
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Docking-guided identification of protein hosts for GFP chromophore-like ligands†
Natalia V.
Povarova
a,
Nina G.
Bozhanova
a,
Karen S.
Sarkisyan
a,
Roman
Gritcenko
a,
Mikhail S.
Baranov
ab,
Ilia V.
Yampolsky
ab,
Konstantin A.
Lukyanov
a and
Alexander S.
Mishin
 *a
*a
aShemyakin-Ovchinnikov Institute of Bioorganic Chemistry, Miklukho-Maklaya 16/10, 117997 Moscow, Russia. E-mail: mishin@ibch.ru
bPirogov Russian National Research Medical University, Ostrovitianova 1, Moscow 117997, Russia
First published on 23rd February 2016
Abstract
Synthetic analogs of the Green Fluorescent Protein (GFP) chromophore emerge as promising fluorogenic dyes for labeling in living systems. Here, we report the computational identification of protein hosts capable of binding to and enhancing fluorescence of GFP chromophore derivatives. Automated docking of GFP-like chromophores to over 3000 crystal structures of Escherichia coli proteins available in the Protein Data Bank allowed the identification of a set of candidate proteins. Four of these proteins were tested experimentally in vitro for binding with the GFP chromophore and its red-shifted Kaede chromophore-like analogs. Two proteins were found to possess sub-micromolar affinity for some Kaede-like chromophores and activate fluorescence of these fluorogens.
Introduction
The synthetic analogue of the chromophore of green fluorescent protein (GFP) is about three orders of magnitude dimmer in solution than its natural archetype buried within the protein β-barrel. It is established that this drastic decrease in fluorescence quantum yield is a result of the photoinduced isomerisation of its core.1 The ability to fluoresce could be restored by hindering the isomerisation by means of metal ion complexation,2 tight binding to specifically designed RNA aptamers,3 binding to protein hosts,4,5 introduction of chemical conformational lock,6 or aggregation-induced emission in the solid state.7 The fluorescence increase upon binding to protein hosts is of particular interest as it could be used in imaging and other cell biology applications.1,8 However, the only known protein host4 capable of restoring GFP chromophore fluorescence belongs to the albumin family, abundant plasma proteins with unique ability to bind a wide variety of hydrophobic small-molecule ligands.9 This protein was found as a result of high-throughput screening in vitro, laborious experiments that require significant quantities of tested compounds.An alternative approach – molecular docking – is a potent tool for the identification of interacting pairs of small molecules and protein hosts, and even for the computational design of such pairs from the known binding mode.10 Unfortunately, judging by the vast experimental evidence on the role of the molecular properties of small-molecule ligands in receptor–ligand interaction,11 the GFP chromophore lacks sufficient size, flexibility, and the number of hydrogen-bonding groups to be a good candidate for specific protein binding. Therefore, very accurate docking with full-atom scoring functions and flexible receptor geometry would be necessary to identify protein hosts for the GFP chromophore, increasing computational costs of such a screening. These costs can be lowered if chromophores with auxochromic groups in the core are considered, as these groups increase ligand quality and simplify the search of high-affinity protein–chromophore pairs. But nevertheless, the tendency of docking algorithms to converge towards the lowest interface energy often leaves the specific requirements necessary for the fluorescence recovery out of the equation.
Here, we performed automated docking of GFP-like chromophores to available crystal structures of Escherichia coli proteins and tested selected candidate proteins experimentally.
Experimental section
Cloning, protein expression and purification
Genes corresponding to PDB IDs 3HO2, 1DOS, and 2QRY were PCR-amplified (primers are listed in the ESI† Table S4) from E. coli XL1 Blue genomic DNA and cloned into the pBAD expression vector by self-assembly cloning.12For protein expression E. coli strain BW25113 was used. Expression was induced by 0.1% arabinose at 25 °C for 16 h. Cells were centrifuged, sonicated in PBS pH 7.4 (10 mM phosphate buffer, 137 mM NaCl, 2.7 mM KCl) with the PMSF protease inhibitor (Thermo Fisher Scientific), and then purified using TALON metal-affinity resin (Clontech). Finally, the protein was dialyzed against 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000× volume of PBS pH 7.4 supplied with 5 mM β-mercaptoethanol.
000× volume of PBS pH 7.4 supplied with 5 mM β-mercaptoethanol.
Spectral properties
A Varian Cary 100 UV/VIS Spectrophotometer and a Varian Cary Eclipse Fluorescence spectrophotometer were used to measure absorption and excitation–emission spectra.Fluorescence quantum yields (QYs) of the 3HO2–A12H protein–fluorogen pair (20 μM protein, 0.5 μM chromophore, PBS pH 7.4, 23 °C) were determined by direct comparison with EGFP (QY = 0.6). For unbound chromophores, compound 4c from ref. 13 was used as a reference for QY measurements. The solubility of the chromophores was determined by absorption measurements.
Molecular docking and DFT studies
The B3LYP DFT functional has already been successfully applied to related GFP-based systems,14–16 and it was selected for present studies. Ligands were optimized at the B3LYP/def2-SVP level of theory17 using ORCA 3.0.3 software,18 all obtained geometries had no imaginary frequencies. Time-dependent density functional theory (TDDFT) studies, with Tamm–Dancoff approximation, were performed at the PBE0/def2-TZVP (COSMO: H2O) level of theory. The zeroth order regular approximation (ZORA) in conjugation with the corresponding basis set19 was used for TDDFT calculations to take into account relativistic effects (for A12 and A12H series). RIJCOSX approximation20 was used in order to significantly speed up geometry optimization, computing of analytical Hessian,21 and TD-DFT studies.22The ligand and receptor files for docking with rigid protein geometry were prepared using AutoDockTools23 version 1.5.6. The bounding box for the docking region was calculated automatically using PyMol. Docking was performed using AutoDock Vina as described24 with exhaustiveness set to 20 and the maximum number of collected distinct binding modes set to 20.
Docking with flexible geometry of the ligand-binding pocket was performed using Rosetta software (weekly release 2015.05.57576). The crystal structures, PDB IDs: 1PVS and 3HO2 (the residue A163 was manually changed into the native C) were preminimized using relax application25 with the following flags: -flip_HNQ -no_optH false -relax:constrain_relax_to_start_coords -nstruct 50 -ex1 -ex2 -use_input_sc. The output structures with the lowest total score were used for further calculations. For ligand docking we used the RosettaLigand docking algorithm.26 Three rounds of docking were made for each protein–ligand pair. During the first round, 5000 structures were generated using the transform mover with move_distance = 5 Å and angle = 360°. Fifty structures with the lowest interface_delta score among 2000 structures with the best total score were selected for the second round to generate another 5000 structures (100 from each of the selected) decreasing the move_distance to 1 Å and the angle to 45°. The third round of ligand docking was performed using the best 50 structures of the second round and the transform mover with move_distance = 0.2 Å and angle = 5°.
In vitro fluorescence bead assay
Purified proteins (1 mg ml−1 solution) were immobilized on a 1/100 volume of TALON metal-affinity beads, washed with PBS pH 7.4 and placed in 200 μl chambers with cover glass at the bottom. The chromophores were added to a final concentration of 10 μM from EtOH stock solution. A Leica AF6000 fluorescence microscope (Wetzlar, Germany) was used for imaging with GFP (excitation BP470/40, emission BP525/50) and TxRed (excitation BP560/40, emission BP645/75) filter sets.Results and discussion
Since the high quantum yield of fluorescence of the GFP chromophore is determined mainly by the sterical hindrance of its isomerization, we hypothesized that placing emphasis on the geometrical match between the GFP chromophore and the potential binding pocket within a protein is important. To separate affinity optimization and geometrical matching of the core GFP chromophore, we devised a two-step docking approach. First, we selected potential hosts for the GFP chromophore. Second, we docked a wider library of GFP-like chromophores to these protein hosts.We applied molecular docking of the GFP chromophore to a number (over 3000) of crystal structures of E. coli proteins available in the Protein Data Bank with resolution better than 2.0 Å and the chain length shorter than 500 amino acid residues. We chose one of the fastest available docking tools24 and performed docking in a blind and automatic manner: ligands were stripped out from PDB files and only the first protein chain was examined. This massive docking analysis allowed us to select top-scoring structures that can putatively bind the GFP-like chromophore (Table S1, ESI†). We then performed molecular docking of larger GFP-like chromophore derivatives – so called Kaede-like chromophores27 – against top 500 structures from previous round of docking. As a result, chromophores A5, A12, A12H, A24, A26, A27, and A28 (ESI† Methods) were selected for further tests. From the candidate protein list, we succeeded in cloning and expression of 4 proteins with a high (Table 1) GFP or Kaede docking rank: (here and further named by PDB IDs) 3HO2 (β-ketoacyl-acyl-carrier-protein synthase II), 1DOS (fructose-bisphosphate aldolase), 2QRY (thiamine binding protein), and 1PVS (3-methyladenine glycosylase II).
Purified proteins were immobilized on beads and examined under a fluorescence microscope. However, no fluorescence increase was observed upon incubation with the 100 μM GFP chromophore. At the same time, the addition of chromophores A5, A12, A12H, and A24 resulted in a considerable increase of bead fluorescence (Fig. 1).
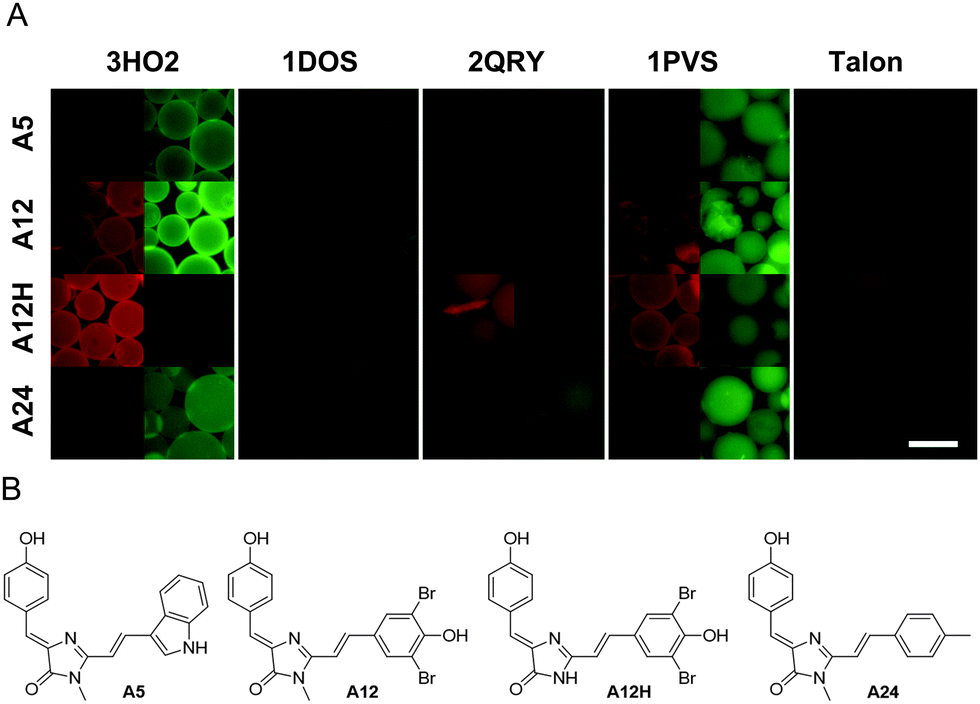 | ||
| Fig. 1 Bead-based fluorescence assay. (A) Montage of fluorescence microscopy images of bead-immobilized proteins in chromophore solutions. Rows: chromophores; columns: proteins, with TxRed and GFP filter sets interleaved. Talon designates beads with no immobilized protein (negative control). Scale bar – 100 μm. (B) Structures of the chromophores mentioned in panel A. | ||
Next, we studied spectral changes upon mixing of the chosen chromophore–protein pairs in solution. Some of the tested pairs exhibited submicromolar Kd (Fig. 2A). Interestingly, in the case of binding of chromophores A5, A12, and A24 to 3HO2 protein in solution, the fluorescence intensity increase was minor, less than 2-fold, under protein or chromophore saturating conditions (Fig. 2A). Thus, the corresponding signal increase in the bead-based assay can probably be attributed to chromophore accumulation uncoupled from the increase in fluorescence quantum yield. In contrast, A12H and A12 showed a strong fluorescence increase upon binding to 3HO2 and 1PVS in solution (Fig. 2B) demonstrating truly fluorogenic behavior. Indeed, the binding of the chromophore to the protein host resulted in two orders of magnitude increase in fluorescence quantum yield of the chromophore (A12H–3HO2 pair: QY increase from 0.0003 to 0.052 and A12–3HO2 pair: QY increase from 0.0005 to 0.026). The observed fluorescence enhancement in the A12H–3HO2 fluorogen–protein pair is stronger than that for the human serum albumin protein host bound to GFP-like chromophore analogs identified by high-throughput protein screening5 and further directed chemical modifications.4
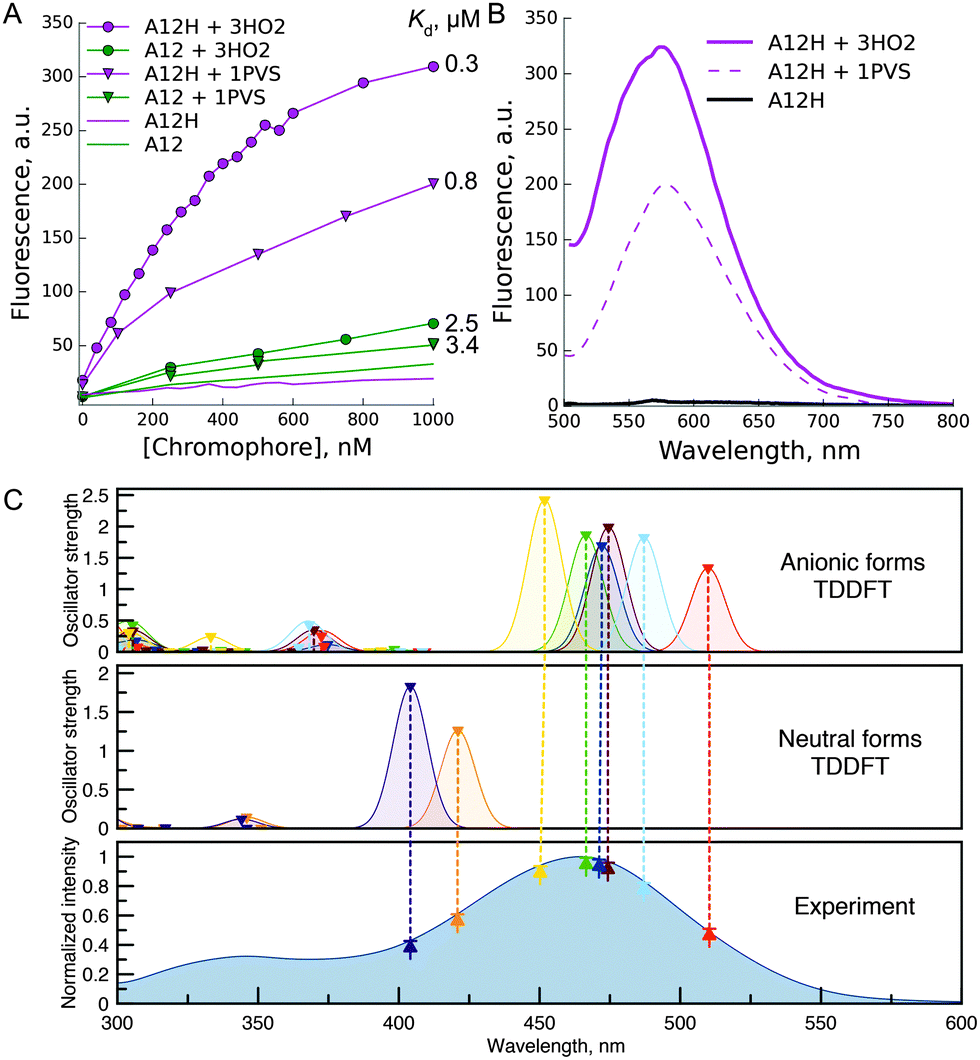 | ||
| Fig. 2 Fluorescence increase in solution upon binding of the chromophore to the protein host. (A) Representative titration curve of 1 μM 3HO2 or 1PVS protein solutions with chromophores A12 or A12H; (B) emission response of the A12H chromophore upon binding to protein hosts 3HO2 or 1PVS; (C) TDDFT studies of the A12H chromophore. Dashed lines show the correspondence between the experimental absorption spectrum and theoretically obtained S0 → S1 transitions at the ZORA-PBE0/def2-TZVP (COSMO: H2O) level of theory. See ESI† (Fig. S3) for the description of the neutral and anionic species, which were taken into account. | ||
The fluorescence of A12H in the complex with proteins 3HO2 and 1PVS is spectrally similar to the dim fluorescence of the free chromophore in water solution (Fig. 2B) and is likely to arise from one of the multiple possible anionic states. As reported previously, spectral shifts of the maxima of Kaede-like chromophores are determined by the electron-donating and withdrawing properties of the aryl substituent at the ethylene double bond.28,29 Thus, the smallest bathochromic shifts were observed (Table S2, ESI†) for the neutral substituent (A24 with tolyl group, absorbance maxima 436 nm) while the hydroxy-phenyl or indolyl shifted the absorption and emission maxima for 40 nm. The absence of the alkyl group in the first position of the imidazolone ring leads to the small hypsochromic shift (15 nm for A12H in comparison to the A12) being in good agreement with the literature30 and resulting from the better electron donating properties of the alkyl group. Deprotonation of the hydroxyl group in all the compounds resulted in ∼80 nm red-shifts of absorption and emission maxima. The process is characterized by the pKa of 7–8, and it can take place in water buffer solutions with neutral pH.28,29 This suggests that in some cases (A12 and A12H) in protein complexes we might have observed the emission of the anionic form.
In order to further investigate this point, we performed time-dependent density functional theory (TDDFT) calculations on the free chromophores. According to these studies, the main peak in the experimental absorption spectrum of the A12H chromophore (Fig. 2C), at 465 nm, along with the 485 nm excitation peak in the complex with the 3HO2 protein host can be attributed to S0 → S1 excitations of different anionic species (Fig. S3, ESI†). Similar red shifting of anionic species in comparison to neutral species was found for all the studied chromophores (see ESI†, Fig. S4–S6).
In order to address the observed strong fluorescence increase in pairs 3HO2/A12H and 1PVS/A12H despite the inconsistencies with ranking in rigid-residue docking, we performed docking analysis with flexible residues and the full-atom Rosetta scoring function. High-resolution docking resulted in highly converged docking poses of the A12H chromophore within a tight binding pocket of the 3HO2 host (Fig. 3B) that was present in top-50 out of 5000 last-round-optimized structures (Fig. S2, ESI†). Also, one of the anionic forms of A12H provided a slightly better overall docking score than the neutral one.
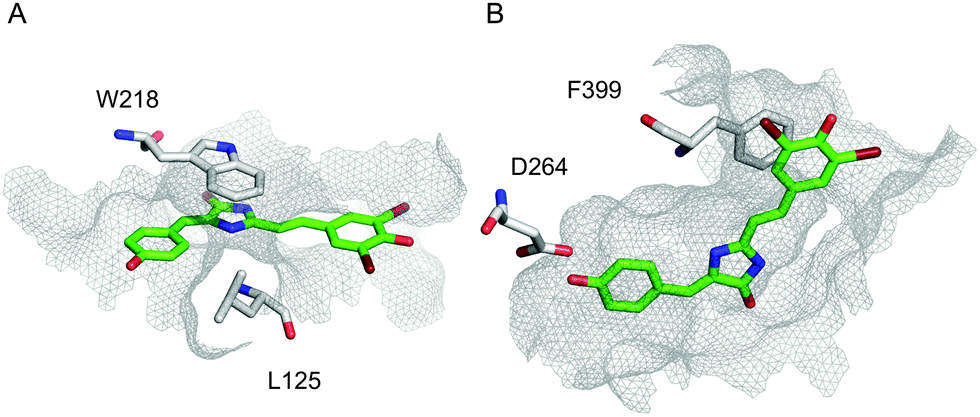 | ||
| Fig. 3 High-resolution docking. Top-scoring docking modes for A12H–1PVS (A), and A12H–3HO2 (B) interactions, correspondingly. Residues with significant contribution are shown, A12H is shown in green, and the surface of the binding pocket is outlined in grey mesh. | ||
Among amino acid residues of 3HO2 with significant contribution to the ligand docking (ΔΔG < −1) stacking interactions of F399 (PDB numbering) with the chromophore occurred in all cases (A12H, A12, A24, A5) and might have been the major factor for hindering of the chromophore isomerization. Importantly, it was found that the fluorescence recovery of the GFP-like chromophores by Spinach RNA aptamer binding occurs by stacking interactions too.31,32 Additional stacking and H-bonding interactions are present in some cases (Table S3, ESI†). Apparently, the convergence of docking modes should be used as an additional metric for the computational design of fluorogen chromophore–protein pairs. It should be noted that both 3HO2 and 1PVS showed a fluorescence increase in bead assay with four out of seven chromophores selected by initial computational screening, therefore less computationally extensive rigid-residue docking could indeed be applied for narrowing down the search of suitable ligand candidates before accurate high-resolution analysis.
Conclusions
For the first time we applied molecular docking for the identification of the candidate protein host for binding of GFP chromophore analogs. Two out of four proteins that were cloned and tested in vitro (3HO2, 1PVS) showed fluorogenic behavior and sub-micromolar Kd towards Kaede-like chromophores.We believe that the present approach could be further applied in various fields. First, the fluorescence increase of fluorogenic dyes upon binding to a protein host provides an easy and reliable way of the experimental verification of various docking protocols. Second, bacterial proteins found in this work or analogous protein hosts and their mutant variants can potentially be used as fluorescent tags in heterologous expression models (e.g., mammalian cells), similar to antibody-based Fluorogen Activating Proteins8 and the recently developed Yellow Fluorescence-Activating and absorption-Shifting Tag (Y-FAST).33 Finally, this method can provide a way to visualize native endogenous proteins (e.g., 3HO2) in living cells.
Acknowledgements
This work was supported by the Russian Foundation for Basic Research (grant 15-33-20518-mol-a-ved) and by the Molecular and Cell Biology program of Russian Academy of Sciences. Experiments were partially carried out using the equipment provided by IBCH core facility (CKP IBCH).References
- C. L. Walker, K. A. Lukyanov, I. V. Yampolsky, A. S. Mishin, A. S. Bommarius, A. M. Duraj-Thatte, B. Azizi, L. M. Tolbert and K. M. Solntsev, Curr. Opin. Chem. Biol., 2015, 27, 64–74 CrossRef CAS PubMed.
- A. Baldridge, K. M. Solntsev, C. Song, T. Tanioka, J. Kowalik, K. Hardcastle and L. M. Tolbert, Chem. Commun., 2010, 46, 5686–5688 RSC.
- J. S. Paige, K. Y. Wu and S. R. Jaffrey, Science, 2011, 333, 642–646 CrossRef CAS PubMed.
- A. Baldridge, S. Feng, Y.-T. Chang and L. M. Tolbert, ACS Comb. Sci., 2011, 13, 214–217 CrossRef CAS PubMed.
- J.-S. Lee, A. Baldridge, S. Feng, Y. SiQiang, Y. K. Kim, L. M. Tolbert and Y.-T. Chang, ACS Comb. Sci., 2011, 13, 32–38 CrossRef CAS PubMed.
- M. S. Baranov, K. A. Lukyanov, A. O. Borissova, J. Shamir, D. Kosenkov, L. V. Slipchenko, L. M. Tolbert, I. V. Yampolsky and K. M. Solntsev, J. Am. Chem. Soc., 2012, 134, 6025–6032 CrossRef CAS PubMed.
- S. Fery-Forgues, S. Veesler, W. B. Fellows, L. M. Tolbert and K. M. Solntsev, Langmuir, 2013, 29, 14718–14727 CrossRef CAS PubMed.
- C. Szent-Gyorgyi, B. F. Schmidt, B. A. Schmidt, Y. Creeger, G. W. Fisher, K. L. Zakel, S. Adler, J. A. J. Fitzpatrick, C. A. Woolford, Q. Yan, K. V. Vasilev, P. B. Berget, M. P. Bruchez, J. W. Jarvik and A. Waggoner, Nat. Biotechnol., 2008, 26, 235–240 CrossRef CAS PubMed.
- P. A. Zunszain, J. Ghuman, T. Komatsu, E. Tsuchida and S. Curry, BMC Struct. Biol., 2003, 3, 6 CrossRef PubMed.
- C. E. Tinberg, S. D. Khare, J. Dou, L. Doyle, J. W. Nelson, A. Schena, W. Jankowski, C. G. Kalodimos, K. Johnsson, B. L. Stoddard and D. Baker, Nature, 2013, 501, 212–216 CrossRef CAS PubMed.
- W. P. Walters, J. Green, J. R. Weiss and M. A. Murcko, J. Med. Chem., 2011, 54, 6405–6416 CrossRef CAS PubMed.
- A. Matsumoto and T. Q. Itoh, Biotechniques, 2011, 51, 55–56 CrossRef CAS PubMed.
- M. S. Baranov, K. M. Solntsev, N. S. Baleeva, A. S. Mishin, S. A. Lukyanov, K. A. Lukyanov and I. V. Yampolsky, Chem. – Eur. J., 2014, 41, 13234–13241 CrossRef PubMed.
- A. K. Das, J.-Y. Hasegawa, T. Miyahara, M. Ehara and H. Nakatsuji, J. Comput. Chem., 2003, 24, 1421–1431 CrossRef CAS PubMed.
- Y. Ma, Q. Sun, Z. Li, J.-G. Yu and S. C. Smith, J. Phys. Chem. B, 2012, 116, 1426–1436 CrossRef CAS PubMed.
- M. Wanko, P. García-Risueño and A. Rubio, Phys. Status Solidi B, 2012, 249, 392–400 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CrossRef CAS.
- D. A. Pantazis, X.-Y. Chen, C. R. Landis and F. Neese, J. Chem. Theory Comput., 2008, 4, 908–919 CrossRef CAS PubMed.
- F. Neese, F. Wennmohs, A. Hansen and U. Becker, Chem. Phys., 2009, 356, 98–109 CrossRef CAS.
- D. Bykov, T. Petrenko, R. Izsák, S. Kossmann, U. Becker, E. Valeev and F. Neese, Mol. Phys., 2015, 113, 1961–1977 CrossRef CAS.
- T. Petrenko, S. Kossmann and F. Neese, J. Chem. Phys., 2011, 134, 054116 CrossRef PubMed.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 30, 2785–2791 CrossRef CAS PubMed.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455–461 CAS.
- L. G. Nivón, R. Moretti and D. Baker, PLoS One, 2013, 8, e59004 Search PubMed.
- S. DeLuca, K. Khar and J. Meiler, PLoS One, 2015, 10, e0132508 Search PubMed.
- I. V. Yampolsky, A. A. Kislukhin, T. T. Amatov, D. Shcherbo, V. K. Potapov, S. Lukyanov and K. A. Lukyanov, Bioorg. Chem., 2008, 36, 96–104 CrossRef CAS PubMed.
- W.-T. Chuang, B.-S. Chen, K.-Y. Chen, C.-C. Hsieh and P.-T. Chou, Chem. Commun., 2009, 6982–6984 RSC.
- N. S. Baleeva, K. A. Myannik, I. V. Yampolsky and M. S. Baranov, Eur. J. Org. Chem., 2015, 5716–5721 CrossRef CAS.
- M. S. Baranov, K. M. Solntsev, K. A. Lukyanov and I. V. Yampolsky, Chem. Commun., 2013, 49, 5778–5780 RSC.
- H. Huang, N. B. Suslov, N.-S. Li, S. A. Shelke, M. E. Evans, Y. Koldobskaya, P. A. Rice and J. A. Piccirilli, Nat. Chem. Biol., 2014, 10, 686–691 CrossRef CAS PubMed.
- K. D. Warner, M. C. Chen, W. Song, R. L. Strack, A. Thorn, S. R. Jaffrey and A. R. Ferré-D'Amaré, Nat. Struct. Mol. Biol., 2014, 21, 658–663 CAS.
- M.-A. Plamont, E. Billon-Denis, S. Maurin, C. Gauron, F. M. Pimenta, C. G. Specht, J. Shi, J. Quérard, B. Pan, J. Rossignol, N. Morellet, M. Volovitch, E. Lescop, Y. Chen, A. Triller, S. Vriz, T. Le Saux, L. Jullien and A. Gautier, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 497–502 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary data and figures. See DOI: 10.1039/c5tc03931b |
| This journal is © The Royal Society of Chemistry 2016 |