
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A combined approach for predicting the cytotoxic effect of drug-nanoaggregates†
M.
Wojnilowicz‡
a,
M.
Tortora‡
b,
B. G.
Bobay
d,
E.
Santiso
c,
M.
Caruso
b,
L.
Micheli
b,
M.
Venanzi
b,
S.
Menegatti
*c and
F.
Cavalieri
*ab
aDepartment of Chemical and Biomolecular Engineering, The University of Melbourne, Parkville, Melbourne, Victoria 3010, Australia. E-mail: francesca.cavalieri@unimelb.edu.au
bDipartimento di Scienze e Tecnologie Chimiche, Universita' degli Studi di Roma “Tor Vergata”, via della Ricerca Scientifica 1, 00173, Roma, Italy
cDepartment of Chemical and Biomolecular Engineering, NC State University, Raleigh, NC 27606, USA. E-mail: smenega@ncsu.edu
dDuke University NMR Center, Duke University Medical Center, Durham, NC 27710, USA
First published on 12th September 2016
Abstract
We present a combined spectroscopic and computational approach aimed to elucidate the mechanism of formation and activity of etoposide nanoaggregates upon release from dextran–etoposide conjugates. Etoposide is an anticancer drug that inhibits cell growth by blocking Topoisomerase II, the key enzyme involved in re-ligation of the DNA chains during the replication process. In silico and spectroscopic analysis indicate that released etoposide nanoaggregates have a different structure, stability, and bioactivity, which depend on the pH experienced during the release. Molecular dynamics simulation and in silico docking of etoposide dimers suggest that the aggregation phenomena inhibit etoposide bioactivity, yet without drastically preventing Topoisomerase II binding. We correlated the diminished cytotoxic activity exerted by dextran–etoposide conjugates on the A549 lung cancer cells, compared to the free drug, to the formation and stability of drug nanoaggregates.
Introduction
Under aqueous conditions, amphiphilic and hydrophobic drug molecules self-assemble into supramolecular nanostructures.1–3 Drug aggregation under physiological conditions gives rise to a variety of physicochemical and biochemical effects that play a significant role in determining the efficacy of therapeutic formulations.4 Analytical and clinical evidence demonstrates that the often-neglected drug self-assembly phenomena are a major cause of inconsistency and poor reproducibility of published data, and can jeopardize the translation of a drug from screening assays to advanced preclinical and clinical trials.4 Indeed, even minor deviations in the physiological environment can lead to the coexistence of a monomeric drug with a wide spectrum of aggregates with different transport and biorecognition behaviors. Such an assortment of aggregates affords a combination of advantageous and detrimental effects, which, while relevant in determining the success of a drug, renders the accurate estimation of the therapeutic efficacy extremely challenging.The key molecular and environmental parameters governing drug self-aggregation have been comprehensively reviewed by Sosnik.4 Accordingly, the size, structure, surface charge and hydrophobicity of aggregates, all differently affect permeation across cellular membranes and the interaction with surface receptors or intracellular targets. Yet, there is a striking dearth of studies, at both in vitro and in vivo levels, on the relationship between drug self-assembly and therapeutic activity. Currently, in fact, most of the studies are based on the implicit assumption that the drug molecules in solution are in a non-aggregated form. Understanding and controlling the self-assembling behaviour of many drugs with poor water solubility holds great promise to improve the pharmaceutical design, specifically to enhance therapeutic efficacy while reducing side effects.
To this effect, polymer–drug conjugates (PDCs) provide considerable insight into the study and tuning of drug aggregation phenomena towards therapeutic efficacy. Originally, PDCs have been conceived for co-delivering and releasing drugs to a target site, and have been the focus of extensive research throughout the last two decades.5–7 To design systems capable of site-selective drug release, a wide variety of polymers and drug coupling strategies have been proposed, many of which are currently under clinical trials or have recently received approval.7–11 As compared to the pure drug, PDCs show improved solubility and therapeutic efficacy, as well as reduced side effects and multi-drug resistance.12–14
While conjugation to a polymer carrier was expected to prevent drug aggregation owing to slow and controlled drug release from the polymer backbone, it has been found that the polymer carrier does promote assembly of drugs, prior to or upon release, and play a role in tuning the architecture of drug nanoaggregates. We have experienced this effect while working on dextran–etoposide conjugates. Etoposide (ETO) is a poorly soluble drug, widely employed for treating leukaemia, as well as testicular, bladder, prostate, lung, stomach, and uterine cancers.15,16 Etoposide induces cell apoptosis by inhibiting the ability of Topoisomerase II to re-ligate nucleic acids cleaved during the double-stranded DNA passage reaction.17,18 Due to its hydrophobicity, etoposide suffers from major limitations in bio-distribution due to uncontrolled extravasation from the blood vessel to healthy tissues, resulting in a low administrable dose. To overcome these issues, etoposide has been loaded into solid lipid nanoparticles,19 polymeric micelles20 and coupled to a hydrophilic polymer carriers such as dextran.21 Dextran is a naturally occurring biocompatible polymer consisting of α(1 → 6)-linked glucose units. The hydroxyl groups on the dextran chain can be activated and utilized for coupling drugs and fluorescent probes. In prior work, we have studied the activity of dextran–etoposide conjugates in U937 human leukaemia cells.21 Notably, the dextran–etoposide conjugates, while soluble in a much wider range of equivalent drug concentrations, exhibited a slower cytotoxic activity as compared to the free drug at the same dose (50 μM).21 Thus, we postulate that the reduced cytotoxic activity of dextran–etoposide conjugates is to be ascribed to drug aggregation processes.
Following on from these biological studies, we have resolved to investigate the tendency of etoposide to form nanoaggregates upon release in solution from the dextran carrier. To this end, we devised a combined spectroscopic and computational approach to study how the physiological pH and ionic strength determine the formation of ETO nanoaggregates. Our in silico and spectroscopy findings indicate that ETO nanoaggregates have a different structure depending on the pH experienced during the release process. In particular, the released nanoaggregates can switch from a more bioactive to a less bioactive configuration with a change in pH from 5 to 7. We finally present a case study on the activity of dextran–etoposide conjugates in the A549 lung cancer cell line. We correlated the diminished cytotoxic activity of dextran–etoposide conjugate in the A549 lung cancer cells, compared to the free drug, to the formation and stability of drug nanoaggregates. This finding highlights the potential of polymeric nanocarriers to control the self-aggregation properties of drugs to predict their therapeutic behaviour.
Results and discussion
Structural characterization of etoposide nanoaggregates
pH sensitive dextran–etoposide conjugates, hereafter referred to as “Dex–ETO”, were initially prepared following the procedure described in the Experimental section and developed in prior work.21 Briefly, etoposide was activated with 1,1′-carbonyldiimidazole (CDI) and coupled to dextran to form a hydrolysable carbonate bond between the 2,6-dimethoxyphenyl moiety of etoposide and the polysaccharide backbone. A schematic representation of the conjugate preparation is reported in Fig. 1. The conjugation resulted in a single ETO molecule per dextran chain (40 kDa). When dissolved in buffers mimicking the extracellular and cytosolic (PBS, pH 7.4) or intracellular lysosomal (PBS, pH 5.0) environments, the Dex–ETO showed 80–90% release of etoposide by carbonate linkage hydrolysis within 24 h, irrespective of the pH. The analysis of etoposide released after 24 h incubation of Dex–ETO (50 μM, pH 7 and pH 5) was performed by reverse phase high-performance liquid chromatography (HPLC) (Fig. 2) and fluorescence spectroscopy (Fig. 3). The chromatogram (Fig. 2) shows the peak of the etoposide monomer at 5 min and the aggregates formed at both pH 5.0 and 7.4. In particular, lower molecular weight aggregates are eluted between 6 and 12 min, whereas higher molecular weight aggregates, hereafter referred to as NETO5 and NETO7, are observed at higher elution times, that is, between 16 and 21 min.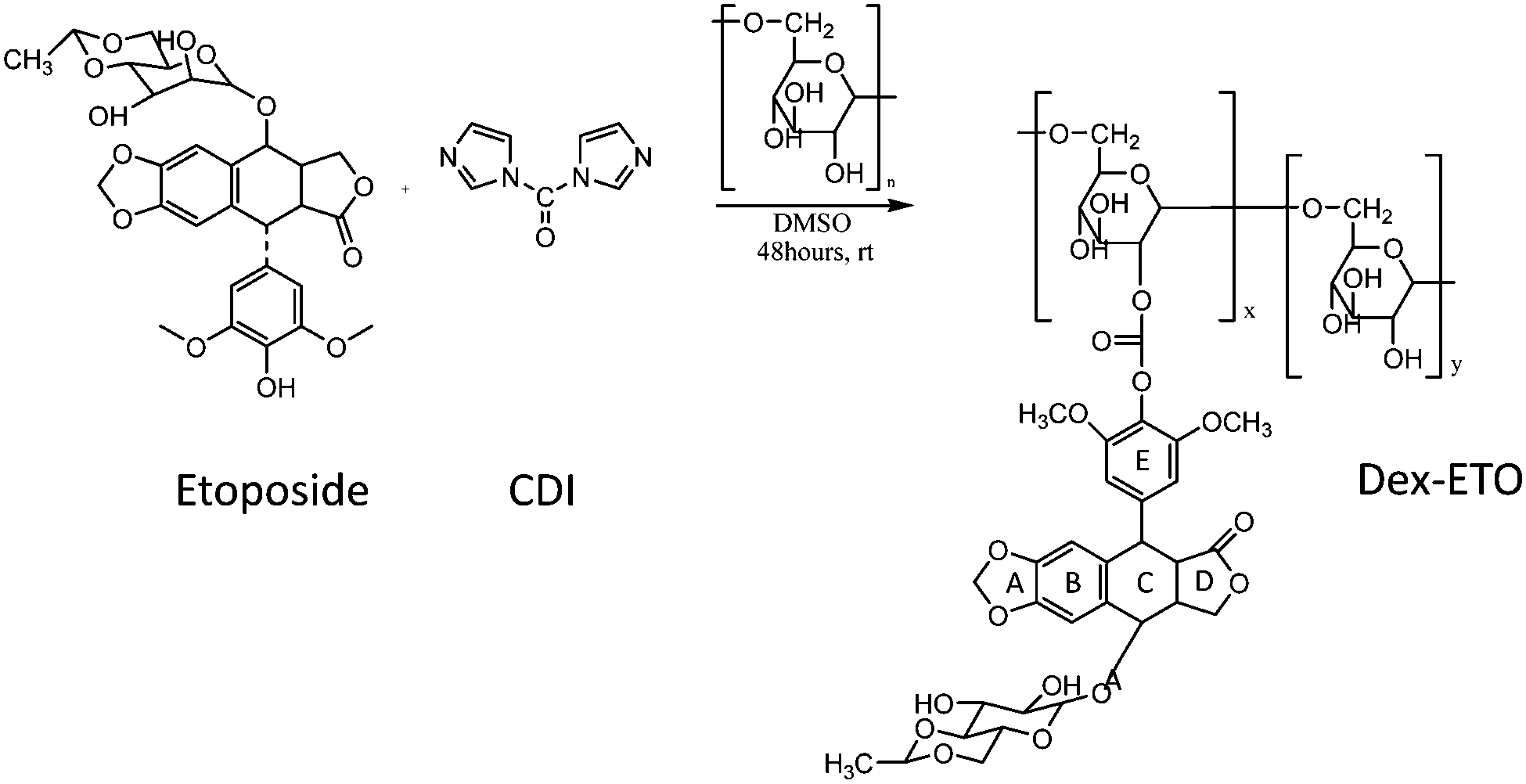 | ||
| Fig. 1 Synthetic procedure for conjugating etoposide to a dextran chain using 1,1′-carbonyldiimidazole (CDI), to form a hydrolysable carbonate bond between the 2,6-dimethoxyphenyl moiety of etoposide and the dextran backbone. | ||
 | ||
| Fig. 2 HPLC chromatograms of free ETO, NETO7 and NETO5 released from Dex–ETO after 24 h, and NETO7 treated with acetonitrile. | ||
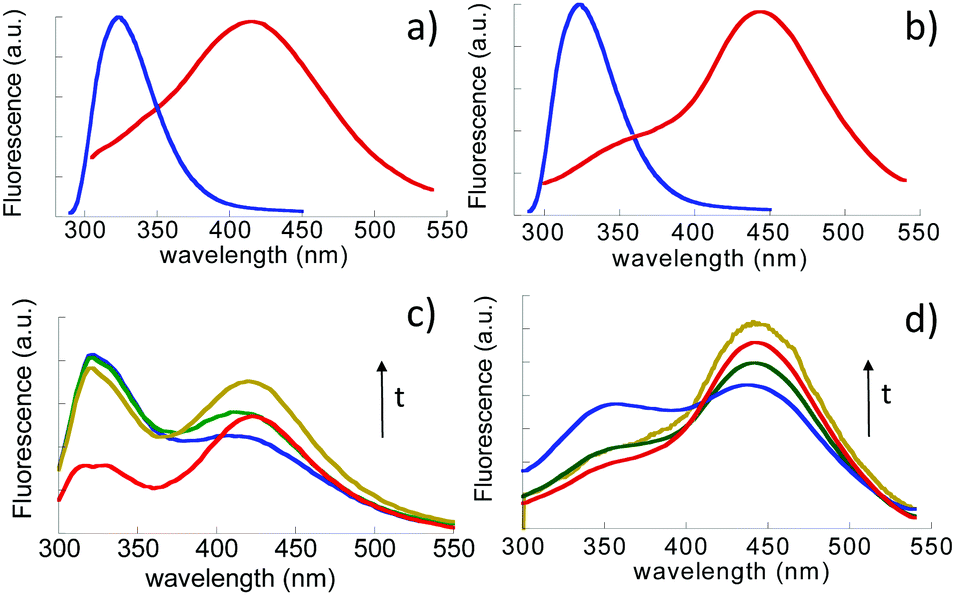 | ||
| Fig. 3 Fluorescence emission spectra of: (a) free ETO (blue line) and NETO 7 at pH 7.4; (b) free ETO and NETO5 at pH 5.0; (c) temporal evolution NETO7 formation over 3 h; (d) temporal evolution of NETO5 formation over 3 h. Both free ETO and NETOs are dissolved in buffer solutions at the same concentration, 50 μM. | ||
The nanoaggregates can be dissolved into monomers in acetonitrile. Yet, pure ETO dissolved in PBS, pH 7.4, at a concentration 50 μM did not show aggregation, as shown by HPLC analysis (Fig. 2). By contrast, when covalently linked to dextran, ETO was shown to self-associate upon release and form small soluble aggregates, e.g. dimers, trimers, and oligomers, thereby indicating that the polymer carrier plays a crucial role in the formation of the aggregates.
Further insight into the aggregation of conjugated ETO induced by the release process was obtained by fluorescence spectroscopy. The fluorescence emission spectra of free ETO (50 μM) measured in buffered solutions at both pH 7.4 and pH 5.0 (Fig. 3a and b) showed only one peak, characteristic of the monomer species, centered at 324 nm, which it is not influenced by pH. On the other hand, NETO5 and NETO7 showed broad red-shifted emission bands at 415 nm and 443 nm, respectively (Fig. 3a and b). To study the temporal evolution of the aggregation process, Dex–ETO were dissolved in saline buffers (PBS, pH 7.4 and pH 5.0) at a concentration of 50 μM and the fluorescence spectra of both solutions were recorded for up to three hours (Fig. 3c and d). The same spectral features, i.e. a monomer emission centered at 320 nm and a red-shifted emission band in the 400–450 nm wavelength region, ascribable to aggregated species, were obtained. A common trend is observed at both pH values, wherein the intensity of the monomer band progressively decreases and that of the aggregates concomitantly increases. Fluorescence emission data indicate a strong tendency of etoposide released from Dex–ETO to undergo aggregation, as the aggregate–monomer equilibrium is clearly shifted toward the aggregated state. Yet, Dex–ETO in MilliQ solution did not induce the formation of aggregates, as the fluorescence spectrum shows only the monomer form (data not shown).
The UV spectra of NETO5 and NETO7 (Fig. S1, ESI†) show a marked blue-shift and strong hypochromism of the lowest energy transition with respect to the ETO spectrum. The observed blue-shift of the absorption band, i.e. from 286 (ETO) to 268 nm (Dex–ETO), is associated with an energy stabilization of 6.7 kcal mol−1 (according to the equation ΔE = hΔν, wherein ΔE is the difference in energy associated with a variation in frequency Δν, and h is the Planck constant), typical of weak intermolecular interactions (van der Waals, π–π stacking, hydrogen bonding). Overall, these data suggest that the NETO aggregation process is driven by π–π stacking interactions between the aromatic moieties of etoposide. The difference in the emission band positions of approximately 30 nm strongly suggests that the aromatic moieties in NETO5 and NETO7 attain a different relative orientation, giving rise to electronic distributions characterized by a net different polarity. The ground-state nature of this interaction is confirmed by the excitation spectra of NETO5 and NETO7 (Fig. S2, ESI†), which show very similar excitation bands at 270–275 nm (monomer emission), and excitation bands peaked at 240 and 295 nm for NETO5 and 265 nm for NETO7 (aggregates emission).
To confirm the structural identity of NETOs, time-resolved fluorescence (TRF) measurements were performed (Table 1). When the emission signal was collected at λem = 330 nm, TRF measurements showed mainly a very fast monomer emission (τ1 = 0.1 ns, α1 = 0.99). The emission signal recorded at λem = 430 nm showed a bi-exponential time decay under the different pH conditions. While the longer time component (τ2 = 3.8 ns) was found to be the same at the two pH values, the shorter lifetime was around 1.4 ns (α1 = 0.90) at pH 7.4 and 0.5 ns (α1 = 0.70) at pH 5.0, indicating a different arrangement of the fluorescent moieties in the two nanoaggregates. Red-shifted emissions, characterized by relatively long-lifetime components, have already been found in aggregation processes driven by the stacking of aromatic units.22 Taken together, the chromatographic and spectroscopy studies indicate that the conjugation of etoposide to the polymer carrier and the environmental conditions, i.e. ionic strength and pH, play an important role in determining the formation and properties of NETOs. The aggregation processes driven by the hydrophobic effect are typically favored with increasing ionic strength, as the latter promotes the association of aromatic moieties. Ion-driven hydrophobic interactions, commonly referred to as “salting-out”, are a purely entropic effect associated with ions of high charge density.23
| Sample | τ 1 (ns) | α 1 | τ 2 (ns) | α 2 |
|---|---|---|---|---|
| NETO7 (λem = 330 nm) χ2 1.7 | 0.11 | 0.99 | 1.57 | 0.01 |
| NETO5 (λem = 330 nm) χ2 2.6 | 0.10 | 0.99 | 2.12 | 0.01 |
| NETO7 (λem = 430 nm) χ2 1.6 | 1.37 | 0.90 | 3.88 | 0.10 |
| NETO5 (λem = 430 nm) χ2 1.7 | 0.52 | 0.70 | 3.75 | 0.30 |
Notably, the conjugation to the carrier polymer, in combination with the pH at which the release is performed, has a crucial role in etoposide aggregation. Fluorescence data suggest that prior to hydrolysis, the dextran chains enable a partial assembly of etoposide molecules. A “crowding effect” exerted by the dextran24 chains can lead to an aggregation process which is not observed in the pure drug solution. Once formed and cleaved from the carrier, the nanoaggregates can further act as nucleation points for more etoposide molecules to assemble into multimers.
The ability of NETO5 and NETO7 to switch from one configuration to another was also studied by monitoring the fluorescence emission of NETO5 when conditioned at pH 7.4. The resulting fluorescence profiles are reported in Fig. 4.
 | ||
| Fig. 4 Fluorescence profiles of NETO5 maintained in pH 7.4 PBS buffer up to 24 hours. | ||
As mentioned, NETO5 showed an emission band peaked at λmax = 440 nm. This band, after conditioning the sample at pH 7.4 for one hour, turned into a structured emission featuring two main peaks at 440 and 425 nm. The latter band is the one observed for NETO7. This indicates a partial conversion of NETO5 to NETO7. After 24 hours at pH 7.4, no further change in the emission spectrum was observed, indicating that the two species reached equilibrium. Conversely, NETO7 does not show any spectroscopic change after 24 h incubation at pH 5.0. This suggests that etoposide aggregates formed at pH 5.0 are less stable than those formed at pH 7.
Molecular dynamics simulation of etoposide dimers
To evaluate the molecular structure and stability of NETOs formed at different pH values, molecular dynamics simulations were also performed. While the aggregates are likely to comprise several (>2) etoposide molecules, we limited our simulations to the formation of dimers, for simplicity. To model the drug behavior when exposed to pH 5.0, we resolved to protonate the dimethoxyphenyl moiety of etoposide. As the pKb of etoposide (4.2) almost coincides with that of dimethoxyphenol (syringol, 4.02) and as dimethoxyphenol is known to protonate at low pH values, we contend that protonation of the dimethoxyphenyl moiety well represents the outlook of etoposide at pH 5.0. The simulations were performed using the NAMD 2.725 molecular dynamics package. The resulting dimer structures are reported in Fig. 5. | ||
| Fig. 5 In silico structure of etoposide dimers formed at (a) pH 5.0 and (b) pH 7.4 obtained via molecular dynamics simulations using NAMD2.7. | ||
The in silico generated structures suggest that the dimer formed at pH 7.4 is more stable than that formed at pH 5.0. The dimer size is approximately 2 nm. Dihedral angles defined by methoxy groups are 39° and 57° in the dimers formed at pH 7.4 and pH 5, respectively. The dissociation constants of the etoposide–etoposide complexes are in fact KD = 5.3 μM and 47.9 μM, respectively. The dimer formed at pH 7.4 shows a symmetric structure stabilized by π–π stacking, hydrogen bonding, and van der Waals interactions, while that formed at pH 5.0 shows a rather irregular structure lacking sandwich-like aromatic interaction and H-bonds. Nevertheless, interactions between aromatic units in different orientations (slipped-out as in J-aggregates) are also possible, resulting in a more polar dimer. These results are in agreement with the fluorescence results, confirming the strong tendency of etoposide to form stable aggregates when a specific arrangement of the aromatic moieties is attained and mediated by the polymer chains and pH conditions.
Molecular dynamics simulation and in silico docking of etoposide dimers
The structural arrangement of NETO aggregates released by Dex–ETO in the intracellular environment can affect the drug ability to bind the topoisomerase target. The structure–activity relationship of NETO7 and NETO5 is expected to depend on (i) the ability of the nanoaggregates to bind directly to the target and (ii) the dissociation of drug nanoaggregates to active monomers.To ascertain whether NETO5 and NETO7 are capable of binding Topoisomerase II, we performed in silico binding studies using the docking software HADDOCK (v.2.1), which simulates protein–drug interaction and estimates the free energy of binding in solution through built-in scoring functions.26 Following a previously published procedure,27 we performed docking simulations against the crystal structure of the Topoisomerase II–DNA complex (PDB ID: 3QX3) using (i) etoposide monomer, (ii) etoposide dimer formed at pH 7.4, and (iii) etoposide dimer formed at pH 5.0. The drug monomer was docked against the residues (Lys456, Glu477, Gly478, Asp479, Arg503, Gly504, Gln778, Met781, Met782, Pro819 on both subunits A and B of Topoisomerase II) known to interact with etoposide, as reported by Wu et al.28 Similarly, the dimers formed at pH 5.0 and 7.4, the structures of which were those obtained through the above reported molecular dynamics simulations, were docked against the same region. The resulting structures are shown in Fig. 6. The binding free energy for each complex was calculated using scoring methods comprising empirical functions that estimate the binding affinity of a given protein–ligand complex. These functions account for van der Waals interactions, hydrogen bonding, deformation penalty, hydrophobic effects, atomic contact energy, softened van der Waals interactions, partial electrostatics, and additional estimations of the binding free energy and dipole–dipole interactions. The resulting dissociation constants (KD) for the interaction with the Topoisomerase II–DNA complex are reported in Fig. 6.
 | ||
| Fig. 6 Competitive mechanism of etoposide (ETO) aggregation and binding to Topoisomerase II (TopoII) as predicted by in silico modeling. The mechanism of ETO aggregation at different pH values (central panel) occurs between the ETO monomer and two dimers. The ETO monomer interacts with TopoII at higher affinity (left magenta panel) than the ETO dimers (right magenta panel). | ||
Notably, the structure of etoposide docked on the Topoisomerase II–DNA complex shows the same features highlighted in the experimental analysis.28 This depicts a model where etoposide binding to Topoisomerase II comprises contributions from its A, B, and E rings. The dimethoxyphenyl group, while not involved in the binding process, seems to be very important for the drug function. The interactions between etoposide and DNA in the ternary complex are based on the sugar moiety of etoposide, as confirmed in the predicted structure. Further, the calculated value of KD obtained for the etoposide monomer (KD = 13.2 μM) is very close to that determined experimentally, thus confirming the accuracy of our docking strategy. The binding constant calculated for both dimers (Fig. 6) contains a significant penalty component associated with steric hindrance, which lowers the affinity of any aggregate comprising two or more etoposide molecules for Topoisomerase II as compared to the monomer. This indicates that etoposide, when in an aggregate form is unlikely to possess any therapeutic activity. Incidentally, this also justifies our choice of using dimers as representative models for drug aggregates, since higher (>2) multimers most likely possess lower affinity than dimers. Overall, these results suggest that the bioactivity of etoposide released from the carrier is controlled by the combined and competitive effect of three concomitant phenomena, namely (i) the binding of the etoposide monomer to Topoisomerase II, (ii) aggregation of ETO into NETO5 and NETO7, and (iii) binding of NETO5 and NETO7 to Topoisomerase II.
The predicted affinity constants (KD) (Fig. 6) indicate that the ETO monomer, while having a higher affinity for Topoisomerase II as compared to the aggregates, also shows a pronounced tendency to form aggregates in an aqueous environment. The crowding effect induced by the carrier is likely to promote this phenomenon, eventually leading to a reduced bioactivity. The dissociation process of NETOs leading to the binding of the etoposide monomer to Topoisomerase II is energetically favored in NETO5 compared to NETO7. These results indicate that the therapeutic activity of Dex–ETO in the intracellular environment may be correlated to (i) the formation of ETO nanoaggregates, (ii) the pH dependent structural arrangements of ETO nanoaggregates and (iii) the dissociation of ETO nanoaggregates into the monomeric bioactive units.
Cytotoxicity of Dex–ETO in the A549 lung cancer cell line
The biological activity of Dex–ETO in A549 lung cancer cells was assessed in vitro using cell viability MTT assay, after 48 hour incubation at 50 and 150 μM concentrations. Free etoposide (12.5–200 μM) was used as a control. Dextran is internalized by endocytosis and trafficked via the lysosomal route,21 whereas the free drug can diffuse through the cell membrane.The cytotoxicity data reported in Fig. 7a indicate that Dex–ETO are less toxic (50% of cell mortality) compared to ETO (70% of cell mortality), when used at a 50 μM concentration. The apoptotic effect exerted by ETO and Dex–ETO on A549 lung cancer cells was also compared using the annexin V/propidium iodide (PI) apoptosis flow cytometry assay (Fig. 7b–d). In particular, free ETO and Dex–ETO at a 150 μM concentration were incubated with cells for 48 hours, followed by staining with the probes. Annexin V binds to phosphatidylserine at early stage of apoptosis, while PI is able to bind with DNA only upon disruption of the membrane, thus only indicating late apoptosis and/or necrosis of the cells. Phosphatidylserine is a phospholipid membrane component that plays a crucial part in cell signalling. Once apoptosis is initiated, phosphatidylserine becomes exposed for binding with annexin V. With progress of apoptosis, the cell membrane starts to be permeable and PI is able to diffuse into the nucleus and bind the DNA.
 | ||
| Fig. 7 Cytotoxicity of Dex–ETO and free ETO in A549 lung cancer cells; (a) cell viability evaluated by MTT assay after 48 h of incubation. Flow cytometry probability counter plots of fluorescence intensity [a.u.] after annexin V staining (y-axis) and propidium iodide staining (x-axis) of (b) untreated cell control; (c) cells incubated with free ETO (48 h, 150 μM); (d) cells incubated with Dex–ETO (48 h, 150 μM). Cross-hairs represent population frequency distribution indicating stages of apoptosis: Q1 – high annexin V positive signal (early apoptosis); Q2 – high annexin V and PI positive signal (late apoptosis); Q3 – high PI positive signal (late apoptosis and/or necrosis); Q4 – low annexin V and PI signal (healthy cells). Populations were gated against untreated control. | ||
The majority of cells incubated with free ETO (Fig. 7c) show a higher positive signal from both annexin V and PI (late apoptosis – Q2 region) (14.5%), compared to Dex–ETO (6.36%) (Q2 region in Fig. 7d). A larger number of cells in early stages of apoptosis (positive annexin V only – Q1 region) were also observed in the case of the free drug (Q1 region, Fig. 7c). However, complete death of cells (positive PI only – Q3) is equal in both samples (region Q3 in Fig. 7c and d).
Overall, these results suggest that compared to free ETO, Dex–ETO are less cytotoxic because they activate the apoptosis cascade gradually in time. Provided that Dex–ETO release NETO aggregates in the intracellular milieu, we can speculate that, despite the tendency of etoposide monomers to aggregate in dimers and multimers, when sufficient time is given for the aggregate dissociation and subsequently monomer–Topoisomerase binding, cell apoptosis is eventually achieved.
Conclusions
This work offers a contribution towards a better understanding and control of the mechanism of formation of drug aggregates upon release from a delivery carrier and their effect on the therapeutic activity of the released drug. While there is increasing awareness on the issue of drug aggregation upon release, the field is to date poorly explored and necessitates fundamental studies of aggregate formation, transport, and biorecognition behavior. The combined experimental and computational approach presented herein is the first of its kind in linking the physicochemical characterization of the kinetics and thermodynamics of aggregate formation to the understanding of the interplay of drug aggregation and binding of a target protein, and to the validation of the proposed mechanism via in vitro studies on a model cell line. In particular, we have observed that etoposide aggregates tend to form upon release from the polymer carrier. ETO nanoaggregates have a different structure depending on the pH experienced during the release process.In the intracellular environment, the monomer and aggregate species are further involved in the binding mechanism towards Topoisomerase II, thereby framing the monomer–aggregate equilibrium against the drug–protein binding equilibrium. The different affinities modeled in silico predict the aggregation phenomena to delay the drug bioactivity, yet without drastically preventing protein binding. This prediction is fully confirmed by in vitro studies on a model A549 lung cancer cell line. Through the presented case study, this work provides a strategy for better understanding the structure–function relationship in polymer–drug conjugates, while seemingly peculiar, these properties are in fact shared by many cancer therapeutics, which are commonly hydrophobic and possess proton acceptor groups. Thus, in the long run, these studies will enable a strategy for a more effective design of therapeutic formulations based on the combinations of small synthetic drugs and polymer carriers.
Experimental
Materials and methods
Etoposide, dextran 40 from Leuconostoc spp. (DEX), anhydrous dimethylsulfoxide (DMSO), N,N′-carbonyldiimidazole (CDI), acetonitrile, deuterated dimethylsulfoxide, phosphate buffer saline, sodium acetate and acetic acid were purchased by Sigma Aldrich and used as received. Dextran 40 was dried in an oven at 110 °C overnight before use.Dex–ETO, NETO5 and NETO7 preparation
12 mg (0.020 mmol) of etoposide were dissolved in 900 μL of DMSO; then 300 μL of a 68 mM CDI solution in DMSO were added and allowed to react for 5 hours. 150 mg of dextran were dissolved in 3.6 mL of DMSO; then the ETO/CDI solution was slowly added to the above solution, under argon bubbling. The mixture was stirred for 30 minutes under argon purging and stirred for two days at room temperature. The resultant solution was precipitated in 10 mL of acetone and the white precipitate was washed three times with acetone to remove the unreacted etoposide. Dex–ETO were collected and freeze-dried. The degree of substitution (DS = mol eto/mol dex × 100) of the conjugates was determined by UV spectroscopy. The UV absorbance (286 nm) of etoposide dissolved in DMSO was determined at different concentrations ranging from 0.3 mM to 2.5 mM, and used to generate a standard curve. Dex–ETO were dissolved in DMSO and the UV absorbance at 286 nm was determined.The kinetics of etoposide release from Dex–ETO at pH 7 and pH 5 were investigated as follows: 25 mg of Dex–ETO were dissolved in 250 μL of PBS buffer at pH 7 and pH 5 and placed in a spin column (10 kDa molecular weight cutoff). The released ETO was recovered by centrifugation and analyzed at fixed time intervals up to 24 h.
Absorption measurements were carried out on a Cary 100 SCAN (Varian, Palo Alto, CA) spectrophotometer. All of the experiments were carried out in quartz cells of 0.1 cm optical length. To prepare NETO5 and NETO7, a stock solution of Dex–ETO (3 mM) dissolved in MilliQ water was prepared. 50 μM Dex–ETO solutions at pH 5 and pH 7.4 were prepared by diluting the stock solution in acetate and phosphate buffer (140 mM NaCl), respectively. The solutions were maintained at room temperature under stirring for 24 h. The released NETO5 and NETO7 were separated from dextran by using a mini dialysis device (10k MWCO Thermo Scientific) and recovered with acetate and phosphate buffer, respectively.
Steady-state fluorescence spectroscopy
Emission spectra were obtained on a Spex-Fluoromax II (Horiba Jobin-Yvon Instruments, Longjumeau, France) spectrofluorimeter, equipped with a 450 W xenon lamp operating in single photon counting (SPC) mode. 50 μM Dex–ETO solutions in PBS and acetate buffer were prepared. Samples were excited at 280 nm and the fluorescence spectra were recorded from 300 to 530 nm using a bandwidth of 3 nm for both excitation and emission slits. The excitation spectra of NETO5 and NETO7 at λem = 430 nm and λem = 320 nm were collected from 230 to 310 nm using a bandwidth of 3 nm for both excitation and emission slits.Time resolved fluorescence spectroscopy
The fluorescence time decays of NETO5 and NETO7 solutions were obtained on an EAI Life-Specs (Edinburgh Analytical Instruments, Edinburgh, UK), operating in the SPC mode (λexc = 280 nm; λem = 330 nm and 440 nm; time interval = 50 ns; channels = 1024; excitation/emission bandwidths = 8 nm). Excitation at λ = 280 nm was obtained by an IBH NanoLED light emitting diode (Horiba Jobin-Yvon Instruments, Longjumeau, France) with 1 nanosecond pulse duration. Experimental time decays were deconvoluted by the pulsed excitation profile using a standard software provided by EAI and based on discrete multiexponential analysis.HPLC analysis
The HPLC system consisted of a modular CHROMQUEST spectra system from THERMOQUEST (San Jose, CA, USA), equipped with two LC-10AT Vp pumps and a Schimadzu UV-Vis spectrometer model (SPD-10AV) detector. A SCL-10A Vp controller operated the HPLC system working under the control of software included in the CHROMQUEST module. The chromatographic separation was performed using a reverse phase stainless steel column (5 μm spherical particle size, 150 × 4.6 mm I.D) C18 (VYDACTM, W.R. Grace &Co, cat. 210TP54). The composition of the mobile phase was CH3CN![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O:CH3COOH (74:25:1%) with a flow rate of 0.7 mL min−1 and using a detection wavelength equal to λ = 250 nm. Samples preparation: 50 μM Dex–ETO solutions in PBS/acetate were maintained at room temperature. After 24 hours, dextran extraction was obtained using a 10 K AmiconUltra-0.5 mL device by centrifugation for 20 minutes at 8000 rpm. The filtered solutions were diluted to 5 μM with the mobile phase solution and injected in the HPLC column.
:H2O:CH3COOH (74:25:1%) with a flow rate of 0.7 mL min−1 and using a detection wavelength equal to λ = 250 nm. Samples preparation: 50 μM Dex–ETO solutions in PBS/acetate were maintained at room temperature. After 24 hours, dextran extraction was obtained using a 10 K AmiconUltra-0.5 mL device by centrifugation for 20 minutes at 8000 rpm. The filtered solutions were diluted to 5 μM with the mobile phase solution and injected in the HPLC column.
Molecular dynamics simulations of etoposide dimerization
All simulations in this work were performed using the NAMD 2.7 molecular dynamics package.28 Adaptive biasing force (ABF) calculations29 were performed using the NAMD collective variables module and free energy profiles were determined using the ABF integrate tool included in the collective variables package. Two etoposide molecules were randomly placed in a 50 × 50 × 50 Å box that was then filled with water molecules to reproduce the liquid density. Interaction parameters were determined using the CHARMM generalized force field30 (CGenFF). The TIP3P water model31 was used. The solvated etoposide system was initially equilibrated using a molecular dynamics simulation at a constant pressure of 1 atm and a constant temperature of 300 K. An adaptive biasing force (ABF) was then applied between the center of mass of etoposide molecules to accelerate sampling and determine a free energy profile. The center of mass distance with the lowest free energy was determined and the configurations of possible etoposide dimers with that center of mass distance were found. A new, unbiased simulation was run for each possible dimer at 1 atm and 300 K, for 10 ns. Only the configurations shown remained associated for the extent of the simulation time. The ABF results were validated by using Laio–Parrinello metadynamics.32Docking of etoposide monomer and dimers on Topoisomerase II–DNA complex
The coordinate files of Topoisomerase II in complex with DNA and the etoposide monomer were obtained from the RCSB Protein Data Bank (PDB ID: 3QX3), while the coordinate files of the etoposide dimers were obtained from the above-described molecular dynamics simulations. The residues Lys456, Glu477, Gly478, Asp479, Arg503, Gly504, Gln778, Met781, Met782, Pro819 on both subunits A and B of Topoisomerase II, which define the concave surface of the enzyme targeted by etoposide, were defined as “active” and used as targets for docking. Molecular modeling was performed using the program HADDOCK (version 2.1). Default HADDOCK parameters (e.g., temperatures for heating/cooling steps, number of molecular dynamics sets per stage, etc.) were used in the docking procedure. The resulting docked structures were grouped in clusters by assigning a minimum cluster size of 4 and an RMSD (root-mean-square-distance) lower than 2.5 using the program ProFit (http://www.bioinf.org.uk/software/profit/). The top cluster was analyzed using the scoring function XScore (–log(Kd), and DiG).Cell viability assay
A549 human lung epithelial carcinoma cells (A549, ATCC CCL-185) were plated on a 96-well plate (Costar 3596, Corning, MA, USA) with a seeding density of 5.0 × 103 cells per well in 100 μL of DMEM medium supplemented with 10% fetal bovine serum. After incubation (37 °C, 5% CO2) for 24 h, Dex–ETO and free etoposide were added to culture media at various concentrations. Cell viability was determined by MTT assay after 48 hours incubation, measuring the absorbance at 554 nm and 670 nm as the reference using an Infinite M200 microplate reader (Tecan, Switzerland).Annexin V/propidium iodide assay
A549 lung cancer cells were plated on a 6-well plate (Costar 3516, Coring, MA, USA) with a seeding density of 3.0 × 105 cells per well in 1 mL of DMEM media supplemented with 10% fetal bovine serum and incubated for 24 h (37 °C, 5% CO2). Dex–ETO and free etoposide were added to a final drug concentration of 150 μM. After 48 h incubation, the cells were stained using a Dead Cell Apoptosis Kit with annexin V Alexa Fluor 488 and propidium iodide (PI) (ThermoFisher Scientific, MA, USA) following the manufacture's protocol. The cells were analyzed using a BD Accuri™ C6 flow cytometer, collecting 10000 cells/treatment.
Acknowledgements
This research was partially funded by the Australian Research Council, Future Fellowship 2014 and Establishment Grant, The University of Melbourne (F. Cavalieri project number FT140100873).Notes and references
- Z. Fülöp, R. Gref and T. Loftsson, Int. J. Pharm., 2013, 454, 559–561 CrossRef PubMed.
- I. Nabiev, F. Fleury, I. Kudelina, Y. Pommier, F. Charton and J.-F. Riou, et al. , Biochem. Pharmacol., 1998, 55, 1163–1174 CrossRef CAS PubMed.
- S. V. Balasubramanian, J. L. Alderfer and R. M. Straubinger, J. Pharm. Sci., 1994, 83, 1470–1476 CrossRef CAS PubMed.
- A. Sosnik, Prog. Mater. Sci., 2016, 82, 39–82 CrossRef CAS.
- R. Duncan, Nat. Rev. Cancer, 2006, 6, 688 CrossRef CAS PubMed.
- N. Larson and H. Ghandehari, Chem. Mater., 2012, 24, 840–853 CrossRef CAS PubMed.
- J. Singh, S. Desai, S. Yadav, B. Narasimhan and H. Kaur, Curr. Pharm. Des., 2016, 22, 2821–2843 CrossRef CAS PubMed.
- X. Pang, Y. Jiang, Q. Xiao, A. Wingnang Leung, H. Hua and C. Xu, J. Controlled Release, 2016, 222, 116–129 CrossRef CAS PubMed.
- G. L. Beretta and F. Cavalieri, Curr. Med. Chem., 2016, 23, 3–22 CrossRef CAS PubMed.
- F. Cavalieri, G. L. Beretta, J. Cui, J. A. Braunger, Y. Yan, J. J. Richardson, S. Tinelli, M. Folini, N. Zaffaroni and F. Caruso, Biomacromolecules, 2015, 16, 2168–2178 CrossRef CAS PubMed.
- E. Markovsky, H. Baabur-Cohen, A. Eldar-Boock, L. Omer, G. Tiram, S. Ferber, P. Ofek, D. Polyak, A. Scomparin and R. Satchi-Fainaro, J. Controlled Release, 2012, 161, 446 CrossRef CAS PubMed.
- E. Koziolová, O. Janoušková, L. Cuchalová, Z. Hvězdová, J. Hraběta, T. Eckschlager, L. Sivák, K. Ulbrich, T. Etrych and V. Šubr, J. Controlled Release, 2016, 233, 136–146 CrossRef PubMed.
- K. M. Camacho, S. Menegatti, D. R. Vogus, A. Pusuluri, Z. Fuchs, M. Jarvis, M. Zakrewsky, M. A. Evans, R. Chen and S. Mitragotri, J. Controlled Release, 2016, 229, 154–162 CrossRef CAS PubMed.
- K. M. Camacho, S. Menegatti and S. Mitragotri, Nanomedicine, 2016, 11, 1139–1151 CrossRef CAS PubMed.
- K. R. Hande, Eur. J. Cancer, 1998, 34, 1514 CrossRef CAS PubMed.
- E. Syrigou, N. Makrilia, J. Koti, M. W. Saif and K. N. Syrigos, Anticancer Drugs, 2009, 20, 1 CrossRef CAS PubMed.
- D. A. Burden, P. S. Kingma, S. J. Froelich-Ammon, M. Bjornsti, M. W. Patchan, R. B. Thompson and N. Osheroff, J. Biol. Chem., 1996, 271, 29238 CrossRef CAS PubMed.
- C. C. Wu, T. K. Li, L. Farh, L. Y. Lin, T. S. Lin, Y. J. Yu, T. J. Yen, C. W. Chiang and N. L. Chan, Science, 2011, 333, 459 CrossRef CAS PubMed.
- D. Pooja, H. Kulhari, L. Tunki, S. Chinde, M. Kuncha, P. Grover, S. S. Rachamalla and R. Sistla, RSC Adv., 2015, 5, 49122–49131 RSC.
- J. Varshosaz, F. Hassanzadeh, H. Sadeghi-Aliabadi and F. Firozian, BioMed Res. Int., 2014, 708593 Search PubMed.
- M. De Nicola, M. Tortora, F. Cavalieri, L. Micheli, S. Kaliappan, E. Bruni, S. Licoccia, E. Traversa, L. Ghibelli, Submitted to Nanomedicine NBM.
- M. Caruso, E. Gatto, E. Placidi, G. Ballano, F. Formaggio, C. Toniolo, D. Zanuy, C. Aleman and M. Venanzi, Soft Matter, 2014, 10, 2508 RSC.
- R. Zangi, M. Hagen and B. J. Berne, J. Am. Chem. Soc., 2007, 129, 4678–4686 CrossRef CAS PubMed.
- A. Christiansen and P. Wittung-Stafshede, FEBS Lett., 2014, 3, 811–814 CrossRef PubMed.
- J. C. Phillips, R. Braun, W. Wang, J. Gumbart, E. Tajkhorshid, E. Villa, C. Chipot, R. D. Skeel, L. Kalé and K. Schulten, J. Comput. Chem., 2005, 15, 1781–1802 CrossRef PubMed.
- N. Duhem, F. Danhier, V. Pourcelle, J. M. Schumers, O. Bertrand, C. S. LeDuff, S. Hoeppener, U. S. Schubert, J. F. Gohy, J. M. Brynaert and V. Préat, Bioconjugate Chem., 2014, 25, 72–81 CrossRef CAS PubMed.
- S. Huo, S. Jin, X. Ma, X. Xue, K. Yang, A. Kumar, P. C. Wang, J. Zhang, Z. Hu and X. J. Liang, ACS Nano, 2014, 6, 5852–5862 CrossRef PubMed.
- C. C. Wu, T. K. Li, L. Farh, L. Y. Lin, T. S. Lin, Y. J. Yu, T. J. Yen, C. W. Chiang and N. L. Chan, Science, 2011, 333, 459–462 CrossRef CAS PubMed.
- E. Darve, D. Rodríguez-Gómez and A. Pohorille, J. Chem. Phys., 2008, 128, 144120 CrossRef PubMed.
- K. J. Vanommeslaeghe, E. Hatcher, C. Acharya, S. Kundu, S. Zhong and J. Shim, et al. , J. Comput. Chem., 2010, 31, 671–690 CAS.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- G. Bussi, A. Laio and M. Parrinello, Phys. Rev. Lett., 2006, 96, 090601 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6tb02105k |
| ‡ M. W. and M. T. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |