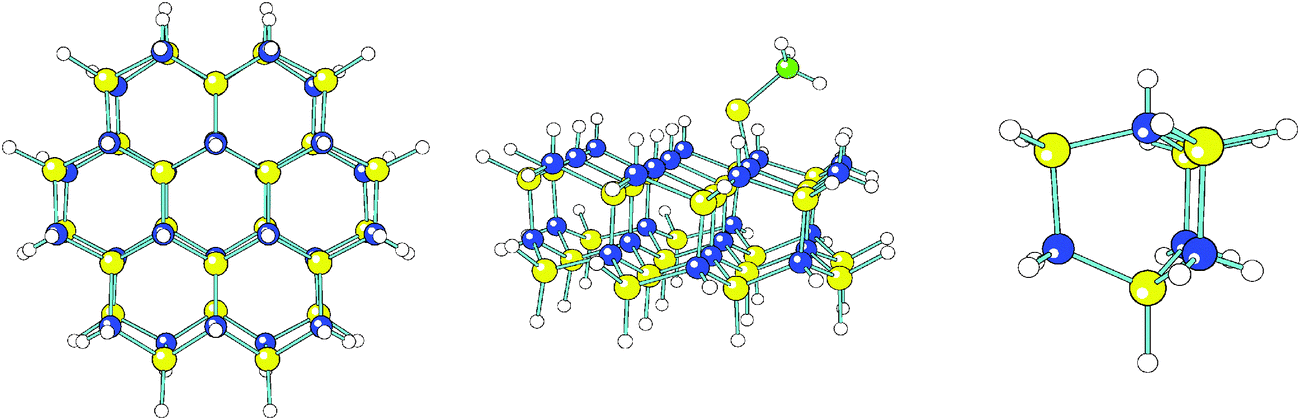 | ||
| Fig. 1 Molecular geometries of the H-saturated (GaN)24 cluster, top view (left), the cluster with an adsorbed GaCH3 molecule at a H vacancy site, side view (middle), and the smaller H-saturated (GaN)4 cluster, side view (right). Yellow: Ga, blue: N, white: H, green: C atoms. | ||
In the following, adsorption reaction energies are given in the tables for reactions at a surface where the appropriate number of H vacancies are already present. The reaction energy for removing one H atom from the H-saturated (GaN)24 cluster is 459 kJ mol−1 and the reaction free energy is 272 kJ mol−1 (the corresponding values for removing two H atoms from the same cluster are 918 kJ mol−1 and 554 kJ mol−1).
2.3 Surface reaction model
The CFD simulations give the gas phase partial pressure for each species in the bulk flow. To study the possible contribution from each species to the carbon doping, we should analyze the composition at the growth surface, which in a CVD system often is different from the bulk flow composition. One way of estimating this is by calculating impingement rates of the species. The impingement rate, Φ, of species i on the substrate surface can be estimated based on the partial pressure using the expression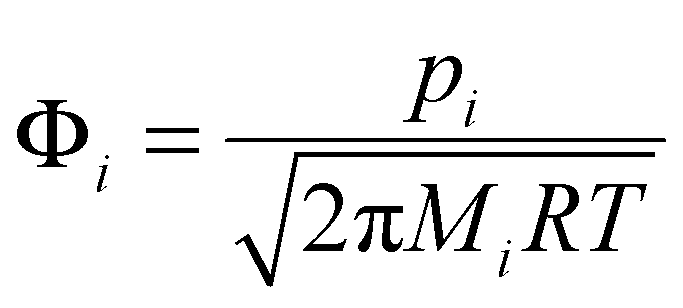 | (1) |
A common way to describe the surface reaction rate is
![[q with combining dot above]](https://www.rsc.org/images/entities/i_char_0071_0307.gif) = γ·e−Ea/RT·Φ = γ·e−Ea/RT·Φ | (2) |
2.4 Experimental growth
The growth of epitaxial layers of GaN took place in a horizontal hot-wall CVD system34 operating at 50 mbar with a carrier gas mixture of palladium membrane purified hydrogen (H2) and nitrogen (N2) (99.999%), at a volumetric H2/N2 ratio of 6.33. Growth was done on nominally on-axis 4H-SiC (0001) substrates where first an epitaxial aluminum nitride (AlN) nucleation layer was grown using trimethyl aluminum (TMAl) (SAFC Hightec EpiPure™ grade) and NH3 (99.9999%, further purified using a Nanochem® purifier down to <1 ppb for important contaminants) with NH3/TMAl = 1043 at 1200 °C. GaN growth was carried out directly on the AlN layer using TMGa (SAFC Hightec EpiPure™ grade) and NH3.To study carbon incorporation from TMGa, GaN layers were grown where either temperature or the growth rate was varied during the deposition to produce multiple layers under different CVD conditions and thereby obtaining different carbon concentrations in each layer. The thickness of the individual layers was approximately 200 nm. The growth temperature was varied from 800 °C to 1050 °C at a constant NH3 flow rate, with a NH3/N2 ratio of 2/3, and a TMGa flow giving a NH3/TMGa = 625. The growth rate was varied at a constant temperature of 1050 °C by changing the TMGa flow rate while keeping the NH3 flow constant, giving NH3/TMGa = 157–625.
The multilayers were analyzed by secondary ion mass spectrometry (SIMS), using Cs+ as primary ions, to obtain the impurity concentrations. The detection limit for carbon in these specific measurements was 1–2 × 1016 cm−3. All of the SIMS measurements were performed on the center part of the samples.
3. Results
3.1 Gas phase composition
The gas mixture composition was studied by Computational Fluid Dynamics (CFD). Due to the chemical kinetics, concentrations of different species vary with residence time in the reactor. In Fig. 2 the calculated molefractions of the most abundant species under the standard process conditions (T = 1050 °C, NH3/TMGa = 625) vs. residence time in the CVD reactor are shown. Calculated molefractions under other process conditions (T = 1050 °C, NH3/TMGa = 157; T = 800 °C, NH3/TMGa = 625) are presented in the ESI† for comparison. Even though a large number of elementary reaction steps were included in the kinetic model, it can be concluded that the formation of the main resulting hydrocarbon species can be described by less than 30 reaction steps, illustrated in Fig. 3. As expected, and in line with previous findings,20–22 negligible concentrations of the TMGa:NH3 adduct and Ga(CH3)2NHx compounds were found at all temperatures and flow rates. Instead, the TMGa quickly decomposes in the gas into GaCH3 and two methyl radicals. GaCH3 takes a while longer to decompose, and its concentration starts to drop after about 1 second at 1050 °C (Fig. 2), but is relatively stable for times >2 seconds at temperatures below 900 °C (see the ESI†). The methyl radicals are converted into mainly CH4. However, some of them instead combine into C2H6, which then decomposes to C2H4 and C2H2 (Fig. 3). Atomic hydrogen, from splitting of H2 molecules, and reactions between hydrocarbons and H2, has concentration levels of the same order as GaCH3 and CH4, and its concentration is fairly stable after 0.05 s. Notable is also that C2H2 does not approach a state of equilibrium within the first two seconds. This could be explained by the relatively long chain of reactions from CH3 to reach C2H2 (Fig. 3), and the relatively low reaction rates for converting C2H3 to C2H2.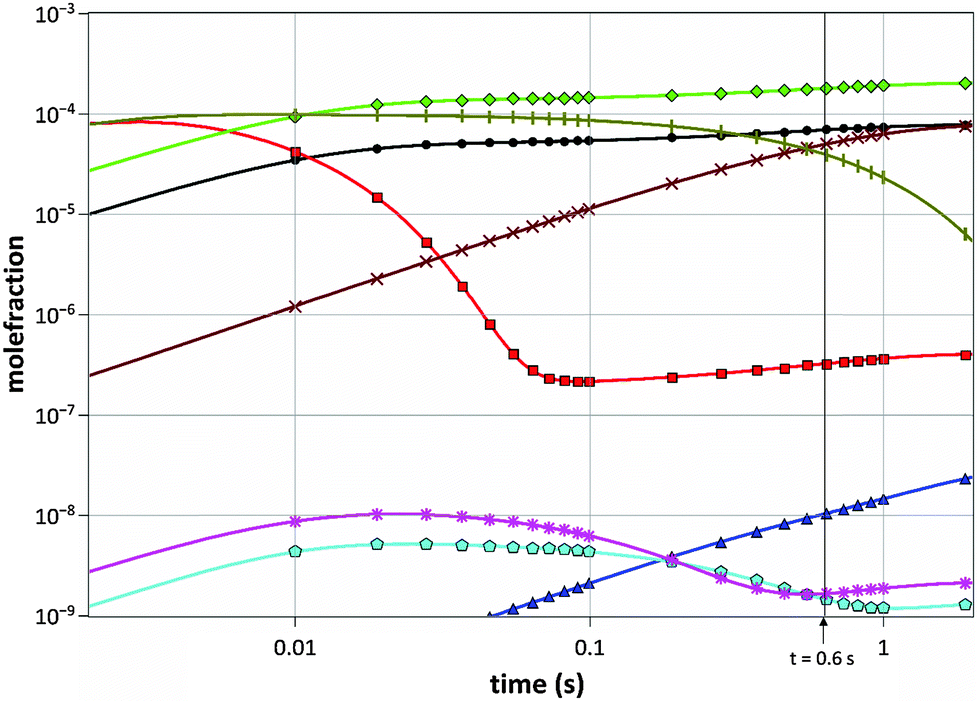 | ||
Fig. 2 Calculated molefractions of the most abundant species in the gas phase. (H2, N2, and NH3 are omitted.) Process conditions: T = 1050 °C, NH3/TMGa = 625. H (●), CH3 (■), CH4 (◆), C2H2 (△), C2H4 (![[pentagon open]](https://www.rsc.org/images/entities/char_e1c1.gif) ), C2H6 (*), Ga (×), GaCH3 (|). ), C2H6 (*), Ga (×), GaCH3 (|). | ||
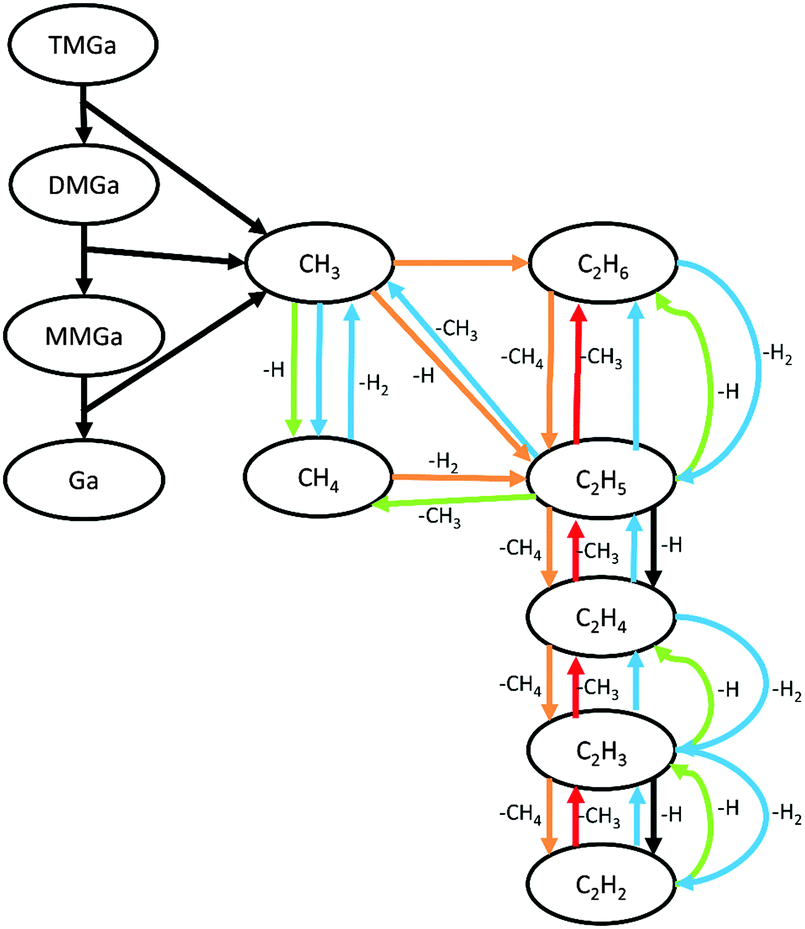 | ||
| Fig. 3 Decomposition reaction paths for TMGa in the gas phase. Unimolecular reactions are indicated by black arrows. Reaction partners in bimolecular reactions are indicated by H (blue), H2 (green), CH3 (orange), and CH4 (red). | ||
3.2 Surface reactions and activation energies
Quantum-chemical calculations were utilized to compute geometries and energies for adsorption of the molecules found to be the most abundant ones in the gas-phase (H, H2, Ga, CH3, GaCH3, C2H2, C2H4 and C2H6). CH4 was excluded from the study since it is assumed that its reactivity with the GaN surface is negligible. Calculations were done both at the GaN surface, using the cluster model described in Section 2.2, as well as for the molecules in the gas phase. Electronic energies (i.e. potential energies for the nuclear motions), enthalpies and Gibbs free energies were calculated for these structures. From the energies obtained, the reaction energies and reaction enthalpies were computed for the reactions between the gas phase species and the GaN surface. In addition, activation energies were computed for the relevant reactions.As pointed out in Section 2.2, adsorption is assumed to take place on a hydrogen terminated surface where one or two “free sites” (i.e. a surface site without the terminating hydrogen atom/atoms) are available for the adsorbing species. Thus, the reaction energies calculated here does not include the removal of surface H. The reaction energies to create a single “free” surface site is 459 kJ mol−1, and two adjacent “free” surface sites is 918 kJ mol−1, respectively.
Atomic Ga adsorbs without activation energy, and the reaction free energy is calculated to be −304 kJ mol−1. GaCH3 can adsorb either with its Ga atom or with the methyl group at a single adsorption site (Reactions (2) and (4) in Table 1). In the first case GaCH3 adsorbs with its Ga atom to a free surface site without activation energy, while keeping its bond to the methyl group. To break the Ga–CH3 bond when the molecule has adsorbed in this way requires about 147 kJ mol−1, as obtained from the calculations, which means that very few of these bonds actually will break at the temperatures used here.
| Surface reaction | Reaction probability, γ | Activation energy, Ea (kJ mol−1) | Reaction free energy, ΔrG0 (kJ mol−1) | |
|---|---|---|---|---|
| Growth | ||||
| 1. | Ga(g) → Ga(ads) | 0.135 | 0 | −304 |
| Growth and carbon adsorption | ||||
| 2. | GaCH3(g) → GaCH3(ads) | 0.135 | 0 | −112 |
| Carbon adsorption | ||||
| 3. | CH3(g) → CH3(ads) | 0.135 | 0 | −117 |
| 4. | CH3Ga(g) → CH3(ads) + Ga(g) | 0.135 | 35 | −3.0 |
| 5. | C2H2(g) → C2H2(ads) | 0.002 | 38 | −270 |
| 6. | C2H4(g) → C2H4(ads) | 0.002 | 28 | −171 |
In the second case, when GaCH3 approaches the surface with the methyl group first, the Ga–CH3 bond will break as the bond between the methyl group and the surface is established (Reaction (4)).
Similarly, C2H6 could react with the surface in the same way (i.e. with one of its CH3’s approaching the surface, while breaking off the other CH3 group). This reaction however, has a positive reaction free energy which indicates that the reaction is disfavored, and the activation energy is relatively large, 188 kJ mol−1.
There is also a possibility that GaCH3 adsorbs on two adjacent sites (adsorbing with both its Ga and C atom at the same time). This reaction is energetically favored (reaction free energy of −307 kJ mol−1), assuming there are two adjacent sites available. However, the probability that the molecule actually finds two adjacent vacancies is considered much smaller than the probability that it will find one single vacancy, and thus the contribution from this type of adsorption should be small in comparison to the other two options.
Free methyl radicals are also present in the gas phase for the whole temperature range studied here. The reaction free energy for CH3 adsorption (Reaction (3)) is similar to that of GaCH3 adsorption (Reaction (2)), and the activation energy is taken to be close to zero considering the highly reactive nature of the methyl radical.
At the higher temperatures C2H2, C2H4 and C2H6 have high enough partial pressures in the gas phase to possibly make a significant contribution to carbon doping. It is assumed that these species will contribute with both their carbon atoms to doping if they adsorb. Adsorption will thus take place when there are two adjacent free sites available. For C2H2 and C2H4 the reaction free energies are −270 kJ mol−1 and −174 kJ mol−1, respectively, and the activation energies are 38 kJ mol−1 and 28 kJ mol−1. When C2H6 approaches the surface the calculations show that the free sites at the surface will “steal” two of the hydrogen atoms from the molecule, resulting in a fully hydrogen terminated surface, and no carbon incorporation will take place.
The Gibbs free energies and activation energies for the reactions chosen to be relevant in the present study are presented in Table 1. The values of the reaction probabilities in Table 1 are further explained in Section 4.
3.3 Experimental results
Experiments were performed to measure the carbon incorporation with varying temperatures and varying TMGa inlet flow rates. The carbon concentration and thickness (from where the growth rate could be calculated) of each layer were measured by SIMS. The variation in carbon concentration measurements is between 4–11% (less variation at higher carbon concentration). The error in growth rate measurements is estimated to be less than 5%. The results are presented in Tables 2 and 3 below.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 sccm H2 and 3000 sccm N2), and the pressure was 50 mbar
000 sccm H2 and 3000 sccm N2), and the pressure was 50 mbar
| Temperature (°C) | 800 | 900 | 1000 | 1030 | 1050 |
|---|---|---|---|---|---|
| Carbon concentration (cm−3) | 1.2 × 1020 | 4.3 × 1019 | 1.9 × 1017 | 6.1 × 1016 | 3.0 × 1016 |
| Growth rate (μm h−1) | 0.65 | 0.80 | 0.99 | 1.12 | 1.18 |
| NH3/TMGa ratio | 625 | 418 | 314 | 251 | 209 | 179 | 157 |
|---|---|---|---|---|---|---|---|
| Carbon conc. (cm−3) | 6.0 × 1016 | 8.1 × 1016 | 1.2 × 1017 | 1.7 × 1017 | 2.4 × 1017 | 3.3 × 1017 | 4.0 × 1017 |
| Growth rate (μm h−1) | 1.29 | 1.32 | 1.83 | 2.38 | 2.82 | 3.17 | 3.99 |
4. Model of carbon incorporation
Based on the results presented above, we propose an empirical model for the chemistry of GaN growth and carbon incorporation, where reaction rates are calculated according to eqn (2), taking into account the reactions in Table 1. Adsorption of NH3 is not included in the model: it is assumed that the growth is limited by supply of Ga.4.1 Calculations of growth rate
It is crucial that the model can give reasonably accurate growth rates if it shall be used to predict doping concentrations. The Ga that contributes to growth comes either from atomic Ga or GaCH3. All other Ga-containing molecules have too low partial pressures in the gas phase to make any significant contribution. GaCH3 can adsorb either with its Ga atom or with its C atom in the methyl group. When it adsorbs with its C atom, the Ga–CH3 bond will break, resulting in a free Ga atom in the gas phase. This Ga atom is then available for adsorption on another surface site. It seems reasonable to assume that the probability for either orientation of the GaCH3 molecule is equal, so that 50% undergoes reaction (2) and the other 50% undergoes reaction (4). Since the Ga atoms resulting from reaction (4) are also available for adsorption, these are included as well when calculating the growth rates.Thus, the growth rate, Ṙg, is calculated by
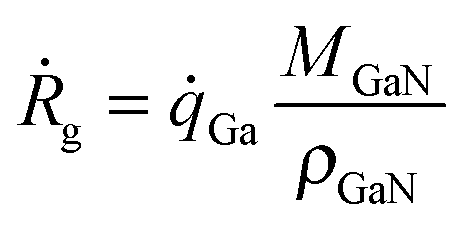 | (3) |
Ga = R1 + R2 + R4, MGaN is the molar mass of GaN, ρGaN is the GaN density, and 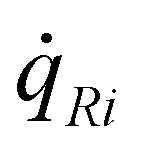 is calculated using eqn (2). The probability parameter, γ, was determined by comparing calculated growth rates against experiments. As mentioned in Section 2, γ is a measure of the probability that a species impinging on the surface actually will participate in an adsorption reaction. Thus, this parameter could be related to the fraction of sites available for adsorption, and here it could also be assumed to include the depletion of precursors upstream of the deposition area, which decreases the amount of available molecules for growth. A value of γ = 0.135 was chosen for the reactions involving Ga, to give reasonably good agreement in growth rates, as compared to experimental data. This value was also used for reaction (3), where CH3 adsorbs directly on the surface, since it seems reasonable to assume that the probability to find a single surface adsorption site would be equal regardless of which molecule that adsorbs. The calculated and measured growth rates are compared in Fig. 4 and 5 below.
is calculated using eqn (2). The probability parameter, γ, was determined by comparing calculated growth rates against experiments. As mentioned in Section 2, γ is a measure of the probability that a species impinging on the surface actually will participate in an adsorption reaction. Thus, this parameter could be related to the fraction of sites available for adsorption, and here it could also be assumed to include the depletion of precursors upstream of the deposition area, which decreases the amount of available molecules for growth. A value of γ = 0.135 was chosen for the reactions involving Ga, to give reasonably good agreement in growth rates, as compared to experimental data. This value was also used for reaction (3), where CH3 adsorbs directly on the surface, since it seems reasonable to assume that the probability to find a single surface adsorption site would be equal regardless of which molecule that adsorbs. The calculated and measured growth rates are compared in Fig. 4 and 5 below.
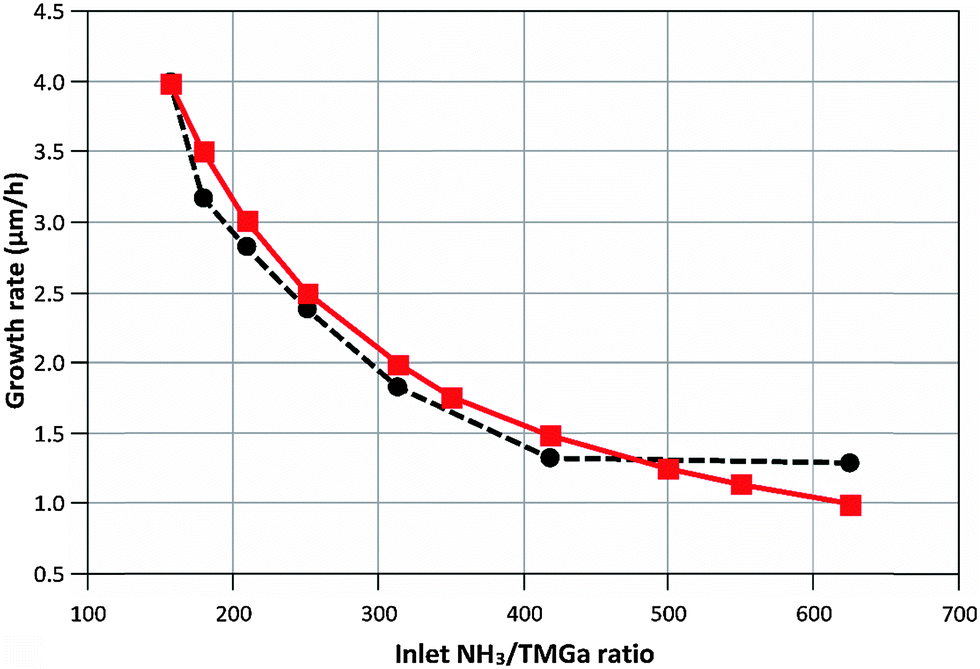 | ||
| Fig. 4 Calculated (–■–) and measured (--●--) growth rates when varying the inlet NH3/TMGa ratio at constant NH3 flow (2000 sccm). Process temperature is 1050 °C. | ||
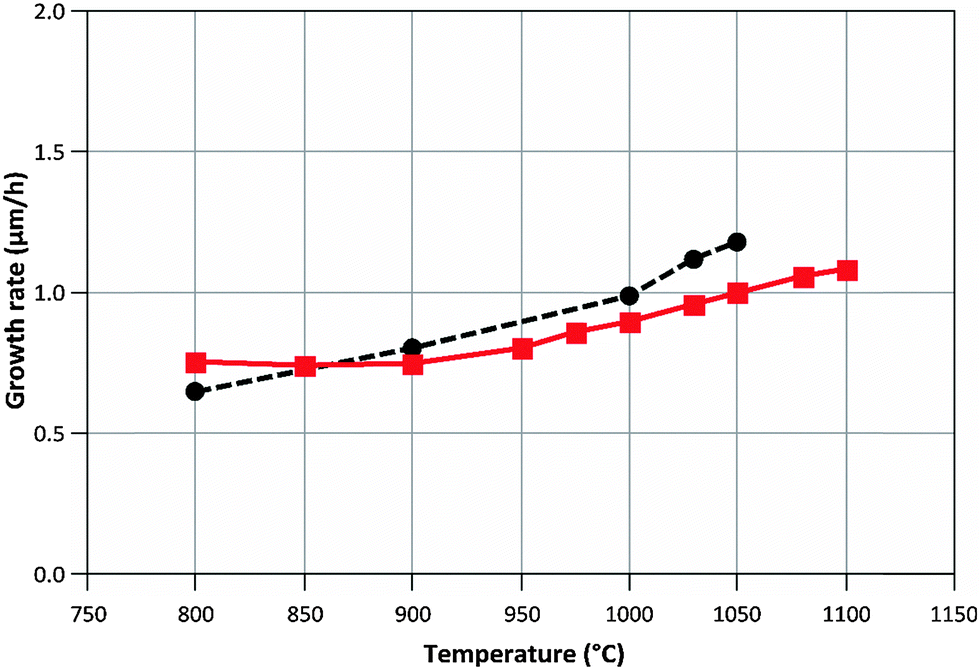 | ||
| Fig. 5 Calculated (–■–) and measured (--●--) growth rates when varying the process temperature. NH3/TMGa ratio = 625. | ||
The model predicts a change in relative contribution to the growth from GaCH3 and atomic Ga, respectively, with temperature. The relative contribution from GaCH3 changes from 99.1% at 800 °C to 27.8% at 1050 °C. This change could be one explanation to the improved morphology and structural quality obtained at higher temperatures, as CH3 has been suggested to degrade both these properties of GaN epitaxial layers.8
4.2 Calculations of doping concentration
The doping concentration (number density), nc, is the fraction of C atoms in the GaN lattice.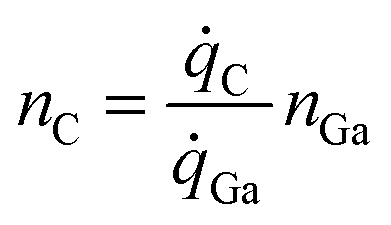 | (4) |
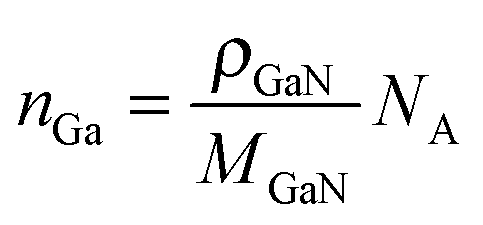 | (5) |
Ga is the same as in eqn (3), and thus eqn (4) can be re-written using the growth rate Ṙg so that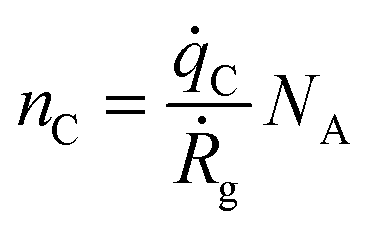 | (6) |
When examining the measured doping variation with temperature it is observed that there is a large drop in doping concentration of two orders of magnitude between 900 °C and 1000 °C (see Table 2). Looking at the simulated concentrations of different carbon containing species in the gas phase we find no obvious reason for this large drop. It is further observed that if all carbon atoms that follow Ga to the surface via GaCH3 would be incorporated into the material, the doping level would be at least two orders of magnitude too high in the lower part of the temperature range, and about 5 orders of magnitude too high at the higher temperatures. Fig. S6 in the ESI† shows the contribution from the different molecules vs. temperature. These results strongly suggest that there must be an efficient mechanism that removes carbon from the surface before it is incorporated into the material. This is consistent with previous findings.4,35 In the literature some mechanisms have been suggested for the removal of carbon from the GaN surface, such as a rehydrogenation process where atomic hydrogen reacts with carbon at the surface, and then desorb as methyl or methane, or blocking of surface sites by H atoms. Lam et al. estimated the activation energy for direct desorption of methyl radicals from the GaN surface to be about 171 kJ mol−1.36 They also found that significant amounts of methane may be produced from reactions involving adsorbed methyl and hydrogen atoms. Koleske et al. estimated an activation energy of about 154 kJ mol−1 for the carbon removal at temperatures between 650 °C and 1100 °C.4
Assuming the gas phase reaction mechanism we have used here is correct, it is mainly carbon coming from GaCH3 and CH3 that should be removed (or not adsorbed at all) for the modeling results to agree with experimental data. When calculating reaction energies for four possible reactions for the removal of adsorbed methyl (Table 4) it can be seen that all of them are exothermic and therefore energetically favored. It can also be concluded that the removal of the CH3 group from adsorbed GaCH3 with the aid of atomic H or CH3 (reactions (7) and (8)) in the gas phase is much more favorable than the corresponding removal of adsorbed CH3 from the surface (reactions (9) and (10)).
| Removal of methyl groups | Reaction energy, ΔE (kJ mol−1) | |
|---|---|---|
| 7. | GaCH3(ads) + H(g) → Ga(ads) + CH4(g) | −328 |
| 8. | GaCH3(ads) + CH3(g) → Ga(ads) + C2H6(g) | −255 |
| 9. | CH3(ads) + H(g) → CH4(g) | −80 |
| 10. | CH3(ads) + CH3(g) → C2H6(g) | −6 |
We have here listed some of the more likely reaction steps for removing CH3 from the surface of the growing GaN crystal. However, we believe that the process is more complex than this, but it is beyond the scope of this paper to go any further in the surface reaction modeling at this point. Instead we assume that we could describe the removal of methyl with one single reaction step, where the reaction rate is given by an Arrhenius expression. By analyzing how many of the methyl groups that needs to be removed, for the modeling results to agree with experimental findings, we find that this single reaction step should have an activation energy of 0.26 kJ mol−1. If we on the other hand would assume that every CH3 in the GaCH3 molecules that adsorb immediately are removed, as soon as adsorption has taken place (e.g. with the aid of H or gaseous CH3 reactions (7) and (8)), then some of the remaining CH3’s must still be removed. The activation energy for removing these remaining methyls should then be 9.49 kJ mol−1, to match the model with experiments. The assumption that all of the CH3’s in the adsorbed GaCH3 are removed could be motivated by the relatively low reaction energies for those reactions.
Adding either of these two desorption reaction mechanisms to the model greatly improves the model’s agreement with experimental data. It is also noted that with this desorption mechanism included, all of the CH3’s will be removed at temperatures above 950 °C under the conditions we use. At higher temperatures (>950 °C) the doping concentration levels are well predicted taking only C2H2 and C2H4 into account as carbon contributors. For the reactions involving C2H2 and C2H4, a value of γ = 0.002 was used. The value was determined by trial-and-error to achieve a good agreement with experimental data when varying the NH3/TMGa inlet ratio, but it seems to give good results also when the temperature varies between 1000 °C–1100 °C. The C2H2 and C2H4 molecules adsorb on two surface sites simultaneously, and it would be safe to assume that the probability to find two adjacent free surface sites is much less than finding a single site. Thus, the value of γ seems reasonable.
Calculated and measured doping concentrations are compared in Fig. 6 and 7 below.
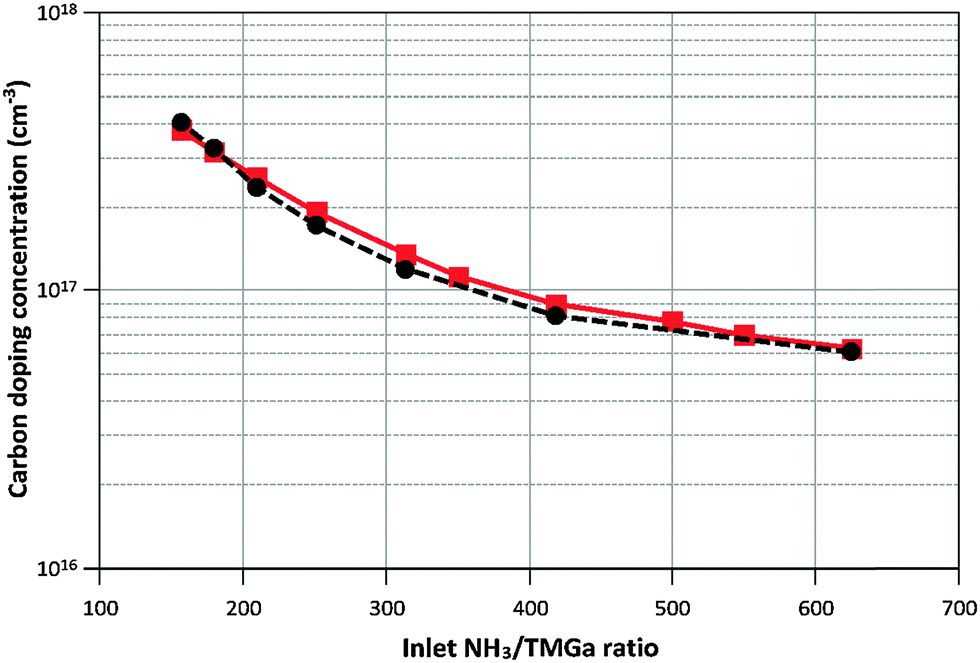 | ||
| Fig. 6 Calculated (–■–) and measured (--●--) carbon doping concentrations when varying the NH3/TMGa ratio at constant NH3 flow (2000 sccm) and T = 1050 °C. | ||
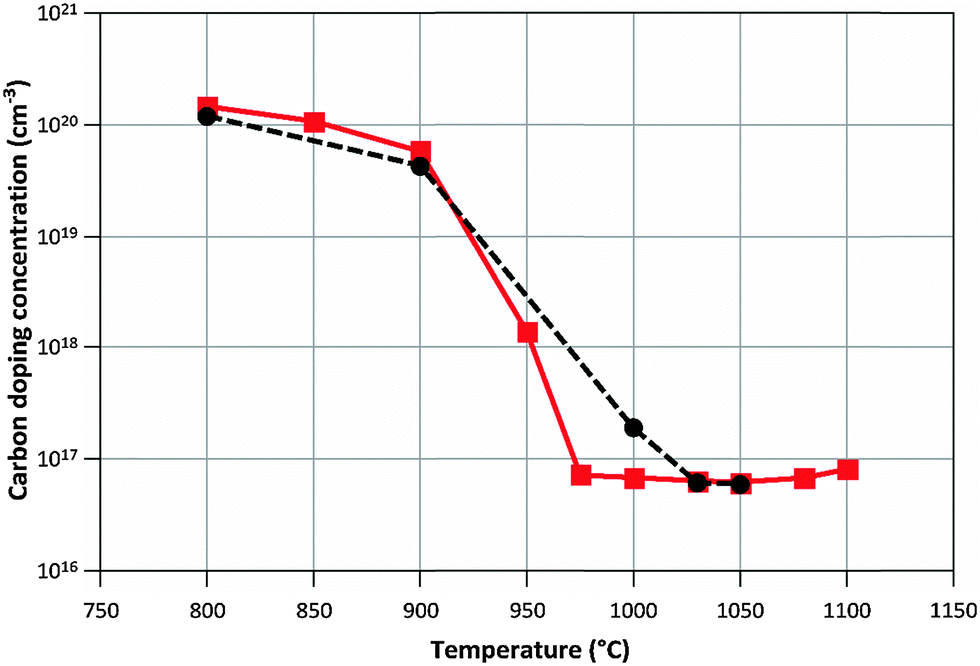 | ||
| Fig. 7 Calculated (–■–) and measured (--●--) carbon doping concentrations when varying the process temperature. NH3/TMGa ratio = 625. | ||
5. Discussion
The proposed model mimics the carbon doping concentrations well under the process conditions studied. At low temperatures carbon is supplied mainly via GaCH3 molecules, which bind to the surface with their Ga atoms, leaving the Ga–CH3 bond intact. The Ga in these GaCH3 molecules are needed to achieve a correct prediction of growth rates over the temperature range studied. It is however evident from the obtained results that most of the carbon from the methyl groups must desorb from the surface to have doping concentration levels in line with measured data. The amount of desorbed methyl ranges from about 99.7% at 800 °C to 100% at temperatures above 1000 °C. According to the quantum chemical calculations, the Ga–CH3 bond is hard to break, and therefore the carbon drawn to the surface by the GaCH3 molecule will stay there if it is not removed by some other reaction mechanism. In the proposed model we suggest that the removal of CH3 could take place through the reaction between a hydrogen atom in the gas and an adsorbed methyl group, eventually forming methane, or through reaction between a CH3 radical in the gas and an adsorbed methyl group, forming C2H6. These reactions are exothermic and therefore energetically favored. There could, of course, be some other mechanism or a combination of several reactions responsible for a lower carbon incorporation, compared to the modeling results. Either way, it can be concluded that, if a good correlation with measured values should be obtained, some desorption mechanism is needed. Another explanation could be that the amount of GaCH3 in the gas-phase is overpredicted by the kinetic model, and that we instead would have more atomic Ga contributing to the growth. This would initially lead to more CH3 in the gas, but since a lot of these molecules will be converted to CH4, the effect of an easier breakage of the GaCH3 molecule will be less active carbon containing molecules contributing to the doping. However, at the moment we find no reason to distrust the gas-phase reaction rates for TMGa decomposition, obtained by Sengupta et al.,16 which were evaluated under relevant process conditions.At higher temperatures, C2H2 and C2H4 are the main species contributing to the carbon doping. This is the result of increased production rates of these molecules in the gas, and the fact that CH3 is removed completely from the GaN surface at higher temperatures, using the assumptions made in the model. With our model, the change in carbon doping concentrations with varying TMGa inlet concentrations and with varying temperatures is well predicted. It should be noted that if CH3 would not be removed completely at the higher temperatures, the prediction of the variation in carbon doping concentration with TMGa inlet concentration would not be as accurate as is obtained now. Thus, we can conclude that there is a relatively rapid switch at about 1000 °C, from CH3 to C2Hx as the most important carbon source.
It should also be noted that to obtain high crystalline quality of the GaN layers, the optimal growth temperature is above 1000 °C, and that lower temperatures often yield layers with high dislocation densities. This agrees well with our findings here, since it has been suggested by others that high amounts of CH3 could degrade the GaN morphology and structural quality.
6. Conclusion
The model for carbon incorporation into GaN epitaxial layers presented here predicts the carbon doping levels very well at the most relevant temperatures and precursor flow rates. Since the methodology is based on ab initio quantum chemical calculations, and geometry independent calculations of the gas-phase composition, the constructed model is essentially reactor independent and it should therefore be possible to use it for any type of GaN CVD equipment.The contribution of gallium to the GaN layer shifts from GaCH3 at low temperatures to pure Ga at higher temperatures. In the same way there is a shift in carbon contribution, from CH3 at low temperatures to C2Hx at higher temperatures.
Acknowledgements
Financial support from the Swedish Foundation for Strategic Research (SSF) and the Swedish Defence Material Administration (FMV) is gratefully acknowledged. The Swedish National Supercomputer Centre (NSC) and SNIC are acknowledged for providing computational resources.References
- M. J. Uren, IEEE Trans. Electron Devices, 2012, 59, 3327 CrossRef CAS.
- A. Armstrong, A. R. Arehart, B. Moran, S. P. DenBaars, U. K. Mishra, J. S. Speck and S. A. Ringel, Appl. Phys. Lett., 2004, 84, 374 CrossRef CAS.
- A. Zado, E. Tschumak, J. W. Gerlach, K. Lischka and D. J. As, J. Cryst. Growth, 2011, 323, 88 CrossRef CAS.
- D. D. Koleske, A. E. Wickenden, R. L. Henry and M. E. Twigg, J. Cryst. Growth, 2002, 242, 55 CrossRef CAS.
- A. Stegmüller, P. Rosenow and R. Tonner, Phys. Chem. Chem. Phys., 2014, 16, 17018 RSC.
- G. Parish, S. Keller, S. P. Denbaars and U. K. Mishra, J. Electron. Mater., 2000, 29, 15 CrossRef CAS.
- A. Ishibashi, H. Takeishi, M. Mannoh, Y. Yabuuchi and Y. Ban, J. Electron. Mater., 1995, 25, 799 CrossRef.
- X. Li, Ö. Danielsson, H. Pedersen, E. Janzén and U. Forsberg, J. Vac. Sci. Technol., B: Nanotechnol. Microelectron.: Mater., Process., Meas., Phenom., 2015, 33, 021208 Search PubMed.
- W. Tsang and R. F. Hampson, J. Phys. Chem. Ref. Data, 1986, 15, 1087 CrossRef CAS.
- W. Tsang, J. Phys. Chem. Ref. Data, 1988, 17, 887 CrossRef CAS.
- W. Tsang, J. Phys. Chem. Ref. Data, 1990, 19, 1 CrossRef CAS.
- W. Tsang, J. Phys. Chem. Ref. Data, 1991, 21, 20 Search PubMed.
- D. L. Baulch, C. J. Cobos, R. A. Cox, C. Esser, P. Frank, T. Just, J. A. Kerr, M. J. Pilling, J. Troe, R. W. Walker and J. Warnatz, J. Phys. Chem. Ref. Data, 1992, 21, 411 CrossRef CAS.
- D. L. Baulch, C. J. Cobos, R. A. Cox, P. Frank, G. Hayman, T. Just, J. A. Kerr, T. Murrells, M. J. Pilling, J. Troe, R. W. Walker and J. Warnatz, J. Phys. Chem. Ref. Data, 1994, 23, 847 CrossRef CAS.
- D. L. Baulch, C. T. Bowman, C. J. Cobos, R. A. Cox, T. Just, J. A. Kerr, M. J. Pilling, D. Stocker, J. Troe, W. Tsang, R. W. Walker and J. Warnatz, J. Phys. Chem. Ref. Data, 2005, 34, 757 CrossRef CAS.
- D. Sengupta, S. Mazumder, W. Kuykendall and S. A. Lowry, J. Cryst. Growth, 2005, 279, 369 CrossRef CAS.
- A. Thon and T. F. Kuech, Appl. Phys. Lett., 1996, 69, 55 CrossRef CAS.
- S. A. Safvi, J. M. Redwing, M. A. Tischler and T. F. Kuech, J. Electrochem. Soc., 1997, 144, 1789 CrossRef CAS.
- T. G. Mihopoulos, V. Gupta and K. F. Jensen, J. Cryst. Growth, 1998, 195, 733 CrossRef CAS.
- U. Bergmann, V. Reimer and B. Atakan, Phys. Chem. Chem. Phys., 1999, 1, 5593 RSC.
- J. Schäfer, A. Simons, J. Wolfrum and R. A. Fischer, Chem. Phys. Lett., 2000, 319, 477 CrossRef.
- D. Sengupta, J. Phys. Chem. B, 2003, 107, 291 CrossRef CAS.
- R. S. Zhu, R. Q. Zhang and K. S. Chan, Chem. Phys. Lett., 2000, 320, 561 CrossRef CAS.
- R. B. Bird, W. E. Stewart and E. N. Lightfoot, Transport Phenomena, New York, Wiley, 2007 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. J. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Wallingford CT, Gaussian, Inc., 2009 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- V. A. Rassolov, M. A. Ratner, J. A. Pople, P. C. Redfern and L. A. Curtiss, J. Comput. Chem., 2001, 22, 976 CrossRef CAS.
- B. Brena and L. Ojamäe, J. Phys. Chem. C, 2008, 112, 13516 CAS.
- M. Kula and L. Ojamäe, Int. J. Quantum Chem., 2012, 112, 1796 CrossRef CAS.
- W. Paszkowicz, S. Podsiadlo and R. Minikayev, J. Alloys Compd., 2004, 382, 100 CrossRef CAS.
- C. Peng, P. Y. Ayala, H. B. Schlegel and M. J. Frisch, J. Comput. Chem., 1996, 17, 49 CrossRef CAS; X. Li and M. J. Frisch, J. Chem. Theory Comput., 2006, 2, 835 CrossRef PubMed.
- E. B. Wilson, Jr., Phys. Rev., 1934, 45, 706 CrossRef; E. B. Wilson, Jr., C. Decius and P. C. Cross, Molecular Vibrations, McGraw–Hill, New York, 1955 Search PubMed.
- D. A. McQuarrie and J. D. Simon, Molecular Thermodynamics, University Science Books, 1999 Search PubMed.
- U. Forsberg, A. Lundskog, A. Kakanakova-Georgieva, R. Ciechonski and E. Janzén, J. Cryst. Growth, 2009, 311, 3007 CrossRef CAS.
- O. Ambacher, M. S. Brandt, R. Dimitrov, T. Metzger, M. Stutzmann, R. A. Fischer, A. Miehr, A. Bergmaier and G. Dollinger, J. Vac. Sci. Technol., B: Microelectron. Nanometer Struct.–Process., Meas., Phenom., 1996, 14, 3532 CrossRef CAS.
- H.-T. Lam and J. M. Vohs, Surf. Sci., 1999, 426, 199 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5tc03989d |
| This journal is © The Royal Society of Chemistry 2016 |