
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent advances in twisted intramolecular charge transfer (TICT) fluorescence and related phenomena in materials chemistry
Shunsuke
Sasaki
a,
Gregor P. C.
Drummen
*b and
Gen-ichi
Konishi
ac
aDepartment of Organic and Polymeric Materials, Tokyo Institute of Technology, Okayama, Tokyo, Japan
bBionanoscience and Bio-Imaging Program, Cellular Stress and Ageing Program, Bio&Nano-Solutions, Bielefeld, Germany. E-mail: gpcdrummen@bionano-solutions.de
cPRESTO (Japan) Science and Technology Agency (JST), Kawaguchi, Saitama, Japan
First published on 11th February 2016
Abstract
Twisted intramolecular charge transfer (TICT) is an electron transfer process that occurs upon photoexcitation in molecules that usually consist of a donor and acceptor part linked by a single bond. Following intramolecular twisting, the TICT state returns to the ground state either through red-shifted emission or by nonradiative relaxation. The emission properties are potentially environment-dependent, which makes TICT-based fluorophores ideal sensors for solvents, (micro)viscosity, and chemical species. Recently, several TICT-based materials have been discovered to become fluorescent upon aggregation. Furthermore, various recent studies in organic optoelectronics, non-linear optics and solar energy conversions utilised the concept of TICT to modulate the electronic-state mixing and coupling on charge transfer states. This review presents a compact overview of the latest developments in TICT research, from a materials chemistry point of view.
 Shunsuke Sasaki | Shunsuke Sasaki was born in Kobe, Japan, in 1992. He received his BSc in polymer chemistry from Tokyo Institute of Technology in 2013 under the supervision of Professor Gen-ichi Konishi. He is currently pursuing his master course studies regarding the structure–property relationship of organic dyes. His research interests include photophysical processes, organic synthesis, fluorescent dyes, photoredox catalysis and material science on strongly-correlated electron systems. |
 Gregor Drummen | Gregor Drummen was born in Maastricht, the Netherlands, in 1968. He received bachelor degrees in Chemical Engineering and Bioprocesstechnology from Zuyd University (NL), post-graduate diplomas in Biotechnology and Biomedical Science from Teesside and Ulster Universities (UK), a MSc in Chemistry, specializing in Cell and Chemical Biology, and a PhD degree in Biochemistry (Professors Henk van den Bosch and Jos Op den Kamp) from the Bijvoet Center for Biomolecular Research (formerly CBLE) and the Institute for Biomembranes of Utrecht University (NL). After post-doctoral and senior scientist appointments with various Multinational and Biotech companies and the University of Duisburg-Essen (Germany), he currently runs Bio&Nano-Solutions and the LAB3BIO project. His research interests include oxidative stress and antioxidants, extracellular vesicles, renal disease, advanced optical imaging techniques and fluorescent probes, and (bio)nanotechnology, including Laser ablation synthesis in solution (LASiS) and non-linear optics. |
 Gen-ichi Konishi | Gen-ichi Konishi was born in Rochester, NY, USA in 1971. He received his BSc degree from Osaka Prefecture University under the supervision of Professor Kazuhiko Mizuno in 1995 and his PhD from Kyoto University, Department of Polymer Chemistry, in 2000 under the supervision of Professor Yoshiki Chujo. Immediately thereafter he joined Shinshu University School of Medicine, where he worked as an assistant professor for 2 years in the Division of Physiology. He became an assistant professor in the Department of Chemistry & Chemical Engineering, Kanazawa University in 2002 and was promoted to associate professor in the Department of Organic and Polymeric Materials, Tokyo Institute of Technology in 2006. He was appointed JST PRESTO researcher (Element Strategy; mentor: Professor Hideo Hosono). His research interests include photochemistry, fluorescent dyes, redox-photosensitised reactions (photoredox catalysis), polymer synthesis, optical materials, and physiology of the autonomic nervous system. |
Introduction
Photophysical properties of some fluorescent molecules are well-known to be sensitive to local steric environments. Fluorescence phenomena that involve energy transfer, partial charge or pure electron transfer, intermolecular proton transfer, excimer/exciplex formation and J- or H- aggregates are sometimes affected largely by, for instance, relative distance/orientation of fluorophores, conformation of individual fluorophores and their mobility. These fluorophores are not only versatile reporters for steric environments at mesoscopic to microscopic scales, but also expected to be a motif of novel functional molecules because certain steric restrictions or well-ordered arrangement of fluorophores may activate unprecedented photophysical properties.We start from energy transfer phenomena such as Förster Resonance Energy Transfer (FRET) because these phenomena are photophysical processes generally known to researchers and are often employed in applications relevant to material and life sciences.1 Though energy transfer processes are different from charge transfer process in terms of underlying mechanisms, they have some phenomenological aspects and potential applications in common. Electron transfer phenomena occur between electronically separated fluorophores, in which their relative distance, orientation and mobility play instrumental roles. Therefore, some of these applications include molecular dynamics measurements, e.g., interaction between molecules or conformational changes within molecules, various reporter assays, bio-imaging, boosting or directing photoluminescence in both organic molecules and inorganic nanostructures such as zinc oxide quantum dots, solar collectors, and in semiconductors such as organic light-emitting diodes (OLEDs).1–5
FRET (Fig. 1a) involves non-radiative energy transfer from the excited state donor fluorophore to an acceptor molecule via Coulomb interaction.6,7 FRET relies on close donor and acceptor (D–A) proximity, scales with an inverse 6th power law, requires spectral overlap of donor emission and acceptor absorption spectra, and depends on the relative orientation of the donor emission and acceptor absorption dipole moments. Next to energy transfer between species, various other processes can also deplete the donor’s excited state, thereby changing the fluorophores’ emission characteristics. Dexter energy transfer (DET)8 is an exchange process in which two molecules (intermolecular) or two parts of the same molecule (intramolecular) bilaterally exchange their electrons (Fig. 1b). DET requires orbital overlap between donor and acceptor and scales with an e−R D–A distance dependence.8 Charge transfer processes include the aforementioned excimer and exciplex formation,1,9,10 which are short-lived homodimers (excimer) or heterodimers (exciplex) of which at least one molecule is in the excited state. Such complexes occur via electrostatic attraction because of partial charge transfer between the individual entities and show red-shifted emission compared with the monomer’s emission.
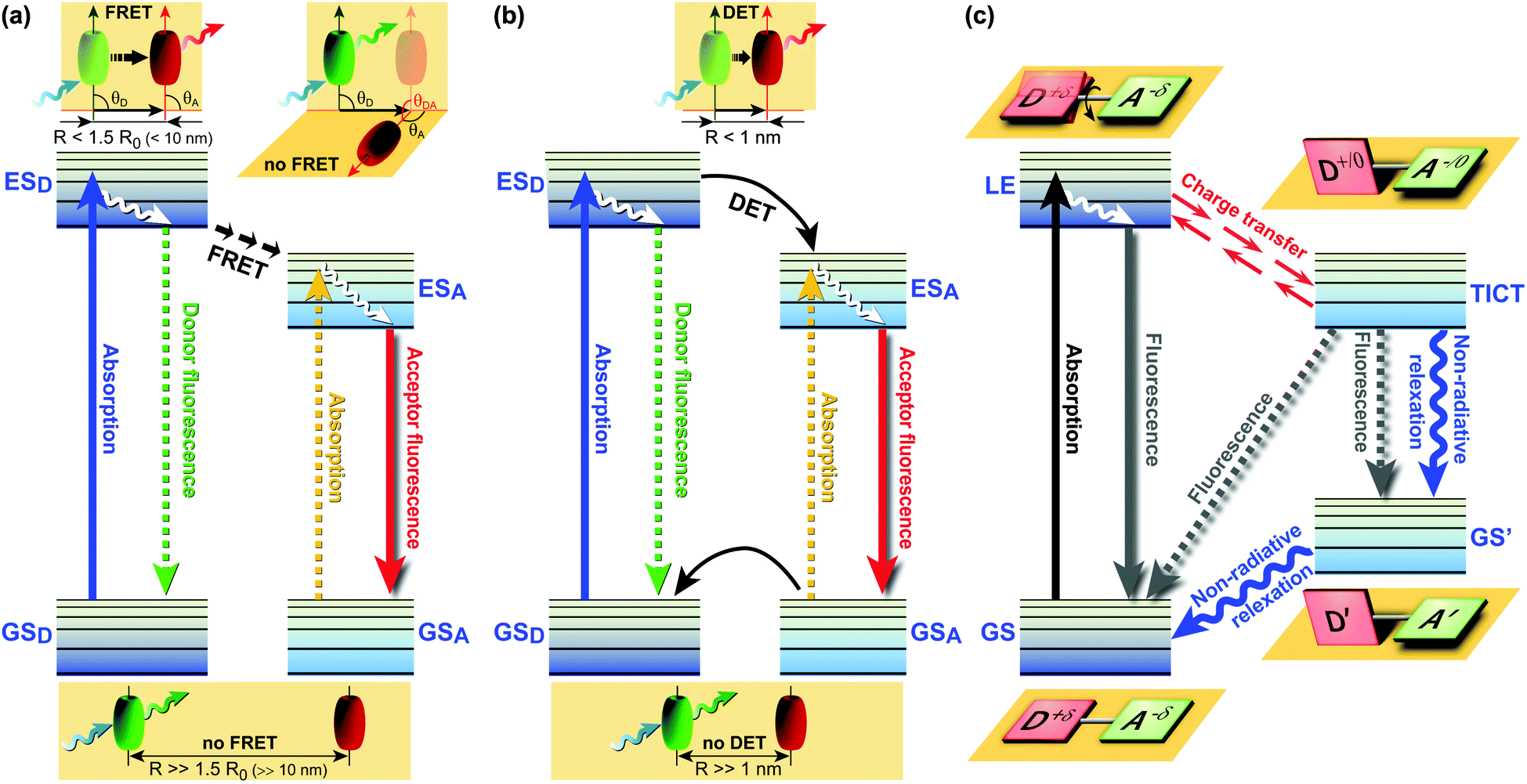 | ||
| Fig. 1 Jabłoński diagrams of various energy/electron donor–acceptor (D–A) systems. (a) Förster Resonance Energy Transfer (FRET). θ: angle between vectors of the donor emission and the acceptor absorption; (b) Dexter Energy Transfer (DET); (c) Twisted Intramolecular Charge Transfer (TICT) dynamics.11 Upon excitation from the GS, the LE state equilibrates rapidly with the TICT state after fast charge transfer. GS = ground state; GSD = ground state donor; GSA = ground state acceptor; ESD = excited singlet state donor; ESA = excited singlet state acceptor; LE = locally excited state; R = effective D–A distance. | ||
While the aforementioned processes all reflect relative arrangements of fluorophores, fluorescence phenomena governed by Twisted Intramolecular Charge Transfer (TICT) involve conformations of an individual fluorophore. TICT is a relatively common phenomenon in molecules that consist of a D–A (be advised that D and A in the context of charge transfer refers to electron donating or accepting groups rather than excitation energy donors or acceptors) linked by a single bond (Fig. 1c).12 In polar environments, such fluorophores undergo fast intramolecular electron transfer from the donor to the acceptor part of the molecule. This electron transfer is accompanied by intramolecular D–A twisting around the single bond (Fig. 1c) and produces a relaxed perpendicular structure. The equilibration between a relaxed perpendicular conformer and a coplanar conformer often results in dual fluorescence, i.e., from a high energy band through relaxation of the locally excited (LE) state and from a lower energy band due to emission from the TICT state. Since the relaxation pathways can easily be modulated by substituents, local polarity and steric restrictions, the TICT process can be exploited for novel design strategies of functional molecules. Therefore, TICT fluorescence holds great promise in applications such as OLEDs, chemosensors, and photovoltaic devices. This compact review examines some of the most recent advances in TICT research from the viewpoint of materials chemistry and potential applications in life science research, biomedical imaging and diagnostics, in optoelectronic devices, and photovoltaic applications.
Fundamentals of TICT
The seminal review by Grabowski, Rotkiewicz and Rettig compiled intuitive, but insightful information and images of TICT formation, and forms an essential basis for engineering various materials that directly exploit connected D–A systems.12 On an adiabatic energy surface of the charge transfer (CT) state of an excited D–A system, two disparate excited states exert opposite forces; the force that twists a D–A junction has an electron transfer (ET) character, whereas the force that prefers a coplanar conformation arises from mixing with a locally excited (1LE) state (Fig. 2). The 1ET state is associated with a pair of frontier orbitals, i.e. the HOMO and LUMO where a single electron is transferred from the HOMO to the LUMO. As for D–A systems, an electron is transferred from the donor orbital (HOMO) to the acceptor orbital (LUMO) upon photoexcitation resulting in a biradicaloid pair (Fig. 2a). Frontier orbital interaction increases the excitation energy required to reach the 1ET state and consequently a perpendicular conformation minimises the excitation energy. At a perpendicular conformation, the relative energy level of the 1ET state can be approximated by subtracting the electron affinity of an acceptor orbital from the ionisation potential of the donor orbital and thus strong donors and acceptors stabilise the 1ET state. The 1ET state is not only ubiquitously found in D–A systems but also in those with isomerisable double bonds. In contrast, mesomeric interaction between π-subsystems (Ψ1 and Ψ2) stabilises the 1LE state in a coplanar conformation (Fig. 2a). Resonance stabilisation and accompanying planarisation between π-subsystems is particularly prompted in the excited state; multiple interactions between whole occupied and unoccupied π-orbitals of Ψ1 and Ψ2 enhance the HOMO level and reduce the LUMO level resulting in a lower 1LE level. These competing two forces divide the 1CT surface to generate either single or multiple minima (Fig. 2b). When the ET character of 1CT outweighs the LE character of S1, the 1CT minimum distinctly becomes a TICT state.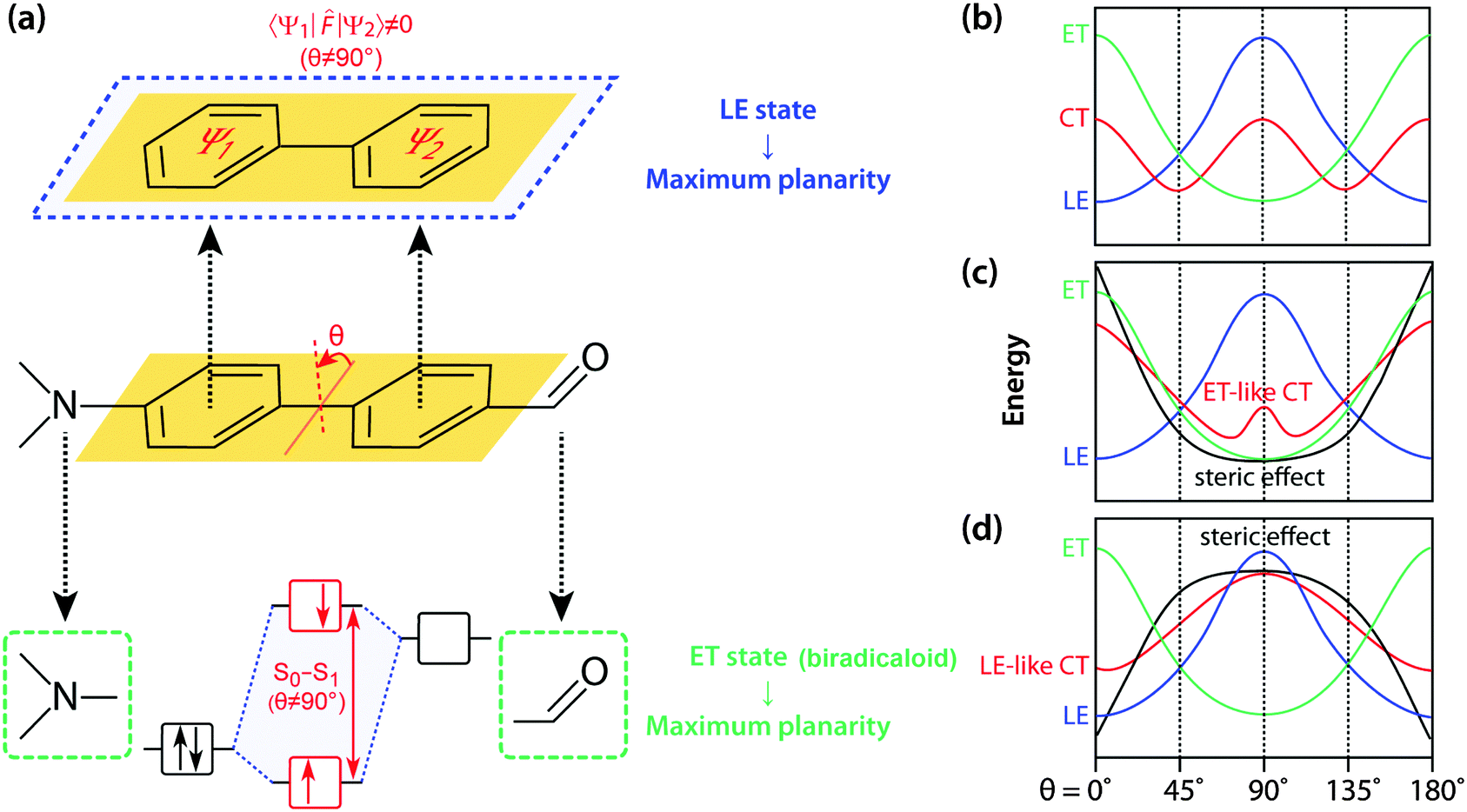 | ||
| Fig. 2 (a) Preferred geometries of 1LE and 1ET states. Schematic energy diagrams of the 1LE (blue), 1ET (green) and 1CT (red) states when: (b) 1LE and 1ET character are comparable to each other, (c) when the steric restriction (black) is introduced to twist the D–A junction (e.g. an alkyl group at ortho-position) and (d) when the D–A junction is made coplanar (e.g. a carbon bridge). | ||
The relatively simple concept that governs TICT opens up a myriad of possibilities for designing novel functional molecules; the competition between LE and ET character of 1CT can easily be manipulated by adjusting several factors, including steric restrictions, polarity environments, and D–A efficacy and strength. It is the sensitivity towards conformation and the surrounding environment that makes D–A systems attractive for various applications. For example, the introduction of steric hindrances such as an alkyl group at the ortho-position of the D–A junction deforms the 1CT surface to locate its minima at a severely twisted conformation (Fig. 2c). In this conformation, the 1LE state is too destabilised to mix efficiently with the 1ET state and therefore, the 1CT minimum becomes a highly twisted and polarised state (i.e. TICT state). On the other hand, when a steric restriction such as a carbon bridge between a donor and an acceptor forces the D–A junction to be coplanar, the 1LE state is sufficiently stabilised so that the 1CT minimum is governed by 1LE character (i.e. coplanar ICT state, Fig. 2d). The polarity of the surrounding environment brings about similar effects with regard to 1ET–1LE mixing and such a polarity effect can be employed to design fluorescent agents for environmental sensing.
Sensing environmental polarity
Because a stronger ET character translates into a more pronounced charge separation, TICT conformations are distinctly induced in polar environments and by substitution of strong donors and acceptors. For example, Nagano and co-workers developed a library of boron-dipyrromethene (BODIPY)-based environmental polarity sensors (1) (Fig. 3), whose donors, when the polarity of an environment surpasses a particular ON/OFF threshold, quench the LE fluorescence of BODIPY via electron-transfer reactions.13 The BODIPY core is generally only moderately sensitive to environmental changes.14 Transitions from fluorescent, but less polarised states dominated by LE states, to non-fluorescent and highly polarised TICT states are also driven by the introduction of steric restrictions; the so called “pretwisting”. Since the transition dipole moment of the 1LE state of BODIPY and polarisation of the 1ET state of (1) are perpendicular to each other, 1ET–1LE mixing does not affect 1LE fluorescence of BODIPY just before the ON/OFF threshold. Exploiting this principle, excited-state dipole moments and the resultant solvatochromisms – the ability to change color in response to changes in solvent polarity – of push–pull biphenyls (2) were modulated simply by imposing torsional restrictions.15 When a LE state cannot be mixed with a 1ET (or 1CT) state for symmetry reasons, the 1LE–1CT interconversion results in fluorescence properties that are highly sensitive to steric environments, i.e., aggregation and rigidification of media, which we recently exemplified in the push–pull binaphthyl (3) shown in Fig. 3.16 However, it is worth mentioning that TICT formations do not always impose weak fluorescence. As comprehensively reviewed by Grabowski et al.,12,17 if the structural relaxation of excited states involves more than one bond simultaneously, the resulting TICT states may acquire sizable fluorescence quantum yields.18 Recently, Yamaguchi’s group reported a striking example of this phenomenon in N-borylated 2,5-diarylpyrroles (4).19 Similarly, we applied this concept by extending the π-conjugation of the widely used dopant for organic light emitting diodes (OLED), 9,10-bis(N,N-diarylamino)anthracene (5); distortion of the donor (N,N-diarylamine) plane destabilises the 1ET state and the resultant mixing with the LE state enhances the oscillator strength of S0←1CT fluorescence.20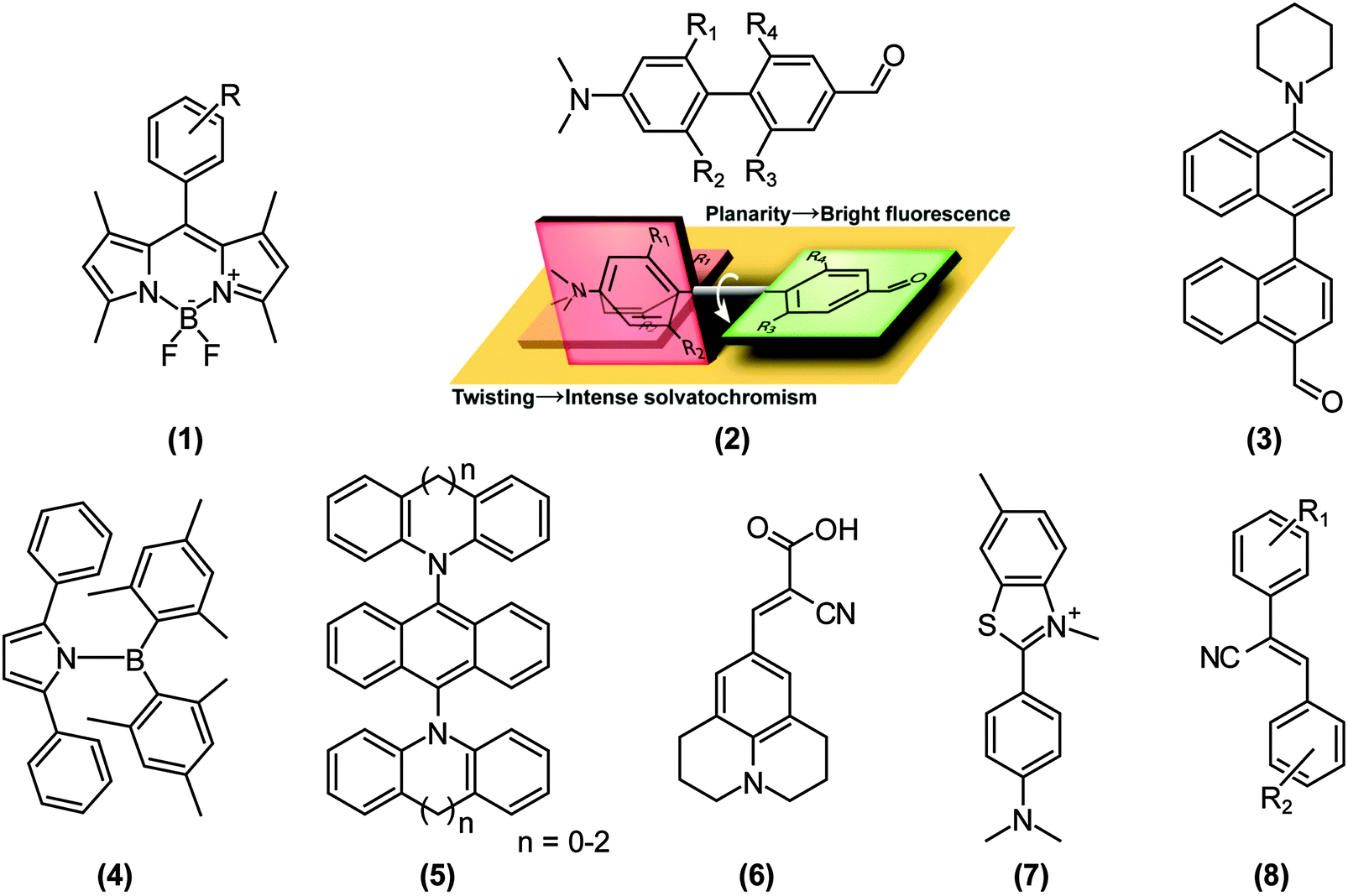 | ||
| Fig. 3 Examples of fluorescent molecules that utilise twisted intramolecular charge transfer (TICT) phenomena, as discussed in this review. (1) 4,4-Difluoro-8-(R-phenyl)-1,3,5,7-tetramethyl-4-bora-3a,4a-diaza-s-indacene; (2) R1,R2,R3,R4-substituted 4-formyl-4′-N,N-dimethylamino-1,10-biphenyl; schematic representation of solvatochromism in push-pull biphenyls; (3) 4-formyl-4′-piperidyl-1,1′-binaphthyl (a dual-mode fluorescent D–π–A-type biaryl dye); (4) 1-[bis(2,4,6-trimethylphenyl)boranyl]-2,5-diphenyl-1H-pyrrole; (5) diarylaminoanthra-cenes; (6) 9-(2-carboxy-2-cyano vinyl)julolidine; (7) thioflavin T (2-[4-(dimethylamino)-phenyl]-3,6-dimethyl-1,3-benzothiazol-3-ium chloride); (8) α-cyanostilbene derivatives. | ||
As mentioned above, torsional restrictions in D–A systems enable precise control over the proportion of the LE and ET character of the D–A system. Therefore, the pretwisting strategy must be effective when designing solvatochromic probes15,21 and photoinduced electron transfer (PET) sensors,22 in spite of the limited number of reports.
Sensing microenvironmental viscosity
Depending on the proportion of ET and LE character, directly connected D–A systems drastically change their fluorescence emission wavelengths, quantum yields, lifetimes, and spectral shapes. These dynamic characteristics are highly useful for fluorescence imaging of microenvironmental viscosity, especially in biological systems. The particular class of TICT-active fluorophores called “molecular rotors” can enhance their fluorescence intensity in sterically restricted environments such as viscous media.23 Viscous environments make TICT-active fluorophores unable to overcome potential barriers against 1LE–TICT interconversions or, in some cases, viscosity hampers internal conversion from the TICT state. Representative molecular rotors (Fig. 3) are 9-(2-carboxy-2-cyanovinyl)julolidine (6) and its analogues,24,25 whose versatilities have been demonstrated not only in molecular biology, such as in peptide–protein interaction studies,26 imaging intracellular microviscosity,27 and amyloid deposit research,28 but also in polymer science,29 contact mechanics,30 and fluid dynamics.31 Thioflavin-T (7) is a well-known TICT-based molecular rotor used in in vitro cell biological and biomedical assays, in particular in drug discovery assays and mechanistical studies with respect to amyloid-related neurodegenerative diseases.32 Though there are other emerging molecular rotors, such as α-cyanostilbene derivatives (8)33 and various PET probes,34 we cannot cover the numerous articles on molecular rotors here, and therefore, we refer the reader to more specialised reviews on molecular rotors.23,24,31,35,36Sensing chemical species
The presence of specific chemical species can also be visualised with TICT-active fluorophores such as RhoNox-1.37 RhoNox-1 (9) is the N-oxide derivative of Rhodamine B (Fig. 4a) and normally exhibits weak fluorescence due to TICT formation, as exemplified in the micrograph of Fig. 4b. However, in the presence of Fe2+ ions, this N-oxide moiety in RhoNox-1 (9) is selectively reduced to yield highly fluorescent Rhodamine B (Fig. 4c). Hirayama et al. determined that the observed fluorescence intensity enhancement directly correlated with the concentration of Fe2+ ions, whilst treatment with various reactive oxygen species did not significantly enhance RhoNox-1’s fluorescence.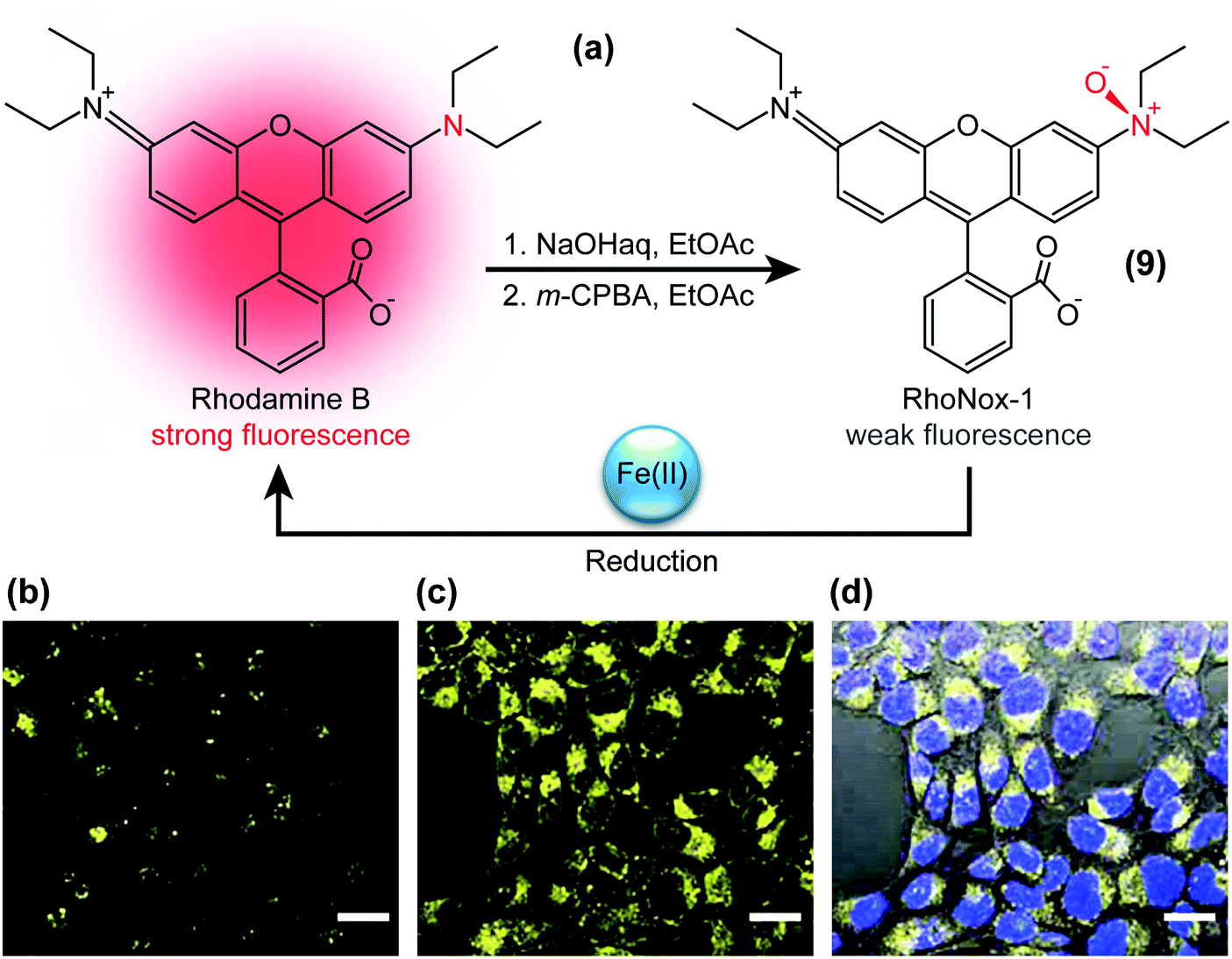 | ||
| Fig. 4 (a) Structure of RhoNox-1 (9), synthesis from Rhodamine B by neutralisation (NaOH) and oxygenation with m-chloroperbenzoic acid (m-CPBA), and mechanism of iron(II)-ion detection. Bottom: Confocal fluorescence microscopy images of Fe(II) detection with 5 μM RhoNox-1 (1 h, 37 °C) in HepG2 cells. (b) Control, (c) preincubated with 100 μM Fe(II) for 30 min, and (d) bright field image merged with (c) and nuclear staining (Hoechst 3334) fluorescence micrographs. Bar: 20 μm. Adapted from ref. 37 with permission from the Royal Society of Chemistry ©2013. | ||
In another approach, Xie and co-workers developed “turn-on” CN− probes (10a–c; Fig. 5a) by introducing dicyanovinyl units at sterically demanding positions of large π frameworks, thereby forcing the moieties to twist out of the anthryl plane.38 On such a highly pretwisted D–A system, the ET character becomes dominant and the system’s fluorescence is severely quenched through TICT formation (Fig. 2c). Nucleophilic addition of CN− to dicyanovinyl groups disables their electron-accepting abilities, and fluorescence is thus drastically and concentration-dependently enhanced (Fig. 5b). In these “turn-on” type fluorescent probes, the strength of either donors or acceptors is determined through chemical reaction with the analyte. Similar strategies, by combining bulky diphenylamino and 9-anthryl units or in a system through H-bonding and charging of aromatic building blocks, have also been adopted in the development of hydrazine (H2NNH2) sensing probes.39,40
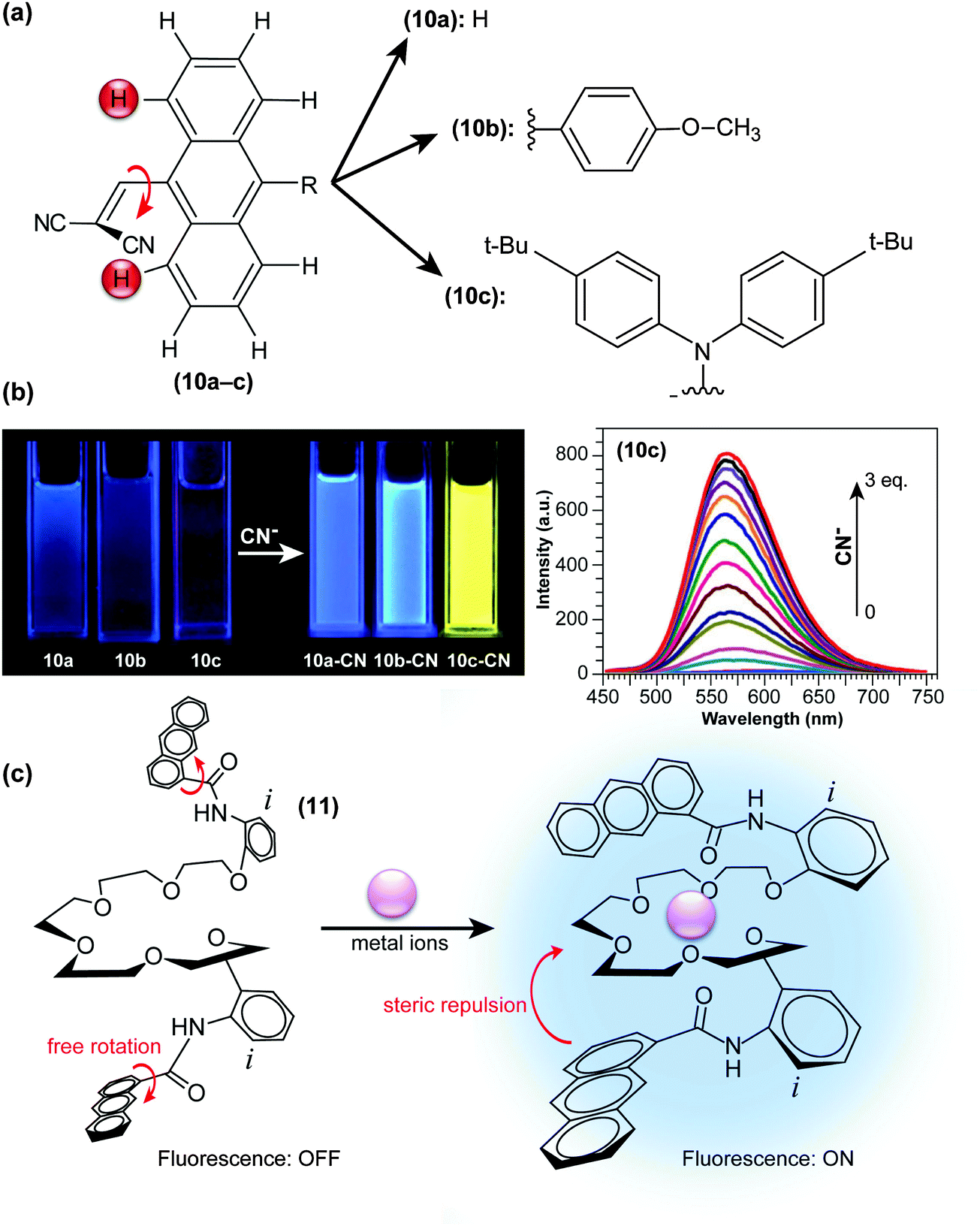 | ||
| Fig. 5 (a) Chemical structures of “turn-on” CN− probes (9-dicyanovinyl anthracene) with various functional groups (10a–10c). (b) Fluorescence emission of 40 μM of compounds 10a–c under UV excitation with a portable lamp in the absence and presence of 3 eq. CN−. Right: Fluorescence changes upon addition of CN− to 10c (20 μM, λex = 396 nm) in CH2Cl2. (c) Proposed structural change of N,N-(ethylenedioxybis(3-oxapentamethyleneoxy-2-phenyl))-bis(1-anthracenecarboxamide) in response to metal ions in the ground state. Adapted from ref. 38, 42 with permission from the Royal Society of Chemistry ©2013 and the Japan Society for Analytical Chemistry ©2009. | ||
Nakamura and co-workers adopted a significantly different approach to detect specific chemical species. They discovered that anthracene anilide derivatives undergo fluorescence quenching via TICT formation, which can partially be suppressed in the presence of alkaline-earth-metal ions.41 To amplify the sensitivity of anthracene anilides, two anthracene anilides were connected with a linear polyether ((11); Fig. 5c) in order to fixate its conformation and suppress torsional motion around the Ph–NH–CO– bond axis upon complexation with metal ions. As a consequence, (11) exhibited a 50–70-fold increase in its fluorescence intensity by the addition of Ca2+, Sr2+ or Ba2+ ions.42 This strategy, wherein complexation prevents TICT formation by imposing steric restriction, is also effective for the detection of neutral molecules such as surfactants.43
Aggregation-induced emission luminogens
Aggregation-induced emission (AIE) luminogens (AIEgens) are a class of fluorophores that display subtle fluorescence when dissolved in good solvents as molecules, but become highly fluorescent when clustered in poor solvents or in solid state as aggregates. Since AIEgens were expected to be promising materials for optoelectronic and bioimaging applications, there has been a vast amount of relevant research, as compiled and comprehensively discussed in recent reviews.44–46 Therefore, in the current review, we will only discuss AIE from the viewpoint of TICT research.Typically, AIE phenomena are predominantly caused by restriction of intramolecular motions (RIM) accompanied by aggregation. Intramolecular motions are also decisive in the photophysics of TICT-active fluorophores; the proportion of LE and ET character at S1 (global or local) minima, potential barriers against LE-TICT interconversions, and non-radiative transition rates of TICT states are all governed by steric environments around fluorophores.12 Consequently, TICT-active fluorophores are ideal candidates for the development of unique AIEgens with exceptional properties (Fig. 6). In fact, tetraphenylethene (TPE; (12)), one of the most famous AIEgen,47 is known to undergo TICT formation in an excited state.12,48,49As mentioned in the previous section, stilbene analogues, such as TPE, often possess strong ET character on their excited state.12 Taking ethylene as the simplest example, its S1 is a weakly polarized π–π* state in a coplanar conformation, whereas in a perpendicular conformation, S1 becomes a p→p orbital electron-transfer (1ET) state, whose excitation energy is equal only to an electronic repulsion experienced by occupying the same orbital. Hence, the excited state of ethylene prefers to be a twisted50 and strongly polarized511ET state. Similarly, when mixing with the 1LE state (or mesomeric interaction between π-subsystems) is ignored and only HOMO–LUMO interaction determines the structure of S1, molecules based on isomerisable double bonds, in principle, should undergo the TICT process. Since mixing from the 1LE state should be weak on the propeller-shaped TPE (12), it is reasonable for TPE to be TICT active. Interestingly, when two phenyl rings of TPE are bridged by ether bonds retaining the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond can freely rotate, the bridged TPE recovers strong fluorescence even in solution state, which implies that the coplanar structure enhances mixing with the 1LE state and thus hampered TICT formation occurs.52 So far, many AIEgens have been designed based on stilbene structures, as exemplified by α-cyanostilbene derivatives (8),33,53–55 triphenylethene carbazole derivatives (13),56 and distyrylanthracene derivatives (14).57 Recently, we developed a novel AIE luminogen (15) based on a simple bis(piperidyl)anthracene structure, but which features unprecedented brightness and is highly pretwisted around D–π junctions.58
C bond can freely rotate, the bridged TPE recovers strong fluorescence even in solution state, which implies that the coplanar structure enhances mixing with the 1LE state and thus hampered TICT formation occurs.52 So far, many AIEgens have been designed based on stilbene structures, as exemplified by α-cyanostilbene derivatives (8),33,53–55 triphenylethene carbazole derivatives (13),56 and distyrylanthracene derivatives (14).57 Recently, we developed a novel AIE luminogen (15) based on a simple bis(piperidyl)anthracene structure, but which features unprecedented brightness and is highly pretwisted around D–π junctions.58
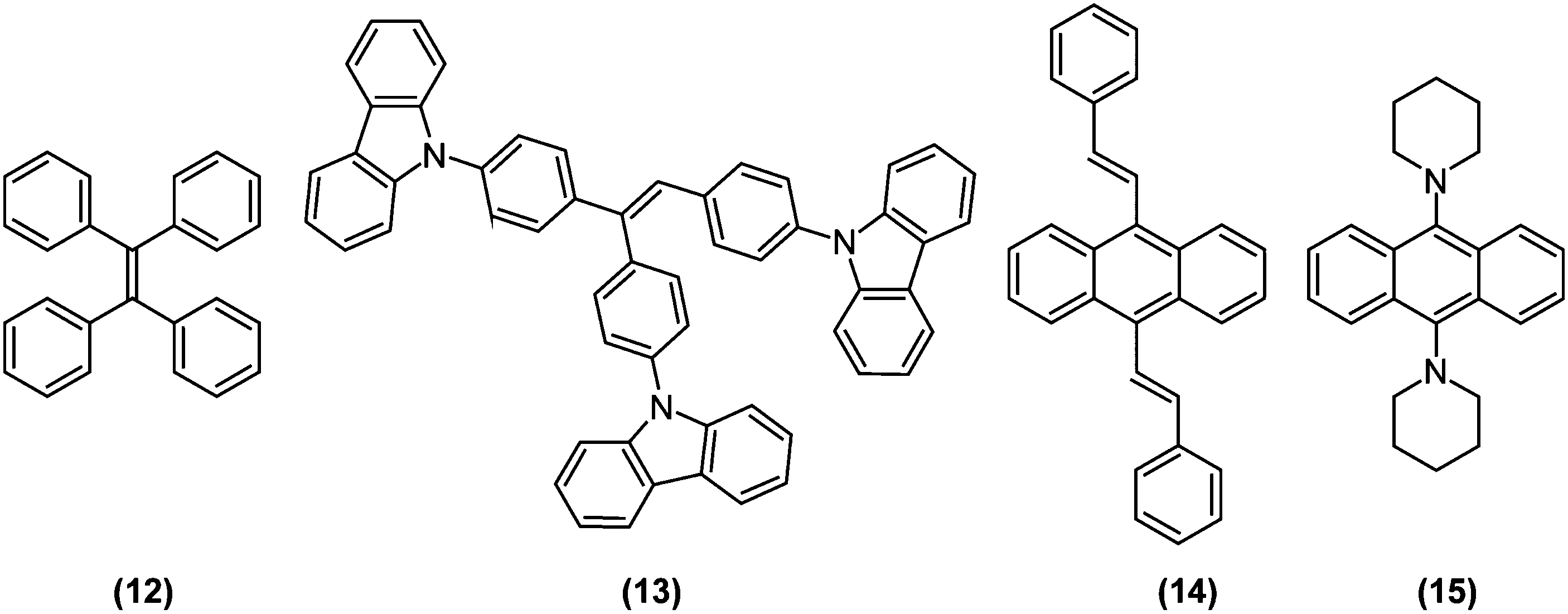 | ||
| Fig. 6 Chemical structures of aggregation-induced emission (AIE) active dyes that utilise twisted intramolecular charge transfer (TICT): (12) tetraphenylethene; (13) 9-(4-(1,2-bis[4-(9H-carbazol-9-yl)phenyl]ethenyl)phenyl)-9H-carbazole; (14) 9,10-distyrylanthracene (DSA); (15) 9,10-bis(piperidyl)anthracene (9,10-BPA). | ||
TICT-active D–A systems also sometimes exhibit AIE and, as mentioned previously, D–A systems based on BODIPY (1) are non-fluorescent in polar environments due to TICT formation, but recover their fluorescence intensity upon aggregation.59 Such a combination of polarity-reduced emissions and AIE was also discovered in push–pull binaphthyl (3) and barbituric acid-functionalised tetraphenylethene derivatives ((16); TPE-HPh-Bar; Fig. 7), and revealed to be useful as optical waveguides.60 TPE-HPh-Bar shows a bright yellow emission at 545 nm upon UV excitation (Fig. 7a), which is quenched above a water (poor solvent) fraction of 60%. Due to the increased solvent polarity, an intensity-reduced (see also Fig. 7b) and red-shifted emission is observed. Above 60%, significant aggregation occurs and the AIE effect dominates the TICT effect with the consequence that the emission rises again (Fig. 7b). It is also noteworthy to mention that several AIEgens based on strong D–A systems, which are presumably TICT-active, have been reported to change their fluorescence properties upon external stimuli such as the exertion of external forces.61,62 These results imply that TICT-active molecules may potentially find applications in the field of fluorescence mechanochromism.
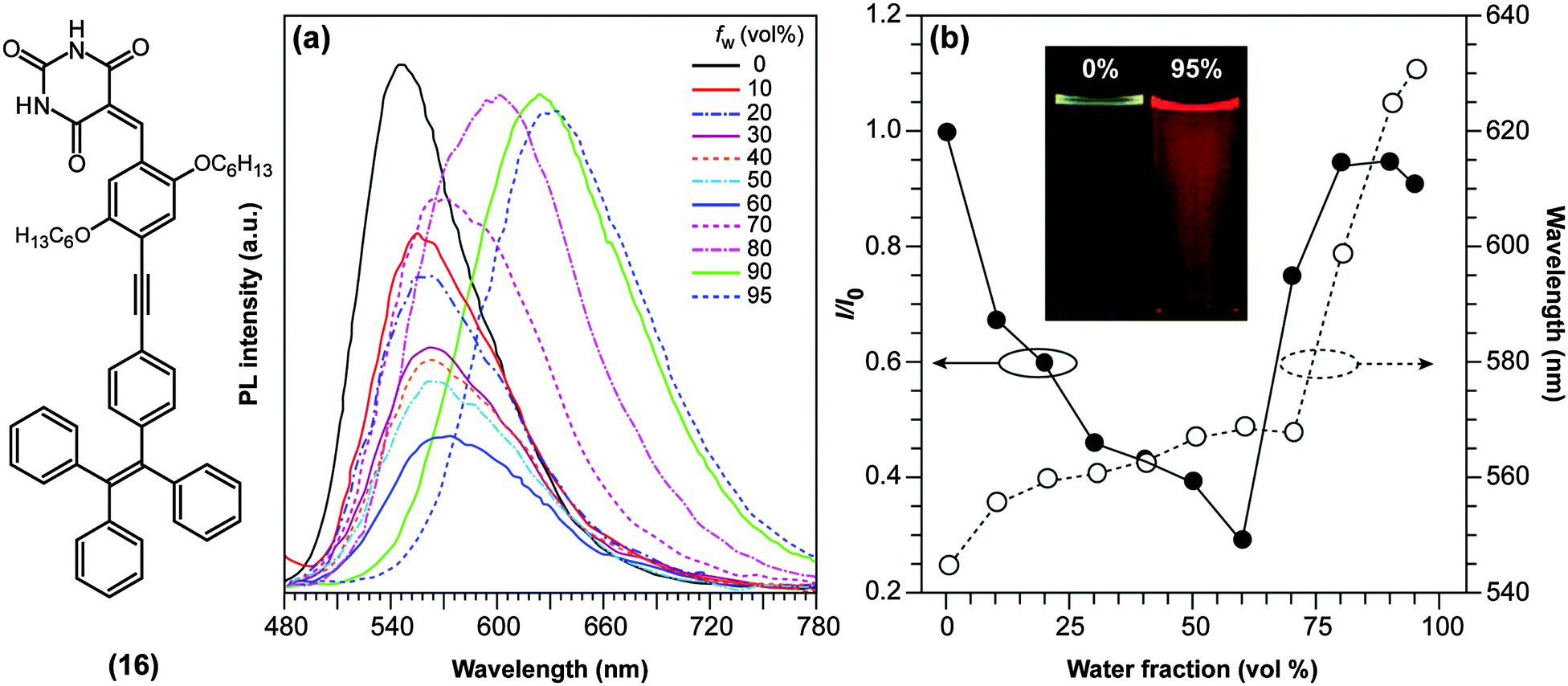 | ||
| Fig. 7 (a) Emission spectra of 10 mM of the red-emissive barbituric acid-functionalised tetraphenylethene derivative ((16); TPE-HPh-Bar; λex = 447 nm) in THF/water mixtures with different water fractions (fw). (b) Plots of relative PL intensities (I/I0) and emission maxima versus the composition of the THF/water mixture of TPE-HPh-Bar. I0 = emission intensity in pure THF solution. Inset: Fluorescence emission of TPE-HPh-Bar in THF/water mixtures (0% and 95%) under UV excitation with a portable lamp. Adapted from ref. 60 with permission from the Royal Society of Chemistry ©2014. | ||
Organic light emitting diodes
The competition between LE character and ET character plays an instrumental role in tuning the electronic structure of dopants. In organic light-emitting diodes (OLED), the recombination of injected holes and electrons produces a so-called charge-transfer (CT) exciton, which decays to generate one photon directly, or relaxes to a low-lying and highly emissive locally excited (LE) exciton. To fully utilise both LE and CT excitons, Ma and co-workers designed the moderately twisted D–A system TPA-PPI ((17); Fig. 8); moderate torsion of TPA-PPI optimises the proportion of 1ET and 1LE character at the 1CT minimum and consequently enables the 1CT state not only to accept CT excitons but also to possess a large oscillator strength of LE excitons.63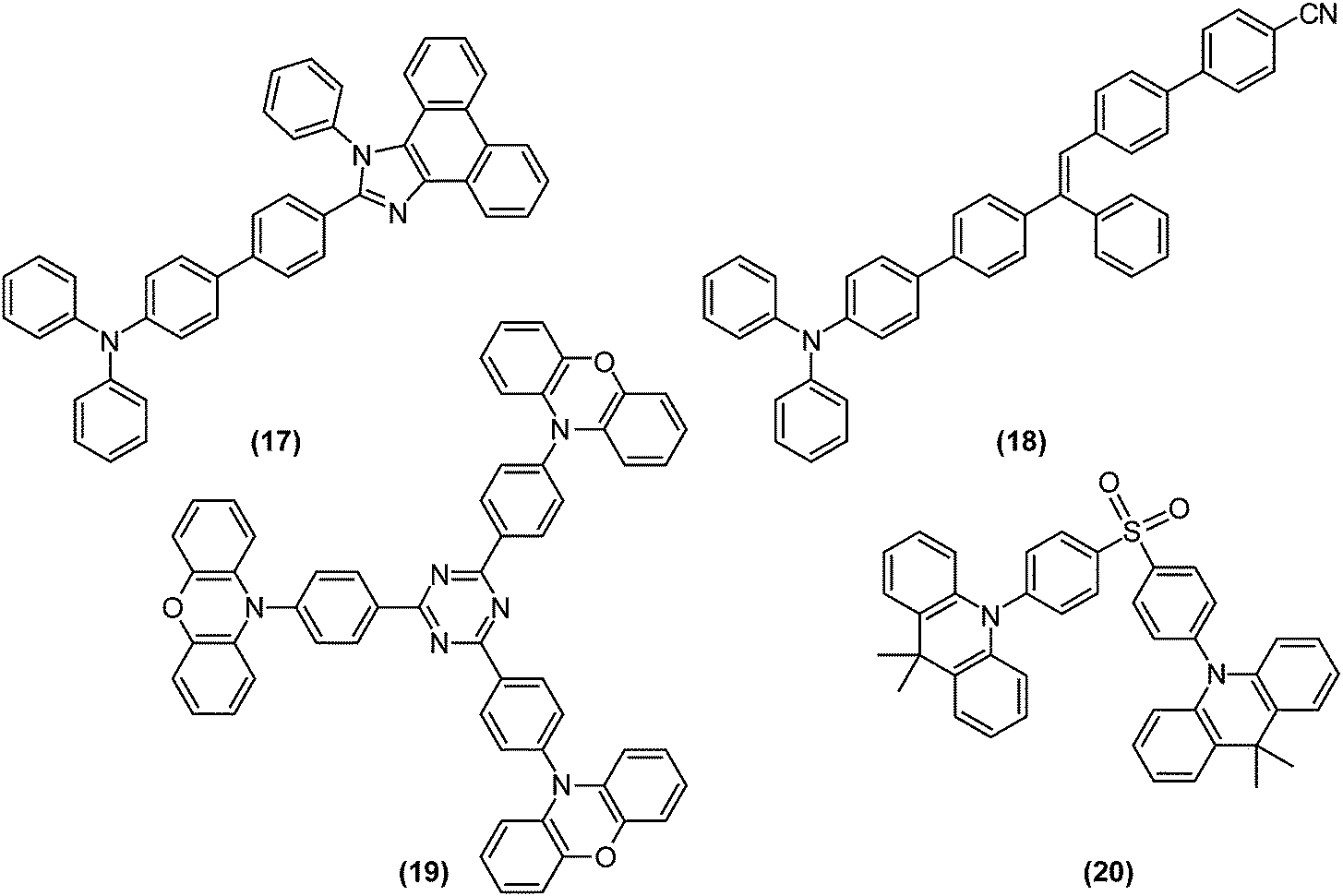 | ||
| Fig. 8 Examples of compounds used in organic light emitting diodes (OLED) whose function is based on twisted intramolecular charge transfer (TICT): (17) N,N-diphenyl-4′-(1-phenyl-1H-phenanthro[9,10-d]imidazol-2-yl)biphenyl-4-amine (TPA-PPI); (18) 4-(4-[(E)-2-(4-[4-(diphenyl-amino)phenyl]phenyl)-2-phenylethenyl]phenyl)benzonitrile (p-TPA-3TPE-p-PhCN); (19) 2,4,6-tri(4-(10H-phenoxazin-10H-yl)phenyl)-1,3,5-triazine (tri-PXZ-TRZ); (20) bis[4-(9,9-dimethyl-9,10-dihydroacridine)phenyl]sulfone (DMAC-DPS). | ||
The strongly mixed state, where the energy levels of LE and CT excitons are closely arranged, is called “hybridised local and charge transfer” (HLCT) state; an important strategy to enhance the electroluminescence efficiencies of OLEDs (Fig. 9a).64,65
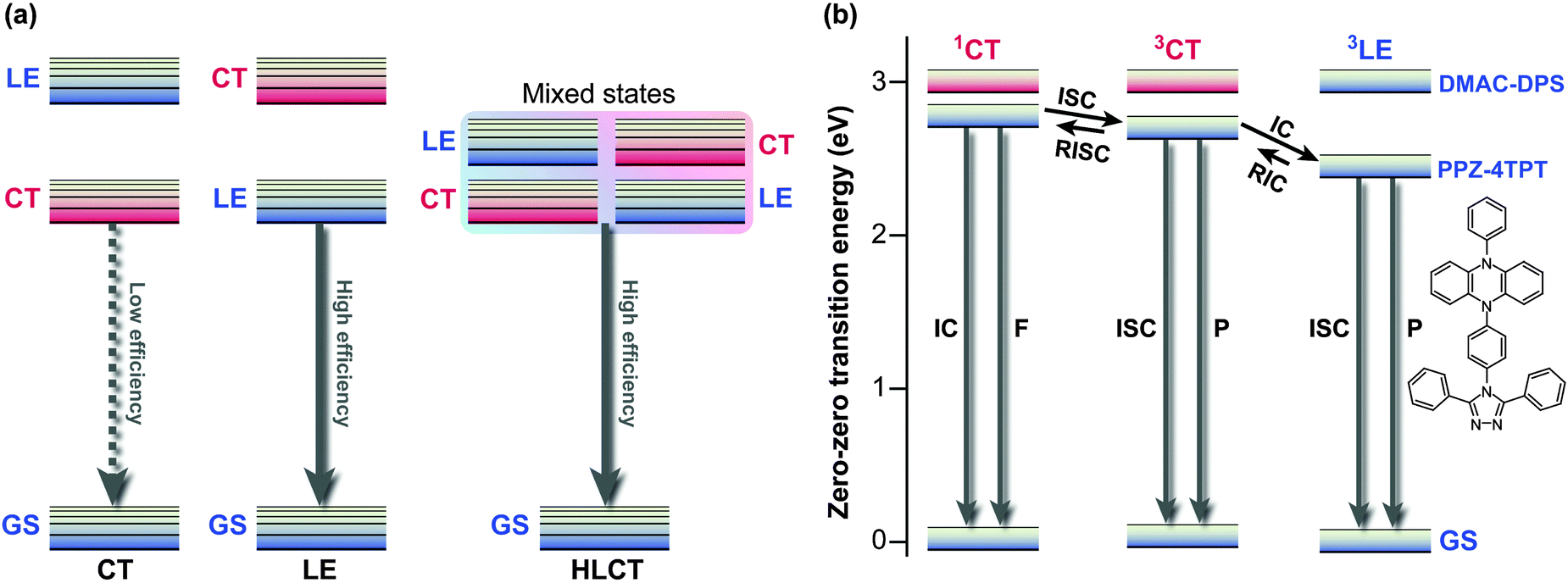 | ||
| Fig. 9 (a) Conceptual illustration of the three possible local excited (LE) and charge transfer (CT) state energy levels in donor–acceptor (D–A) molecules, i.e., emission from CT, LE, and HLCT (hybridised local and charge transfer) states. (b) Energy levels of PPZ–4TPT (4-[4-(5-phenyl-5,10-dihydrophenazine)phenyl]-3,5-diphenyl-1,2,4-triazole) and DMAC–DPS (bis[4-(9,9-dimethyl-9,10-dihydroacridine)phenyl]sulfone) calculated in toluene. F, fluorescence; P, phosphorescence; GS, ground state; IC, internal conversion; RIC, reverse internal conversion; ISC, intersystem crossing; RISC, reverse intersystem crossing. Adapted from ref. 4, 64 with permission from Macmillan Publishers Ltd: Nature Photonics and Wiley-VCH Verlag GmbH & Co. KGaA ©2014. | ||
Electrons and holes possess degrees of freedom not only in orbitals, but also in their spin. Thus, to maximise the quantum efficiency of OLEDs, 25% of singlet excitons and 75% of triplet excitons, generated by arbitrary recombination of carriers, must be utilised as either fluorescence or phosphorescence. Thermally activated delayed fluorescence (TADF) paves the way for solving the problem;66 highly pretwisted D–A systems minimise 1CT–3CT energy gaps due to the absence of electron-exchange interactions. Initially populated 3CT excitons are thus rapidly converted to their singlet counterparts. Therefore, TADF is compatible with TICT-active fluorophores. For example, p-TPA-3TPE-p-PhCN (18), which achieved high external quantum efficiency over its theoretical value when employing the TADF strategy, exhibited weak fluorescence in solution due to TICT formation and was highly emissive in the solid state.67 Recently fluorophores displaying both TADF and AIE were intensively investigated.68,69 Utilising long-wavelength fluorescence of TICT states, colour tuning of TADF emitters were demonstrated on phenoxazine-substituted triphenyl-1,3,5-triazines (19).70
Recently, Adachi et al. achieved fast and efficient blue TADF based on engineering of LE and CT states.4 Since 1CT and 3CT corresponding to blue emissions lie at relatively high energy levels, the lowest triplet state (T1) tends to become a 3LE state and hamper reverse intersystem crossing from 3CT. To solve the problem of blue TADF, they arranged pretwisted D–A junctions at appropriate positions (20) in order to destabilise the 3LE state (Fig. 9b). Taking Fig. 2a as an example, there are many D–A junctions (e.g. N-aryl, aryl–aryl, and aryl-C (formyl) bonds) but only the torsion at the aryl–aryl effectively destabilises 1LE and 3LE states. Thus, the 3LE state of DMAC-DPS (20) was proximal to the 3CT and 1CT states and underwent fast TADF. As a result, the device with (20) offered external quantum efficiencies of up to 19.5%.
Nonlinear optics
Recently, progress in nonlinear optics (NLO) revitalised TICT research. It may sound strange that TICT states, which are formed in excited-state adiabatic reactions, affect NLO properties. However, on highly pretwisted D–A systems, TICT-like states unquestionably exist with near-zero oscillator strengths and consequently affect NLO properties. In 1998, Ratner and co-workers theoretically predicted that highly twisted, but non-perpendicular, D–A systems would exhibit extraordinarily large second-order hyperpolarisabilities due to their minimal LE characters in S1 and large changes in dipole moments induced by photoexcitations.71 This theoretical research had a significant impact on design strategies for NLO materials, because at that time conventional dogma dictated that planar, rigid, and large π-systems were desirable. These nearly perpendicular D–A systems, especially with zwitterionic structures, were named TICTOID chromophores. In 2005, Marks et al. first reported the synthesis of a TICTOID-type dye, i.e., “twisted π-electron system molecular chromophore” TMC-2 ((21); Fig. 10).72 TMC-2 displayed exceptional second-order hyperpolarisability β and electrooptical (EO) response, far surpassing those of existing dyes (μβ = −488![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 × 10−48 esu).72,73 Furthermore, TMC-2 was also shown to possess large third-order hyperpolarisability γ only in real part, equivalent to the nonlinear refractive index.74 The effectiveness of TICTOID dyes is further demonstrated in various molecules, such as cyanine-TICTOID dyads (22)75 and push–pull biphenyls (23).76
000 × 10−48 esu).72,73 Furthermore, TMC-2 was also shown to possess large third-order hyperpolarisability γ only in real part, equivalent to the nonlinear refractive index.74 The effectiveness of TICTOID dyes is further demonstrated in various molecules, such as cyanine-TICTOID dyads (22)75 and push–pull biphenyls (23).76
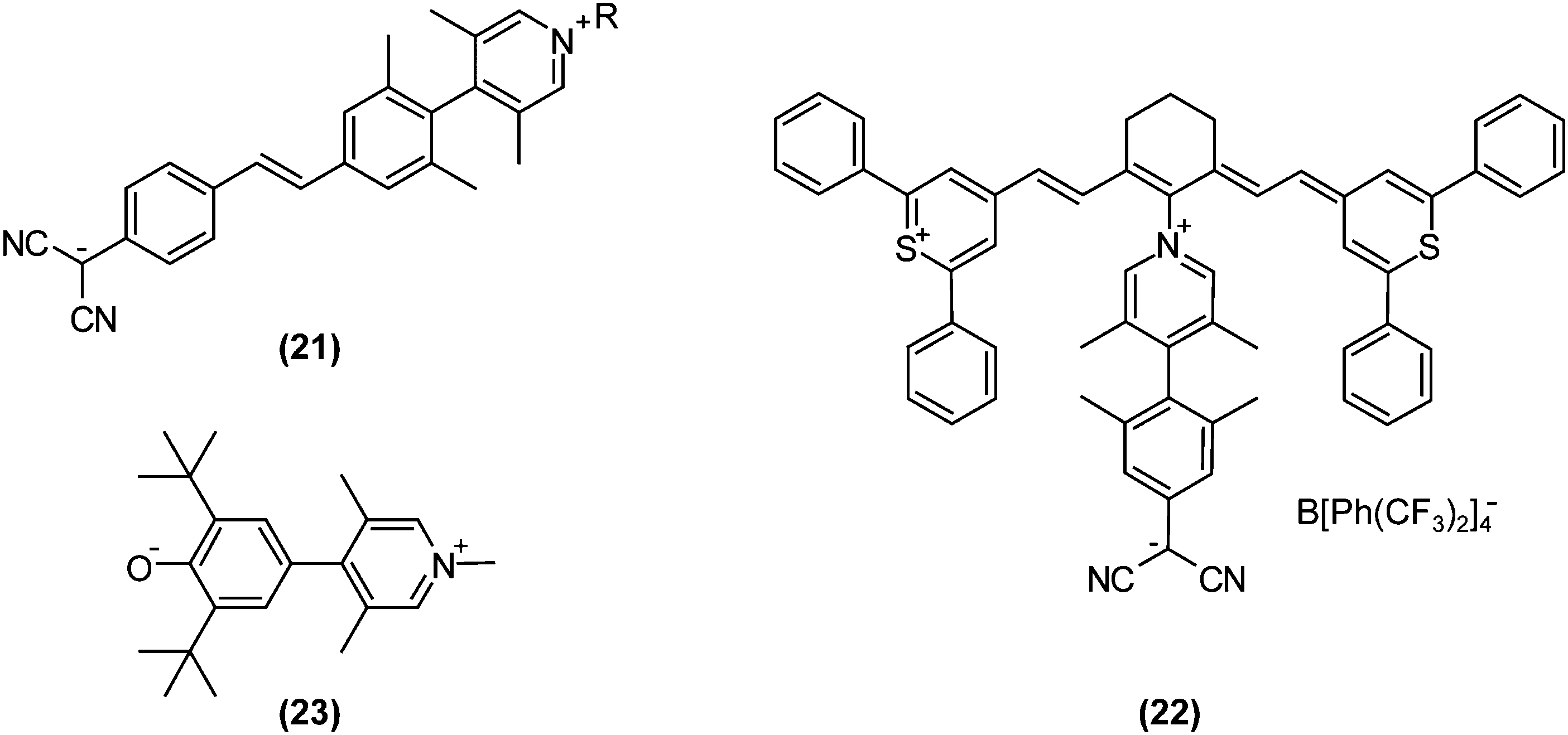 | ||
| Fig. 10 Examples of non-linear optics (NLO) compounds that are based on twisted intramolecular charge transfer (TICT): (21) TMC, twisted π-electron system molecular chromophore: R-substituted 2-(4-[(E)-2-[4-(3,5-dimethylpyridin-4-yl)-3,5-dimethylphenyl]-ethenyl]phenyl)propanedinitrile; (22) Cy-TICT: contains 4-[4-(dicyanomethyl)-2,6-dimethyl-phenyl]-1-[(6E)-2-[(E)-2-(2,6-diphenyl-1λ4-thiopyran-1-ylium-4-yl)ethenyl]-6-[2-(2,6-diphenyl-4H-thiopyran-4-ylidene)ethylidene]cyclohex-1-en-1-yl]-3,5-dimethyl-1λ5-pyridin-1-ylium; (23) 4-(3,5-di-tert-butyl-4-oxidophenyl)-1,3,5-trimethylpyridin-1-ium. | ||
Solar energy conversion with twisted D–A systems
To collect and store solar energy in a stable form, a pair of charges generated by photoexcitation must be collected at electrodes or stored through formation of chemical bonds. Not surprisingly, the long lifetime of photoexcited states enabled by charge separation or triplet state generation plays an instrumental role in solar energy conversions. Typically, charge separations are studied in D–A systems in which the donor and acceptor are separated by an insulating bridge or spacer.Parallel with research and developments in the TICT field, charge separation and transport dynamics were increasingly being studied; also in directly bonded D–A systems. Wasielewski and collaborators examined charge separation dynamics in several twisted D–A systems in which the donor and acceptor were directly bonded or connected by conjugating bridges. As a result, it was discovered that locally excited triplet states could efficiently be generated by charge recombination from highly twisted D–A pairs such as (24)77 and (25),78 as schematically depicted in Fig. 11.
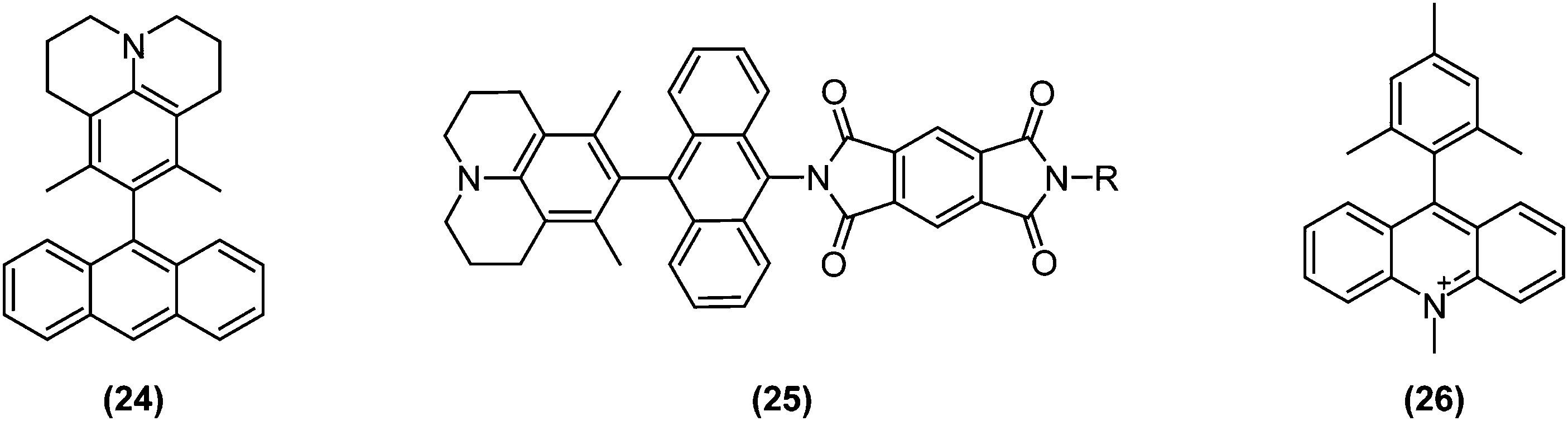 | ||
| Fig. 11 Examples of functional dyes for solar energy conversion based on twisted intramolecular charge transfer (TICT): (24) 3,5-dimethyl-4-(9-anthracenyl)julolidine; (25) 2-(10-(6,8-dimethyl-1-azatricyclo[7.3.1.05,13]trideca-5,7,9(13)-trien-7-yl)anthracen-9-yl)-6-R-1H,2H,3H,5H,6H,7H-pyrrolo[3,4-f]isoindole-1,3,5,7-tetrone; (26) 9-mesityl-10-methylacridinium (Acr+–Mes). | ||
Fukuzumi and co-workers realised efficient generation of a long-lived charge-separation (CS) state with a singly bonded D–A pair, i.e., 9-mesityl-10-methylacridinium (Acr+–Mes; (26)). Severe steric hindrance in Acr+–Mes made the ET character advantageous, and thereby Acr+–Mes kept its perpendicular conformation even in the excited state.79 Furthermore, Acr+–Mes was shown to be active in several photocatalytic reactions.80,81 For instance, Acr+–Mes acts as an efficient photocatalyst in the selective bromination of aromatic hydrocarbons such as 1,2,4-trimethoxybenzene (TMB), with aqueous HBr as a Br source and O2 as an oxidant under visible light irradiation according to:
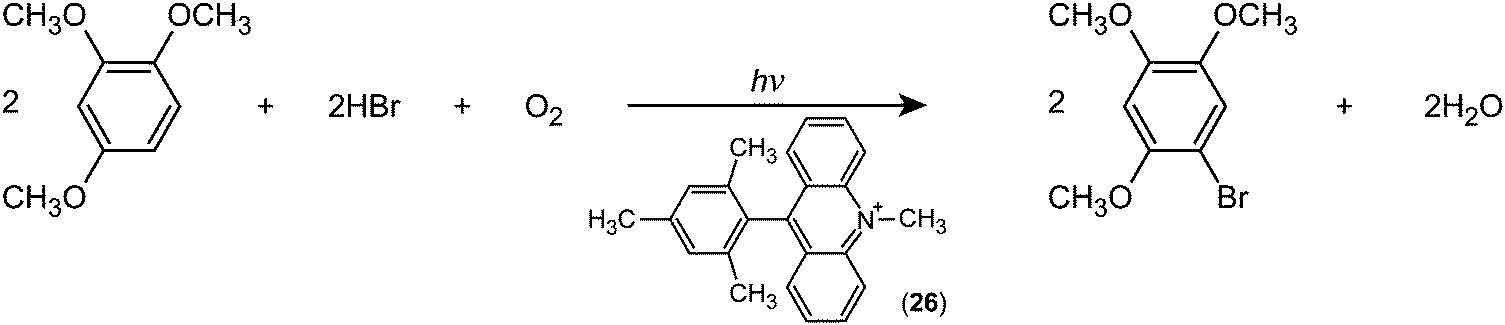 | (1) |
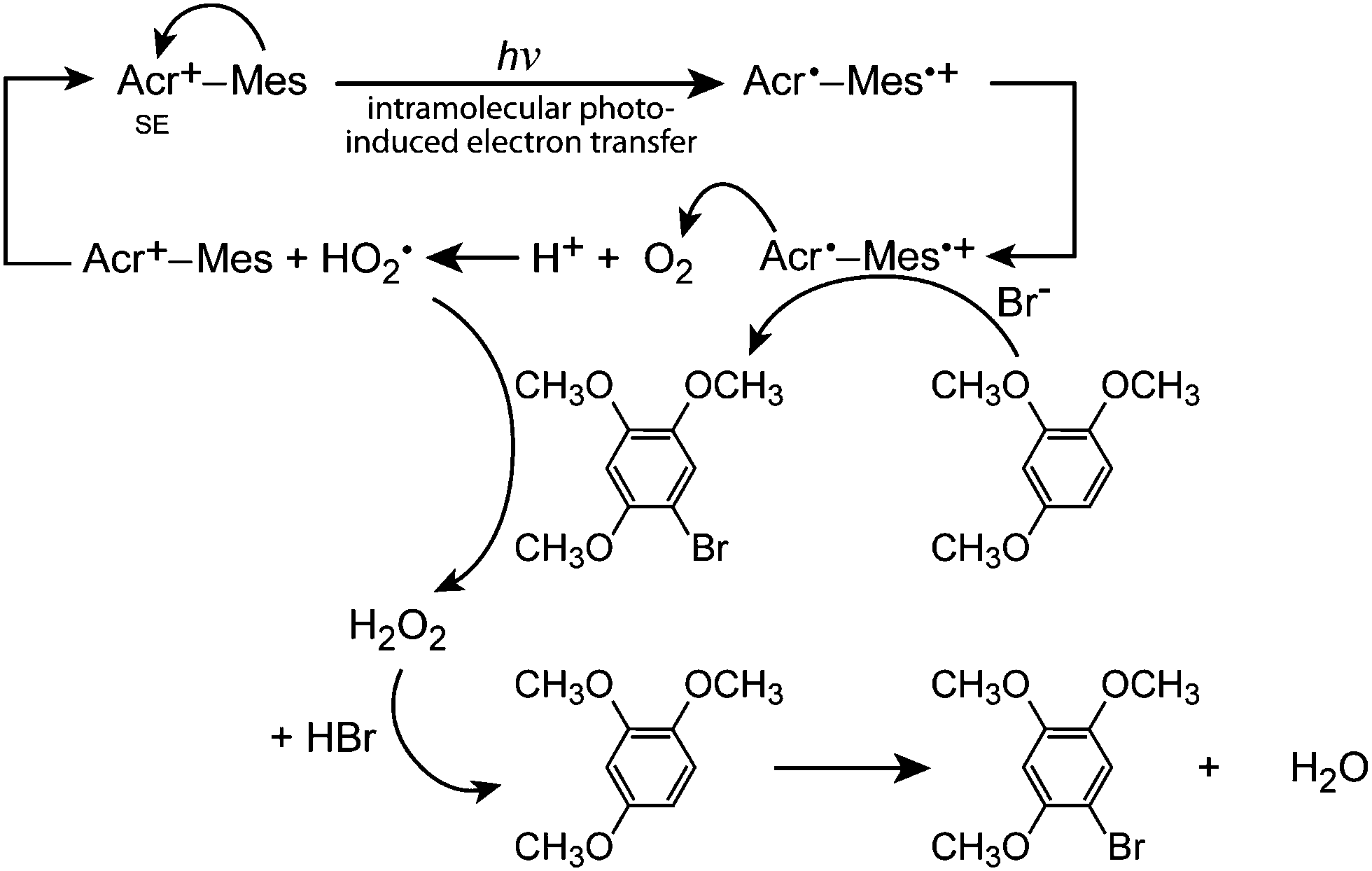 | ||
| Fig. 12 Schematic representation of the photocatalytic bromination of 1,2,4-trimethoxybenzene (TMB) with 9-mesityl-10-methylacridinium (Acr+–Mes) as a catalyst. SE: Singlet excited state. | ||
Conclusions
The foregoing examples have illustrated how researchers with various backgrounds have utilised the TICT phenomenon for purposes ranging from reporter assays and bioimaging to energy harvesting and display/lighting techniques. These examples also illustrate the strength of TICT, namely its tunability when donor, acceptor, and pendant groups are cleverly engineered. Such application-driven chemistry can only be performed well if the basic photophysical properties of TICT are sufficiently understood, as well as their relation to molecular structure. In this sense, TICT has matured significantly thanks to the large number of spectroscopic and theoretical studies that have been performed over the past decades. Nonetheless, from the perspective of materials chemistry, resolving a number of uncertainties would benefit the robustness of design strategies. For instance, in several molecular structures, the extent of pretwisting required to form the TICT state is unclear, data on excited-state CT equilibria are still incomplete due to the difficulty of discerning excited-state species from fluorescence spectra, the influence and contribution of other transfer effects such as exciplexes remains unclear, and various isomeric substitution effects have not yet been fully elucidated. Furthermore, a pile of theoretical insights accumulated so far have not yet been fully translated into design strategies for novel fluorophores. Therefore, it is important for TICT researchers both from material science and chemical physics to cooperate to fill these gaps for the benefit of developing novel fluorophores. Nonetheless, despite all this and the fact that numerous examples of TICT over a wide range of applications exist, it is comforting to know that only a relatively small number of design principles are involved. The future for TICT is bright!Conflicts of Interest
G. P. C. D. is partially exempted from his duties by BNS to pursue fundamental scientific research. The authors declare no further conflicts of interest.Author contributions
S. S drafted the review. G. P. C. D. and G.-i. K. revised and finalised the manuscript and G. P. C. D. made the figures. All authors approved the final manuscript.References
- H. C. Ishikawa-Ankerhold, R. Ankerhold and G. P. Drummen, Molecules, 2012, 17, 4047–4132 CrossRef CAS PubMed.
- J. Wu, W. Liu, J. Ge, H. Zhang and P. Wang, Chem. Soc. Rev., 2011, 40, 3483–3495 RSC.
- Y. Zheng, C. Tan, G. P. Drummen and Q. Wang, Spectrochim. Acta, Part A, 2012, 96, 387–394 CrossRef CAS PubMed.
- Q. Zhang, B. Li, S. Huang, H. Nomura, H. Tanaka and C. Adachi, Nat. Photonics, 2014, 8, 326–332 CrossRef CAS.
- R. Zamiri and G. P. Drummen, in Concise Encyclopedia of Nanotechnology, ed. B. Kharissov, CRC Press, Boca Raton, 2016 Search PubMed.
- T. Förster, Naturwissenschaften, 1946, 6, 166–175 CrossRef.
- T. Förster, Ann. Phys., 1948, 2, 55–75 CrossRef.
- D. L. Dexter, J. Chem. Phys., 1953, 21, 836–850 CrossRef CAS.
- C. Lodeiro and F. Pina, Coord. Chem. Rev., 2009, 253, 1353–1383 CrossRef CAS.
- J. F. Callan, A. P. de Silva and D. C. Magri, Tetrahedron, 2005, 61, 8551–8588 CrossRef CAS.
- G. P. Drummen, Molecules, 2012, 17, 14067–14090 CrossRef CAS PubMed.
- Z. R. Grabowski, K. Rotkiewicz and W. Rettig, Chem. Rev., 2003, 103, 3899–4032 CrossRef PubMed.
- H. Sunahara, Y. Urano, H. Kojima and T. Nagano, J. Am. Chem. Soc., 2007, 129, 5597–5604 CrossRef CAS PubMed.
- G. P. Drummen, L. C. van Liebergen, J. A. Op den Kamp and J. A. Post, Free Radical Biol. Med., 2002, 33, 473–490 CrossRef CAS PubMed.
- S. Sasaki, Y. Niko, A. S. Klymchenko and G.-i. Konishi, Tetrahedron, 2014, 70, 7551–7559 CrossRef CAS.
- S. Sasaki, Y. Niko, K. Igawa and G.-i. Konishi, RSC Adv., 2014, 4, 33474–33477 RSC.
- Z. R. Grabowski, Pure Appl. Chem., 1992, 64, 1249–1255 CrossRef CAS.
- R. Lapouyade, K. Czeschka, W. Majenz, W. Rettig, E. Gilabert and C. Rulliere, J. Phys. Chem., 1992, 96, 9643–9650 CrossRef CAS.
- T. Taniguchi, J. Wang, S. Irle and S. Yamaguchi, Dalton Trans., 2013, 42, 620–624 RSC.
- S. Sasaki, K. Hattori, K. Igawa and G. Konishi, J. Phys. Chem. A, 2015, 119, 4898–4906 CrossRef CAS PubMed.
- S. Sumalekshmy and K. R. Gopidas, J. Phys. Chem. B, 2004, 108, 3705–3712 CrossRef CAS.
- B. Daly, J. Ling and A. P. de Silva, Chem. Soc. Rev., 2015, 44, 4203–4211 RSC.
- M. A. Haidekker and E. A. Theodorakis, Org. Biomol. Chem., 2007, 5, 1669–1678 CAS.
- M. A. Haidekker and E. A. Theodorakis, J. Biol. Eng., 2010, 4, 11 CrossRef PubMed.
- J. Sutharsan, M. Dakanali, C. C. Capule, M. A. Haidekker, J. Yang and E. A. Theodorakis, ChemMedChem, 2010, 5, 56–60 CrossRef CAS PubMed.
- W. L. Goh, M. Y. Lee, T. L. Joseph, S. T. Quah, C. J. Brown, C. Verma, S. Brenner, F. J. Ghadessy and Y. N. Teo, J. Am. Chem. Soc., 2014, 136, 6159–6162 CrossRef CAS PubMed.
- K. Suhling, J. A. Levitt, P. H. Chung, M. K. Kuimova and G. Yahioglu, J. Visualized Exp., 2012, 2925, DOI:10.3791/2925.
- K. Cao, M. Farahi, M. Dakanali, W. M. Chang, C. J. Sigurdson, E. A. Theodorakis and J. Yang, J. Am. Chem. Soc., 2012, 134, 17338–17341 CrossRef CAS PubMed.
- R. D. Priestley, C. J. Ellison, L. J. Broadbelt and J. M. Torkelson, Science, 2005, 309, 456–459 CrossRef CAS PubMed.
- T. Suhina, B. Weber, C. E. Carpentier, K. Lorincz, P. Schall, D. Bonn and A. M. Brouwer, Angew. Chem., Int. Ed. Engl., 2015, 54, 3688–3691 CrossRef CAS PubMed.
- A. Mustafic, H. M. Huang, E. A. Theodorakis and M. A. Haidekker, J. Fluoresc., 2010, 20, 1087–1098 CrossRef CAS PubMed.
- N. Amdursky, Y. Erez and D. Huppert, Acc. Chem. Res., 2012, 45, 1548–1557 CrossRef CAS PubMed.
- L. Zhu and Y. Zhao, J. Mater. Chem. C, 2013, 1, 1059–1065 RSC.
- T. Liu, X. Liu, D. R. Spring, X. Qian, J. Cui and Z. Xu, Sci. Rep., 2014, 4, 5418 CAS.
- M. K. Kuimova, Chimia, 2012, 66, 159–165 CrossRef CAS PubMed.
- M. K. Kuimova, Phys. Chem. Chem. Phys., 2012, 14, 12671–12686 RSC.
- T. Hirayama, K. Okuda and H. Nagasawa, Chem. Sci., 2013, 4, 1250–1256 RSC.
- B. Chen, Y. Ding, X. Li, W. Zhu, J. P. Hill, K. Ariga and Y. Xie, Chem. Commun., 2013, 49, 10136–10138 RSC.
- B. Chen, X. Sun, X. Li, H. Ågren and Y. Xie, Sens. Actuators, B, 2014, 199, 93–100 CrossRef CAS.
- D. Zhou, Y. Wang, J. Jia, W. Yu, B. Qu, X. Li and X. Sun, Chem. Commun., 2015, 51, 10656–10659 RSC.
- T. Morozumi, T. Anada and H. Nakamura, J. Phys. Chem. B, 2001, 105, 2923–2931 CrossRef CAS.
- J. Kim, T. Morozumi, H. Hiraga and H. Nakamura, Anal. Sci., 2009, 25, 1319–1325 CrossRef CAS PubMed.
- Y. Oka, S. Nakamura, T. Morozumi and H. Nakamura, Talanta, 2010, 82, 1622–1626 CrossRef CAS PubMed.
- Y. Hong, J. W. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361–5388 RSC.
- J. Mei, Y. Hong, J. W. Y. Lam, A. Qin, Y. Tang and B. Z. Tang, Adv. Mater., 2014, 26, 5429–5479 CrossRef CAS PubMed.
- R. T. K. Kwok, C. W. T. Leung, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2015, 44, 4228–4238 RSC.
- Y. Dong, J. W. Y. Lam, A. Qin, J. Liu, Z. Li, B. Z. Tang, J. Sun and H. S. Kwok, Appl. Phys. Lett., 2007, 91, 011111 CrossRef.
- C. L. Schilling and E. F. Hilinski, J. Am. Chem. Soc., 1988, 110, 2296–2298 CrossRef CAS.
- T. Tahara and H.-O. Hamaguchi, Chem. Phys. Lett., 1994, 217, 369–374 CrossRef CAS.
- K. B. Wiberg, C. M. Hadad, J. B. Foresman and W. A. Chupka, J. Phys. Chem., 1992, 96, 10756–10768 CrossRef CAS.
- L. Salem, Acc. Chem. Res., 1979, 12, 87–92 CrossRef CAS.
- J. Shi, N. Chang, C. Li, J. Mei, C. Deng, X. Luo, Z. Liu, Z. Bo, Y. Q. Dong and B. Z. Tang, Chem. Commun., 2012, 48, 10675–10677 RSC.
- Y. Gong, Y. Zhang, W. Z. Yuan, J. Z. Sun and Y. Zhang, J. Phys. Chem. C, 2014, 118, 10998–11005 CAS.
- Y. Li, F. Li, H. Zhang, Z. Xie, W. Xie, H. Xu, B. Li, F. Shen, L. Ye, M. Hanif, D. Ma and Y. Ma, Chem. Commun., 2007, 231–233 RSC.
- R. Wei, Y. He, X. Wang and P. Keller, Macromol. Rapid Commun., 2014, 35, 1571–1577 CrossRef CAS PubMed.
- Z. Yang, Z. Chi, T. Yu, X. Zhang, M. Chen, B. Xu, S. Liu, Y. Zhang and J. Xu, J. Mater. Chem., 2009, 19, 5541–5546 RSC.
- J. He, B. Xu, F. Chen, H. Xia, K. Li, L. Ye and W. Tian, J. Phys. Chem. C, 2009, 113, 9892–9899 CAS.
- S. Sasaki, K. Igawa and G.-i. Konishi, J. Mater. Chem. C, 2015, 3, 5940–5950 RSC.
- R. Hu, E. Lager, A. Aguilar-Aguilar, J. Liu, J. W. Y. Lam, H. H. Y. Sung, I. D. Williams, Y. Zhong, K. S. Wong, E. Peña-Cabrera and B. Z. Tang, J. Phys. Chem. C, 2009, 113, 15845–15853 CAS.
- E. Wang, J. W. Y. Lam, R. Hu, C. Zhang, Y. S. Zhao and B. Z. Tang, J. Mater. Chem. C, 2014, 2, 1801–1807 RSC.
- T. Han, Y. Hong, N. Xie, S. Chen, N. Zhao, E. Zhao, J. W. Y. Lam, H. H. Y. Sung, Y. Dong, B. Tong and B. Z. Tang, J. Mater. Chem. C, 2013, 1, 7314–7320 RSC.
- N. Zhao, Z. Yang, J. W. Y. Lam, H. H. Y. Sung, N. Xie, S. Chen, H. Su, M. Gao, I. D. Williams, K. S. Wong and B. Z. Tang, Chem. Commun., 2012, 48, 8637–8639 RSC.
- W. Li, D. Liu, F. Shen, D. Ma, Z. Wang, T. Feng, Y. Xu, B. Yang and Y. Ma, Adv. Funct. Mater., 2012, 22, 2797–2803 CrossRef CAS.
- W. Li, Y. Pan, L. Yao, H. Liu, S. Zhang, C. Wang, F. Shen, P. Lu, B. Yang and Y. Ma, Adv. Opt. Mater., 2014, 2, 892–901 CrossRef CAS.
- Z. Wang, Y. Feng, S. Zhang, Y. Gao, Z. Gao, Y. Chen, X. Zhang, P. Lu, B. Yang, P. Chen, Y. Ma and S. Liu, Phys. Chem. Chem. Phys., 2014, 16, 20772–20779 RSC.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed.
- J. Li, Y. Jiang, J. Cheng, Y. Zhang, H. Su, J. W. Y. Lam, H. H. Y. Sung, K. S. Wong, H. S. Kwok and B. Z. Tang, Phys. Chem. Chem. Phys., 2015, 17, 1134–1141 RSC.
- S. Xu, T. Liu, Y. Mu, Y.-F. Wang, Z. Chi, C.-C. Lo, S. Liu, Y. Zhang, A. Lien and J. Xu, Angew. Chem., Int. Ed., 2015, 54, 874–878 CrossRef CAS PubMed.
- Z. Xie, C. Chen, S. Xu, J. Li, Y. Zhang, S. Liu, J. Xu and Z. Chi, Angew. Chem., Int. Ed., 2015, 54, 7181–7184 CrossRef CAS PubMed.
- H. Tanaka, K. Shizu, H. Nakanotani and C. Adachi, Chem. Mater., 2013, 25, 3766–3771 CrossRef CAS.
- I. D. L. Albert, T. J. Marks and M. A. Ratner, J. Am. Chem. Soc., 1998, 120, 11174–11181 CrossRef CAS.
- H. Kang, A. Facchetti, P. Zhu, H. Jiang, Y. Yang, E. Cariati, S. Righetto, R. Ugo, C. Zuccaccia, A. Macchioni, C. L. Stern, Z. Liu, S.-T. Ho and T. J. Marks, Angew. Chem., Int. Ed. Engl., 2005, 44, 7922–7925 CrossRef CAS PubMed.
- H. Kang, A. Facchetti, H. Jiang, E. Cariati, S. Righetto, R. Ugo, C. Zuccaccia, A. Macchioni, C. L. Stern, Z. Liu, S.-T. Ho, E. C. Brown, M. A. Ratner and T. J. Marks, J. Am. Chem. Soc., 2007, 129, 3267–3286 CrossRef CAS PubMed.
- G. S. He, J. Zhu, A. Baev, M. Samoć, D. L. Frattarelli, N. Watanabe, A. Facchetti, H. Ågren, T. J. Marks and P. N. Prasad, J. Am. Chem. Soc., 2011, 133, 6675–6680 CrossRef CAS PubMed.
- Y. Shi, A. J. T. Lou, G. S. He, A. Baev, M. T. Swihart, P. N. Prasad and T. J. Marks, J. Am. Chem. Soc., 2015, 137, 4622–4625 CrossRef CAS PubMed.
- A. Boeglin, A. Barsella, H. Chaumeil, E. Ay, J. Rotzler, M. Mayor and A. Fort, in Proc SPIE 7774, Linear and Nonlinear Optics of Organic Materials, 2010, pp. 777408–777410 Search PubMed.
- Z. E. X. Dance, S. M. Mickley, T. M. Wilson, A. B. Ricks, A. M. Scott, M. A. Ratner and M. R. Wasielewski, J. Phys. Chem. A, 2008, 112, 4194–4201 CrossRef CAS PubMed.
- M. T. Colvin, A. B. Ricks, A. M. Scott, D. T. Co and M. R. Wasielewski, J. Phys. Chem. A, 2012, 116, 1923–1930 CrossRef CAS PubMed.
- M. Hoshino, H. Uekusa, A. Tomita, S.-y. Koshihara, T. Sato, S. Nozawa, S.-i. Adachi, K. Ohkubo, H. Kotani and S. Fukuzumi, J. Am. Chem. Soc., 2012, 134, 4569–4572 CrossRef CAS PubMed.
- S. Fukuzumi, Phys. Chem. Chem. Phys., 2008, 10, 2283–2297 RSC.
- K. Ohkubo, K. Mizushima, R. Iwata and S. Fukuzumi, Chem. Sci., 2011, 2, 715–722 RSC.
| This journal is © The Royal Society of Chemistry 2016 |