
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePCL–PEG graft copolymers with tunable amphiphilicity as efficient drug delivery systems†
A.
Al Samad
ab,
A.
Bethry
a,
E.
Koziolová
c,
M.
Netopilík
c,
T.
Etrych
c,
Y.
Bakkour
b,
J.
Coudane
a,
F.
El Omar
b and
B.
Nottelet
 *a
*a
aInstitut des Biomolécules Max Mousseron (IBMM) UMR 5247 CNRS-Université Montpellier-ENSCM, Faculté de Pharmacie, 15, avenue Charles Flahault BP14491, 34093 Montpellier cedex 5, France. E-mail: benjamin.nottelet@umontpellier.fr
bLaboratory of Applied Chemistry, Doctoral School of Sciences and Technology, Lebanese University, Lebanon
cInstitute of Macromolecular Chemistry AS CR, v.v.i., Heyrovsky Sq. 2, 162 06 Prague 6, Czech Republic
First published on 30th August 2016
Abstract
The development of flexible drug delivery systems that can be tuned as a function of the drug to be delivered and of the target disease is crucial in modern medicine. For this aim, novel amphiphilic poly(ε-caprolactone)-g-poly(ethylene glycol) (PCL-g-PEG) copolymers with well-controlled design were synthesized by thiol–yne photochemistry. The grafting density and the copolymer amphiphilicity were easily controlled via the reaction parameters: concentration, reaction time, PEG length and the molar ratio between PCL and PEG or the photoinitiator in the reaction mixture. The self-assembling behavior of the copolymers was studied and a correlation between the composition of PCL-g-PEG and the nanoaggregate diameter sizes (28 to 73 nm) and critical aggregation concentrations (1.1 to 4.3 mg L−1) was found. The influence of copolymer amphiphilicity on the drug loading was evaluated with various drugs including anticancer drugs (paclitaxel, ABT-199), drugs to overcome multidrug resistance in cancer cells (curcumin, elacridar), an anti-inflammatory drug (dexamethasone) and an antibacterial drug (clofazimine). Finally, the influence of amphiphilicity on curcumin release and toxicity towards MCF-7 cancer cell lines was studied. The impact of the grafting density, PEG length and the overall EG/CL ratio is discussed in detail. Curcumin loaded PCL-g-PEG with lower EG/CL ratios and shorter PEG chains showed higher toxicity compared to their more hydrophilic counterparts.
Introduction
Amphiphilic copolymers self-assembling into micelles or nanoparticles play a key role in modern drug formulation strategies as a result of their capacity to protect and deliver drugs to the target site, e.g. tumors, with suitable pharmacokinetics.1,2 Drug loaded self-assemblies provide many advantages compared to low-molecular-weight drugs as they can reduce drug toxicity towards healthy tissues and increase the drug stability and the efficacy of drugs by changing the pharmacokinetics to a more favorable one. Among amphiphilic copolymers, aliphatic polyester-based copolymers, in particular PEG-b-PLA and PEG-b-PLGA diblock copolymers, have been widely studied due to their degradability and biocompatibility.3,4 After many years of extensive research and development, some have finally been introduced to the market like the PEG–PLA-based Genexol®5 which paves the way for future products and confirms the potential of polyester-based drug delivery systems.Among the synthetic polyesters, poly(ε-caprolactone) (PCL) is well known for its biocompatibility, its slower hydrolytic degradation compared to PLA or PLGA, its ability to load drugs and its mechanical properties similar to classical polyolefins. These properties make it a valuable candidate for tissue engineering and medical devices.6,7 However, the use of PCL is not limited to macroscopic systems and many examples of micro- or nano-systems for drug delivery, i.e. micelles and particles, are described in the literature.8 All kinds of macromolecular architectures based on PCL and PEG have been proposed from classical diblock copolymers to triblock copolymers, dendrimer-derived, star or miktoarm architectures.9–13 The availability of a variety of macromolecular architectures and compositions has proved the potential of these polymer systems for cancer therapy, and the impact of PCL–PEG copolymer amphiphilicity on the formation and shape of self-assemblies,14,15 drug loading, formulation stability12,16 and cellular internalization17–19 have been studied. It was also demonstrated that PCL–PEG copolymers could act as modulators of the drug efflux transporter P-glycoprotein in vitro and therefore they could be beneficial for the problematic treatment of multidrug resistant (MDR) cancer tumors.20
In this context, graft copolymers are of particular interest, since their architecture allows an easy variation of the number of PEG chains along the polymer backbone and consequently of the resulting amphiphilicity. Graft copolymers also exhibit lower critical aggregation concentrations (CACs) compared to their linear analogs and they assemble into micelles with higher stability in serum and during the freeze-drying process.21–24 As a consequence various polyester-g-PEGs have been synthesized to date following either “grafting through” or “grafting to” strategies. However, most of them are lactide- or glycolide-based polyesters,25–28 whereas only very little has been reported on PCL. For example, the group of Jérôme et al. synthesized PCL-g-PEG via the “grafting to” process by reacting azido-CL and alkynyl end-capped PEG, by a CuAAC click chemistry approach.29,30 A similar approach was utilized by Zhang et al.31 or Parrish et al. with an α-propargyl-δ-valerolactone.32 A “grafting through” approach using a CL end-capped PEG that was copolymerized with CL was also proposed by Rieger et al.33 The main problem of these methods is the use of functional lactones whose synthesis is limited in terms of amount and whose polymerization can be problematic when high molecular weights are targeted. As a consequence, Coudane et al. proposed an alternative “grafting to” approach by using anionic post-polymerization modification of the carbonyl of a commercial PCL to graft chlorocarbonate PEG at the PCL α-position.34,35 This one pot method is rapid and versatile, and the graft copolymer was easily prepared on a multigram scale. However, the grafting density influencing the self-assembly behavior is not well controlled. To overcome this limitation we recently synthesized PCL-g-PEG, thanks to the combination of a scale-up friendly anionic post-polymerization modification of PCL and a subsequent grafting of PEG side chains via the fast and metal-free photoradical thiol–yne reaction, which allowed an easy control of the copolymer amphiphilicity. The prepared PCL-g-PEG polymers with EG/CL ratios ranging from 0.1 to 1.3 formed nanoparticles and frozen micelles of different hydrodynamic diameters.36
The hydrophilicity of the previously prepared systems was however quite low, resulting in PEGylated nanoparticles with low PEG density rather than micelle-like nanoaggregates, which may limit the release properties. In addition, the impact of the copolymer hydrophilic/hydrophobic balance on the ability to deliver various drugs was not investigated. Here, a family of micelles-forming PCL-g-PEGs with higher and controlled hydrophilicities (EG/CL ratios ranging from 2 to 5.5) is synthesized and characterized. The impact of amphiphilicity on the loading of various poorly soluble drugs, including anticancer drugs (paclitaxel, ABT-199), multidrug resistance targeting drugs (curcumin, elacridar), an anti-inflammatory drug (dexamethasone) or an antibacterial drug (clofazimine) is evaluated. Finally, the impact of amphiphilicity on the curcumin release and on the toxicity towards MCF-7 cancer cells is discussed.
Experimental part
Materials
PCL (Mn ≈ 45 kg mol−1), propargyl bromide (80 wt% in toluene), lithium diisopropylamide (LDA, 2 M in THF/heptane/ethylbenzene), 2,2-dimethoxy-2-phenylacetophenone (DMPA, >99%), p-toluenesulfonic acid (>99%), ε-caprolactone (ε-CL, >99%), PBS 1×, clofazimine and curcumin were purchased from Sigma-Aldrich (St-Quentin Fallavier, France). Paclitaxel was obtained from LC Laboratories (Woburn, United States). ABT-199, dexamethasone and elacridar were purchased from Sigma-Aldrich (Prague, Czech Republic). α-Methoxy, ω-hydroxyl poly(ethylene glycol) copolymers with Mn = 2000 and Mn = 5000 g mol−1 (PEG2k and PEG5k) were obtained from Sigma (St-Quentin-Fallavier, France). α-Methoxy, ω-hydroxyl poly(ethylene glycol) with Mn = 750 g mol−1 (PEG0.7k) was obtained from Acros Organics (Noisy-le-Grand, France). Ascorbic acid (purissimum grade) was obtained from Riedel-de Haen. NH4Cl (>99%) was purchased from Acros Organics (Noisy-le-Grand, France), technical grade MgSO4 from Carlo Erba (Val de Reuil, France), and methanol (≥99.8%), dichloromethane (DCM, ≥99.9%), dimethylformamide (DMF, ≥99.8%), toluene (purissimum grade) and anhydrous tetrahydrofuran (THF, ≥99.9%) from Sigma-Aldrich (St-Quentin-Fallavier, France). THF was dried using a Pure Solv Micro Single Unit (Inert®) system. Toluene and ε-CL were distilled over CaH2 prior to use. Spectra/Por® dialysis tubes (cut-off, 6–8 kDa or 3.5 kDa) were obtained from Spectrum Labs. Amicon ultracentrifugal filter units (Ultra-15, 10 kDa) were purchased from Sigma Aldrich (Noisy-le-Grand, France).Characterization
Synthesis of PCL-g-PEG
The PCL-yne/PEG-SH reactions were performed according to our previous work.36 In a typical experiment, PCL-yne (100 mg; 8% alkyne groups with respect to CL units,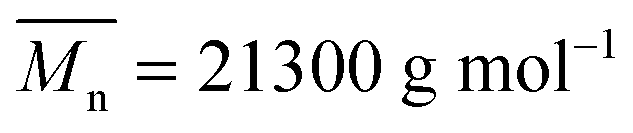 ; Đ = 2.4); and PEG2k-SH (820 mg; 6 eq. with respect to the alkyne group) were dissolved in DMF (10 mL) in a Schlenk flask (Table 1, entry 2). DMPA (9 mg; 0.5 eq.) was dissolved in DMF in another Schlenk flask covered by aluminium foil. The 2 flasks were purged with nitrogen for 10 min before closing with septa. The thiol–yne reaction medium was irradiated for 2 hours under UV light (100 mW cm−2) and DMPA was added by successive additions (4 times) with a syringe at fixed intervals over the reaction time. The mixture was then concentrated under vacuum and centrifuged in water by centrifugal filter units of 10 kDa. Centrifugation was repeated until the complete removal of unreacted PEG chains. The solution was lyophilized and the product was analyzed by SEC to ensure the absence of free PEG.
; Đ = 2.4); and PEG2k-SH (820 mg; 6 eq. with respect to the alkyne group) were dissolved in DMF (10 mL) in a Schlenk flask (Table 1, entry 2). DMPA (9 mg; 0.5 eq.) was dissolved in DMF in another Schlenk flask covered by aluminium foil. The 2 flasks were purged with nitrogen for 10 min before closing with septa. The thiol–yne reaction medium was irradiated for 2 hours under UV light (100 mW cm−2) and DMPA was added by successive additions (4 times) with a syringe at fixed intervals over the reaction time. The mixture was then concentrated under vacuum and centrifuged in water by centrifugal filter units of 10 kDa. Centrifugation was repeated until the complete removal of unreacted PEG chains. The solution was lyophilized and the product was analyzed by SEC to ensure the absence of free PEG.
| Entry | Copolymer | PEGa (eq.) | DMPAa (eq.) | DMF (mL) | Time (h) | EG/CLb | Grafting efficiencyc (%) | M n (g mol−1) | Đ |
|---|---|---|---|---|---|---|---|---|---|
| a Equivalent with respect to alkyne groups. b Calculated from the peak integrations of 1H-NMR spectra. c Calculated by 1H NMR analysis based on the initial content of alkyne groups and the EG/CL ratio and considering diaddition on all alkyne groups as 100% grafting efficiency. d SEC analyses in THF and polystyrene standards. | |||||||||
| 1 | PCL-g-PEG0.7k1.9 | 10 | 1 | 10 | 2 | 1.9 | 56 | 24![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 700 700 |
2.0 |
| 2 | PCL-g-PEG2k2.8 | 6 | 0.5 | 10 | 2 | 2.8 | 28 | 25800 |
2.7 |
| 3 | PCL-g-PEG2k3.6 | 6 | 0.5 | 2 | 1 | 3.6 | 36 | 27000 |
1.8 |
| 4 | PCL-g-PEG2k4.5 | 6 | 0.5 | 2 | 2 | 4.5 | 45 | 25900 |
1.6 |
| 5 | PCL-g-PEG2k4.7 | 6 | 1 | 2 | 2 | 4.7 | 47 | 29000 |
1.8 |
| 6 | PCL-g-PEG2k5.0 | 6 | 2 | 2 | 2 | 5.0 | 50 | 30000 |
2.1 |
1H NMR (300 MHz; CDCl3): δ (ppm) = PCL backbone 4.0 (2H, CH2–O), 2.3 (2H, C(O)CH2), 1.6 (4H, C(O)CH2–CH2–CH2–CH2–CH2–O), 1.3 (2H, C(O)CH2–CH2–CH2–CH2–CH2–O); PEG side chains 4.2 (2H, CH2COO), 3.5 (4H, CH2CH2O), 3.3 (3H, OCH3), 2.8 (2H, CH2SCH2), 2.6 (2H, COOCH2).
Synthesis of PCL-b-PEG
PCL-b-PEG was synthesized according to classical procedures. PEG5k was used a macroinitiator for the ring opening polymerization of CL in anhydrous toluene in the presence of SnOct2 as a catalyst (0.3 eq. with respect to PEG). The amounts of reactant were determined to yield the expected copolymer with EG/CL = 2.6, corresponding to blocks of 5000 g mol−1 for PEG and PCL. ROP was carried out for 48 hours at 100 °C and was stopped by the addition of a few drops of 1 N HCL. The mixture was then concentrated under vacuum prior to precipitation in diethyl ether. The resulting solid was further dissolved in water and centrifuged in water by centrifugal filter units 10 kDa. Centrifugation was repeated until the complete removal of unreacted PEG chains. The solution was lyophilized and the product was analyzed by SEC to ensure the absence of free PEG.Preparation of nanoaggregates
Blank and drug loaded PCL-g-PEG nanoaggregates were prepared by the self-assembly method according to our previous article.36 Typically, the drug (5 mg) and PCL-g-PEG (35 mg) were dissolved in 2 mL acetone. Then 5 mL of PBS were added. The mixture was stirred for one hour using an Ultra-Turrax stirrer (8000 rpm). The remaining acetone was evaporated under vacuum. The solution was then centrifuged for 10 min at 3600 rpm and 20 °C before filtration through a 0.45 μm filter to eliminate the unloaded drug.Determination of drug loading and encapsulation efficiency
The drug loading (DL) and encapsulation efficiency (EE) were determined by UV/Vis spectrophotometry at 425 nm for curcumin, 468 nm for clofazimine and 410 nm for elacridar. Quantification of curcumin and clofazimine was performed with respect to a calibration curve in acetone/water (20:80). Quantification of elacridar was done using a calibration curve in acetone/water (80:20). The amount of loaded dexamethasone and ABT-199 was determined using a high performance liquid chromatography (HPLC, Shimadzu LC-20AD, Japan) instrument equipped with a column (Chromolith® Performance RP-18) and an autosampler (SIL-20AC HT). Fluorescence and PDA detectors (RF10AXL and SPD-M10AVP) were used for the detection of the drugs. A gradient elution mode was applied to detect dexamethasone and ABT-199, a linear gradient from 100% of the mixture (5% ACN (0.1% TFA) + 95% H2O (0.1% TFA)) until 100% of the mixture (95% ACN (0.1% TFA) + 5% H2O (0.1% TFA)) was used. The flow rate was set to 1 mL min−1, the detection wavelength was 240 nm for dexamethasone and 314 nm for ABT-199.
Quantification of paclitaxel (PTX) was done by HPLC (WatersTM 717plus Autosampler), equipped with a C18 column and a water 2998 photodiode array detector (λmaxPTX = 227 nm). The gradient elution applied was ACN/water: 50/50 for 2 min, then ACN/water: 75/25 until the 15th min at 1 mL min−1 flow rate. PTX was extracted from copolymers before injection in HPLC as follows: 1 mL of the PTX-loaded copolymer solution was added to 5 mL of water:DMF (50:50). After centrifugation in Spectra/por® dialysis tubes (cut-off, 6–8 kDa), the solvent was evaporated and PTX was dried under vacuum. PTX was dissolved in a precise volume of ACN and quantified by HPLC.
The DL content and the EE were calculated based on the following formulae:
| DL (%) = 100 × (MD)/(MP + MD) |
| EE (%) = 100 × (MD)/(MDi) |
Drug release kinetics
The release of curcumin from PCL-g-PEG nanoaggregates was assessed in phosphate buffered saline (PBS, pH 7.4) containing 0.1% w/v Tween 80 and 0.015% w/v ascorbic acid at 37 °C under constant orbital shaking (100 rpm; Heidolph Unimax 1010). Tween is classically used to provide water-solubility to curcumin in such assays.37–39 Ascorbic acid is a biomolecule naturally present in plasma, which is used to enhance the stability and avoid the degradation of the released curcumin.40 In a typical release study, 2 mL of curcumin/PCL-g-PEG micellar solution were poured into a dialysis bag (cut-off, 3.5 kDa) that was immersed in 20 mL of buffer solution at 37 °C. At specific time points, the entire release medium was removed and replaced with 20 mL of fresh buffer solution. The collected samples were analyzed by UV-Vis spectrophotometry at 425 nm with reference to a calibration curve of curcumin in acetone:water (20:80). The amount of released curcumin (Rc) was calculated based on the following formula:| Rc (%) = 100 × (MRC)/(MC) |
Degradation of nanoaggregates
7 mg of copolymers PCL-g-PEG0.7k1.2 and PCL-g-PEG2k5.4 were dissolved in 1 mL of acetone in a hemolysis tube, and then 5 mL of PBS were added. After evaporation of acetone, hemolysis tubes were put in an incubator at 37 °C under stirring at 150 rpm. The tubes were removed after 1 week, 1 month, 3 months and 6 months. Samples were freeze-dried, the solid residue was dissolved in THF and the resulting solution was filtered before analysis by SEC. Degradation experiments were done in triplicate.In vitro assay against MCF7 cells
Results and discussion
Synthesis of graft copolymers with controlled amphiphilicity
A convenient thiol–yne photo-addition methodology was used to synthesize the PCL-g-PEG copolymers and to modulate their amphiphilicity. For the sake of clarity, a schematic representation of the preparation and the architecture of the PCL-g-PEG copolymers is provided (ESI,† Scheme S1). Indeed, as shown in our previous work, it is possible to easily control the PEG grafting density by playing with the reaction parameters.36 To evaluate the influence of the PCL-g-PEG hydrophilicity on the potential of copolymers as drug delivery systems, we targeted a broad range of hydrophilicity with EG/CL molar ratios in the range 1.9 to 5.4 (Table 1).An increased number of compositions and higher hydrophilicity of the novel PCL-g-PEGs compared to our previously presented copolymers were obtained, thanks to the precise control of the reactant concentration, the reaction time, the PEG-SH molecular weight, the number of PEG equivalents, and the amount of photoinitiator. The ratio EG/CL in the product was easily increased from 1.9 to 2.8 by replacing PEG0.7k by PEG2k (entries 1 and 2), despite the lower number of PEG chains and DMPA equivalents. As expected and in accordance with our previous study with benzyl mercaptan,36 higher grafting efficiency was obtained for the shorter PEG due to the limited steric hindrance and the higher thiol-end group accessibility for PEG0.7k compared to PEG2k. The impact of the reactant concentration on the grafting density is significant as shown by the increase of the EG/CL ratio from 2.8 to 4.5 for a 4 times lower solvent volume (entries 2 and 4). Moreover, the reaction time also played a significant role as EG/CL increased from 3.6 after 1 hour of reaction to 4.5 after 2 hours (entries 3 and 4). Finally, the equivalent number of DMPA has only a limited impact on the EG/CL ratio: EG/CL varies from 4.5 to 5 despite a 4-fold increase of DMPA equivalents was used (entries 3–5). These results, in conjunction with the previously published ones, confirmed the simplicity of modulation offered by the photo-thiol–yne approach to yield amphiphilic PCL-g-PEG copolymers that are easily changed from highly hydrophobic structures (EG/CL = 0.35, previous work) to more hydrophilic ones (EG/CL = 5).
Molecular weights, measured by SEC, were in the range 25000 g mol−1 to 30000 g mol−1 (Đ = 1.6 to 2.7) and did not vary significantly for various copolymers. Further discussion of the molecular weights of the copolymers with respect to PCL-yne (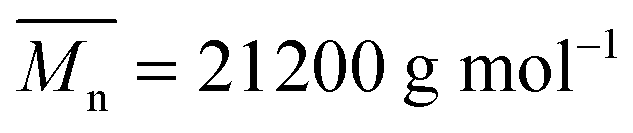 ; Đ = 2.4) or comparison between the copolymers is not provided as the hydrodynamic behavior of the copolymers is known to be strongly impacted by their amphiphilicity and architectures. In particular, underestimation of the molecular weight of graft copolymers was reported in the literature and is believed to be due to the impact of the grafting density on the hydrodynamic radius, which gives more compact structures than their linear counterparts.26,32
; Đ = 2.4) or comparison between the copolymers is not provided as the hydrodynamic behavior of the copolymers is known to be strongly impacted by their amphiphilicity and architectures. In particular, underestimation of the molecular weight of graft copolymers was reported in the literature and is believed to be due to the impact of the grafting density on the hydrodynamic radius, which gives more compact structures than their linear counterparts.26,32
Characterization of PCL-g-PEG nanoaggregates
| Entry | Copolymers | EG/CL | Grafting numbera | CAC (mg L−1) | Sized (nm) | PDI |
|---|---|---|---|---|---|---|
| a The number of PEG chains per PCL chain, calculated by 1H NMR analysis based on the molecular weight of PCL-yne and PEG. b CAC of PCL-g-PEG1.2 was calculated in our previous work using the emission spectra of pyrene.36 c Block copolymer. d Mean size determined by DLS. | ||||||
| 136 | PCL-g-PEG0.7k1.2 | 1.2 | 13 | 1.1b | 32 | 0.24 |
| 2 | PCL-g-PEG0.7k1.9 | 1.9 | 21 | 3.5 | 42 ± 4 | 0.24 ± 0.01 |
| 3 | PCL-g-PEG2k2.3 | 2.3 | 9.5 | 2.6 | nd | nd |
| 4 | PCL-g-PEG2k3.2 | 3.2 | 13 | 2.6 | 28 ± 1 | 0.21 ± 0.01 |
| 5 | PCL-g-PEG2k4.7 | 4.7 | 19 | 4.3 | nd | nd |
| 6 | PCL-g-PEG2k5.4 | 5.4 | 22 | 4.3 | 73 ± 2 | 0.40 ± 0.00 |
| 7 | PCL-b-PEG5k2.6c | 2.6 | 1 | 17 | 59 ± 1 | 0.21 ± 0.01 |
The CAC value was found to be strongly impacted by the composition (EG/CL) and the grafting number (number of PEG chains per PCL backbone) of the copolymers. In more detail, as shown for entries 1 and 2, where PEG0.7k was used, the CAC increased from 1.1 mg L−1 to 3.5 mg L−1 with the increase of hydrophilicity from EG/CL = 1.2 to EG/CL = 1.9, respectively. This is a classical result for amphiphilic self-assembling polymers. The same trend was obtained with PEG2k (Table 2, entries 3 to 6) where the CAC varied from 2.6 to 4.3 mg L−1 for PCL-g-PEG2k2.3 to PCL-g-PEG2k5.4. However, one can note that the EG/CL ratio alone cannot explain the self-assembly behavior of the graft copolymers. The grafting number also has a strong influence on the CAC. PCL-g-PEG with a close architecture, i.e. the same grafting number, presented a similar CAC. This is clearly the case for PCL-g-PEG2k2.3 and PCL-g-PEG2k3.2 that have a similar CAC (2.6 mg L−1), despite different EG/CL ratios due to the presence of ca. 10 PEG chains for both polymers. The same phenomenon is observed in PCL-g-PEG2k4.7 and PCL-g-PEG2k5.4 (ca. 20 PEG chains). At similar EG/CL ratios, the general trend is therefore to observe a lower CAC for a lower grafting number. This is confirmed by the comparison between PCL-g-PEG0.7k1.9 and PCL-g-PEG2k2.3. In this case, close EG/CL ratios are obtained but grafting numbers are very different with 21 PEG0.7k chains for PCL-g-PEG0.7k1.9 against 9.5 PEG2k chains for PCL-g-PEG2k2.3. This leads to a lower CAC for the less substituted PCL-g-PEG2k2.3 although it presents the higher EG/CL ratio. In summary, we have found that all graft PCL-g-PEG copolymers showed low CAC values below 5 mg L−1, whatever the composition and substitution degree.
With the aim of comparing the graft copolymers with their linear block copolymers counterparts, a PCL5k-b-PEG5k diblock copolymer with EG/CL = 2.6 was synthesized. Despite similar EG/CL ratios, this linear copolymer had a much higher CAC (17 mg L−1) than its counterpart PCL-g-PEG2k2.3 (2.6 mg L−1). This result is in agreement with the literature where the lower CAC of grafts copolymers is assigned both to their higher polydispersity and to their reduced mobility as a consequence of the steric hindrance induced by the side chains.23,24 The results obtained for CAC values of all PCL-g-PEG graft copolymers are highly promising, showing that the formed nanoaggregates should be useable as drug delivery systems in medicine as no dissociation is expected upon the high dilution encountered with injection.
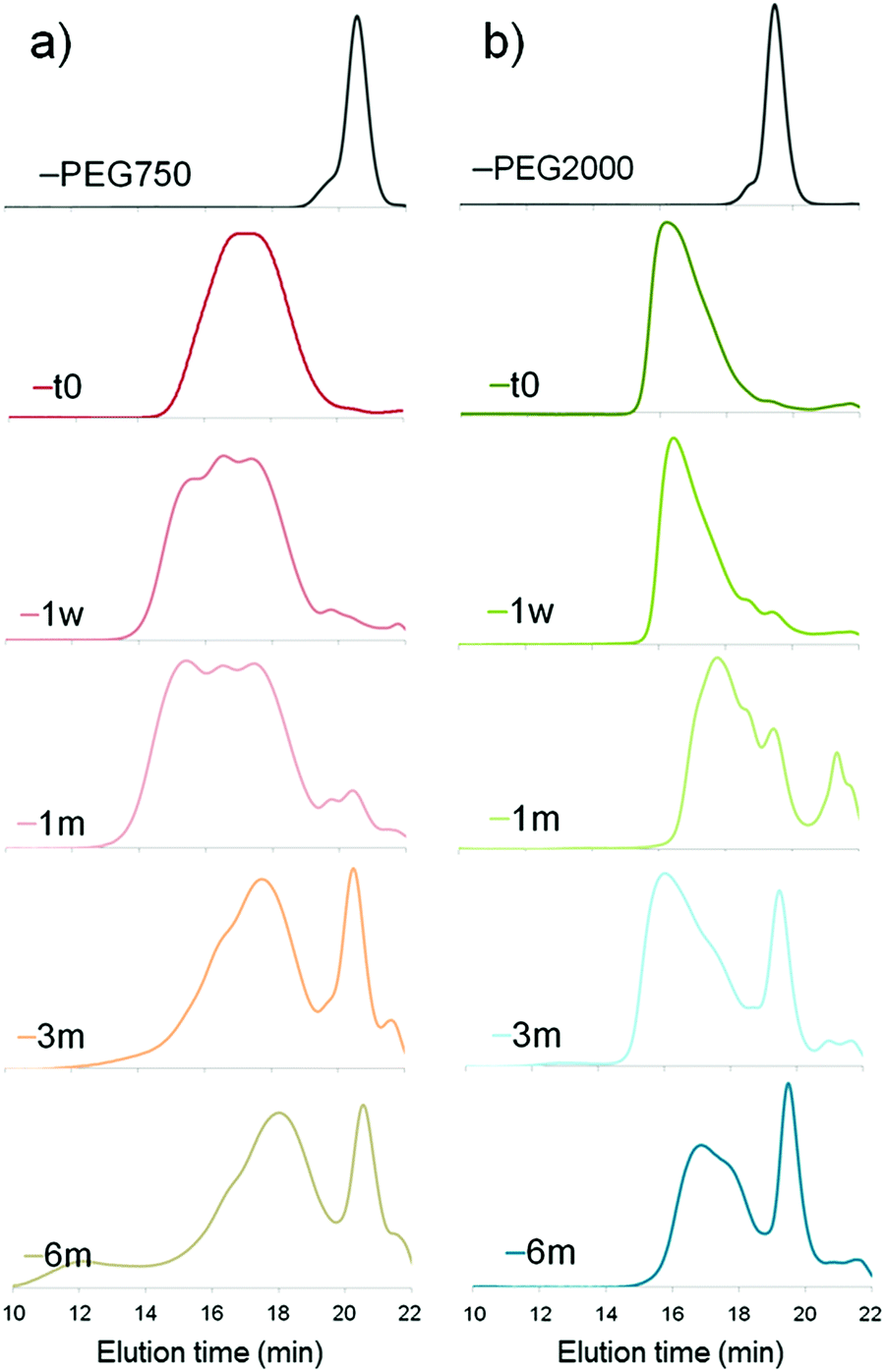 | ||
| Fig. 1 Degradation of PCL-g-PEG copolymers in PBS (pH 7.4, 37 °C) with (a) EG/CL = 1.2 and (b) EG/CL = 5.4 (SEC analyses were run in THF). | ||
For both copolymers, the release of PEG chains was witnessed. It results from the hydrolysis of the ester bond between the terminal hydroxyl-PEG group and the propionic acid used to prepare the PEG-SH. The first appearance of a peak corresponding to free PEG chains was seen after 1 month in the more hydrophobic PCL-g-PEG0.7k1.2 (Fig. 1a) and after only 1 week in the more hydrophilic PCL-g-PEG2k5.4 (Fig. 1b). By comparison of the relative intensities of peaks after 3 and 6 months, it may be concluded that all PEG side chains were hydrolyzed after 3 months in PCL-g-PEG0.7k1.2, whereas PEG release continued between 3 and 6 months in the more substituted PCL-g-PEG2k5.4. In summary, degradation starts earlier for the more hydrophilic copolymer, but due to a higher grafting number, it lasts a longer period compared to the more hydrophobic copolymer.
An additional comment should be made on the initial degradation period of the most hydrophobic copolymer. A high molecular weight population is formed after 1 week, still present after 1 month, to finally disappear after 3 months (Fig. 1a, shoulder peak at ca. 15 minutes). This is confirmed by DOSY-NMR analyses that also show the release of free PEG after 1 month (Fig. 2). Different hypotheses can be done to explain this phenomenon. PCL-g-PEG0.7k1.2 is substituted by 13 PEG0.7k chains on average (Table 2), whereas full substitution of the alkyne groups initially present on the PCL-yne would correspond to ca. 32 PEG0.7k chains. Some unreacted alkyne groups and vinylsulfide groups (thiol–yne monoaddition) are therefore present in the copolymer. Rearrangements between these groups leading to cyclic compounds have been reported in the literature.44 In a same manner, while considering the free alkyne groups and thioether groups, intramolecular chalcogenation has been reported following radical activation that may occur in our case due to the long exposure of degradation medium to day light.45 Finally, another photoactivated side reaction is the direct crosslinking of vinyl sulfide under light during degradation. Taking into account these reactions, it is clear that some rearrangements leading to cross-linked structures may occur in the copolymer during its degradation and can explain the appearance of a population with higher molecular weight. For longer degradation times these cross-linked structures are also degraded and a clear decrease of molecular weight, as well as a lower dispersity, is again observed. A more detailed study on this aspect is however beyond the scope of the present work and is underway. However, one should note that all components of the PCL-g-PEG nanoaggregates will be excreted (released short PEG chains), and/or degraded (hydrolysis of PCL backbone) and thus unwanted accumulation of the carrier structure is not anticipated. Based on the presented results we can therefore conclude that the PCL-g-PEG copolymer nanoaggregates are suitable candidates as long circulating drug reservoirs for treatment of diseases, which need a long term exposure of the drug in the body.
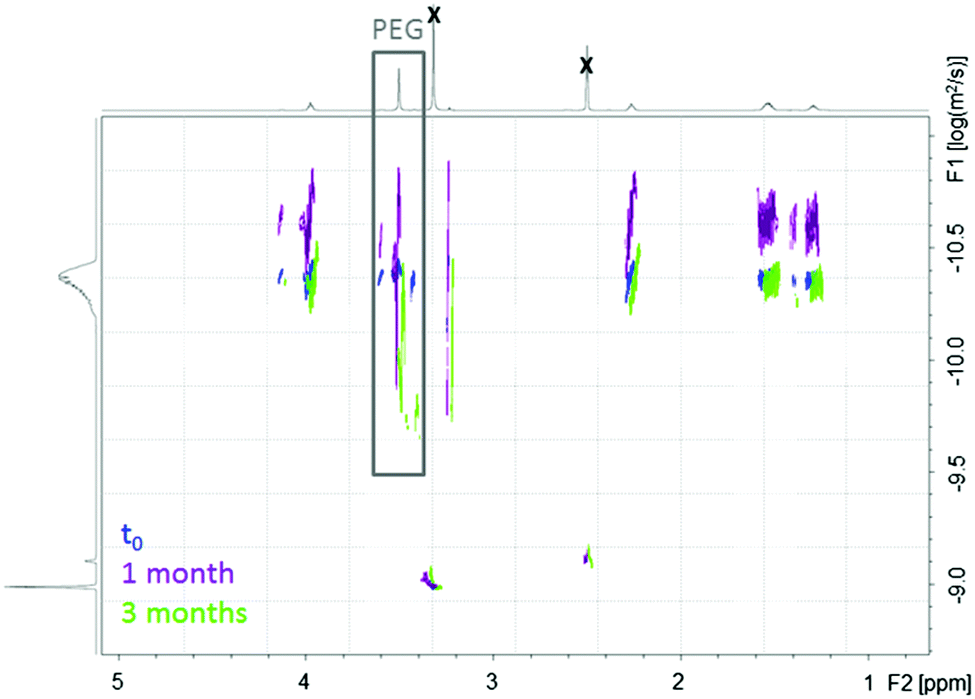 | ||
| Fig. 2 DOSY-NMR spectra of PCL-g-PEG0.7k1.2 as a function of the degradation time in PBS (pH = 7.4) at 37 °C. (DMSOd6, 600 MHz, black crosses correspond to DMSO and water.) | ||
Encapsulation of drugs
For all drugs, higher encapsulation efficiency and higher drug loading were obtained with the more hydrophobic copolymer, as expected (Table 3). The EE was lowered by 22% (curcumin) to 55% (ABT-199, dexamethasone) by changing PCL-g-PEG0.7k1.2 to PCL-g-PEG2k3.2. In more detail, in the case of paclitaxel, the EE was considered too low (<3 wt%) and the DL was not calculated. Similarly, other polyester-based drug delivery systems have shown low efficiency in paclitaxel formulation when using the self-assembly method and large quantities of polymer with respect to PTX (100:1) were therefore required.47 On the other hand, very high EE (92 wt%) and DL (12.1 wt%) were obtained with curcumin. The same phenomenon was observed for clofazimine that was loaded at 11.7 wt% in the copolymer. Finally, ABT-199 and elacridar loadings were found to be very low (1.3 wt% and 2.6 wt%). Nevertheless, these drugs are extremely potent and are still under in-depth pharmaceutical development and evaluation. We suppose that even quite low DLs might be enough to witness a therapeutic effect because of the highly improved pharmacokinetic profile and prolonged accessibility of the drug within the body.
| Copolymer | Paclitaxel | ABT-199 | Curcumin | Elacridar | Dexamethasone | Clofazimine | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| EE (wt%) | DL (wt%) | EE (wt%) | DL (wt%) | EE (wt%) | DL (wt%) | EE (wt%) | DL (wt%) | EE (wt%) | DL (wt%) | EE (wt%) | DL (wt%) | |
| PCL-g-PEG0.7k1.2 | 2.9 | nd | 6.8 | 1.3 | 92.0 | 12.1 | 16.4 | 2.6 | 27.2 | 4.4 | 48.5 | 11.7 |
| PCL-g-PEG2k3.2 | 1.6 | nd | 3.2 | 0.1 | 70.1 | 10.6 | 12.3 | 2 | 12.2 | 2.1 | nd | nd |
| Entry | Copolymer | EE (wt%) | DL (wt%) | Sizea (nm) | PDI |
|---|---|---|---|---|---|
| a Mean size determined by DLS. | |||||
| 1 | PCL-g-PEG0.7k1.2 | 92.0 | 12.1 | 40 | 0.28 |
| 2 | PCL-g-PEG0.7k1.9 | 89.4 ± 7 | 11.6 ± 0.4 | 42 ± 6 | 0.29 ± 0.03 |
| 3 | PCL-g-PEG2k2.3 | 90.8 ± 8.8 | 11.7 ± 0.6 | 23 ± 2 | 0.13 ± 0.01 |
| 4 | PCL-g-PEG2k2.6 | 75.3 ± 5.5 | 10.0 ± 0.6 | 26 ± 4 | 0.30 ± 0.05 |
| 5 | PCL-g-PEG2k3.2 | 70.1 ± 8 | 10.6 ± 0.5 | 34 ± 1 | 0.24 ± 0.01 |
| 6 | PCL-g-PEG2k4.9 | 56.0 ± 2.6 | 9.4 ± 0.7 | 51 ± 7 | 0.50 ± 0.10 |
| 7 | PCL-g-PEG2k5.4 | 57.8 ± 5.4 | 8.2 ± 1.2 | 48 ± 4 | 0.43 ± 0.03 |
| 8 | PCL-b-PEG5k2.6 | 80.3 ± 9.9 | 10.7 ± 0.9 | 56 ± 3 | 0.22 ± 0.04 |
Coming to the comparison between linear and graft copolymers, no influence of the architecture was found for a same EG/CL ratio (EG/CL = 2.6, entries 1 and 8). Similar EEs and DLs were obtained for PCL-g-PEG2k2.6 (PCL = 21300 g mol−1; PEG = 2000 g mol−1) and PCL-b-PEG5k2.6 (PCL = 5000 g mol−1; PEG = 5000 g mol−1). For comparison lower molecular weights PCL-b-PEG copolymers with a same EG/CL = 2.6 ratio are reported in the literature for the encapsulation of curcumin. Liu et al. synthesized PCL2k-b-PEG2k and prepared curcumin-loaded polymer micelles by a solid dispersion method.38 The DL and EE were, respectively, 14.8 ± 0.1 wt% and 98.9 ± 0.7 wt%. Same results were reported by Song et al. (PEG2.4k–PCL2.4k)50 and Gou et al. (PEG2k–PCL2k)51 who used thin-film hydration or nanoprecipitation methods for micelle preparation, respectively. These values are higher than the ones obtained with PCL-g-PEG2k2.6. This can be explained by both the encapsulation method (thin film hydration or solid dispersion method vs. self-assembly method) and the lower molecular weight of the reported diblock copolymers (<5000 g mol−1vs. >10000 g mol−1).
The size of the curcumin loaded nanoaggregates was in the range of 23 nm to 56 nm for all copolymers. Curcumin loaded samples were slightly larger than the blank nanoaggregates, except for the more hydrophilic copolymer PCL-g-PEG2k5.4 whose average size decreased from 73 to 48 nm. This may be due to the fact that curcumin strongly interacts with PCL,51 which may induce a more compacted structure of the initially looser and more swollen nanoaggregates obtained with the most hydrophilic copolymer. As for the blank nanoaggregates (Table 2), for the same reasons, the trend was that larger loaded nanoaggregates were obtained for the more hydrophilic copolymers. The polydispersity of drug loaded samples was large and not very different from the one for non-loaded samples showing that curcumin has little influence on the nanoaggregation behavior of the copolymers. This absence of influence is due to the less controlled self-aggregation of the graft copolymers and, for the lower EG/CL ratios, the rapid folding of the hydrophobic core. One should note that for graft copolymers, a low extent of larger aggregates (<30% in DLS intensity mode) is found with sizes between 100 and 200 nm depending on the copolymer. This was not observed with the well-defined linear PCL-b-PEG5k2.6 giving a more dynamic and homogenous system with better self-assembly compared to the uncontrolled self-aggregation of the graft copolymers. It is noteworthy that PCL-g-PEG2k2.6 (Mn ≈ 25000 g mol−1) yielded nanoaggregates with DH ≈ 26 nm, i.e. of the same size as the micelles generated from low molecular weight PCL-b-PEG2k2.6 (Mn ≈ 4000–5000 g mol−1).50,51 For comparison, PCL-b-PEG5k2.6 with Mn = 10000 g mol−1 yielded larger micelles of DH ≈ 55 nm. This is due to the strong folding of the high molecular weight PCL backbone in PCL-g-PEG2k2.6.
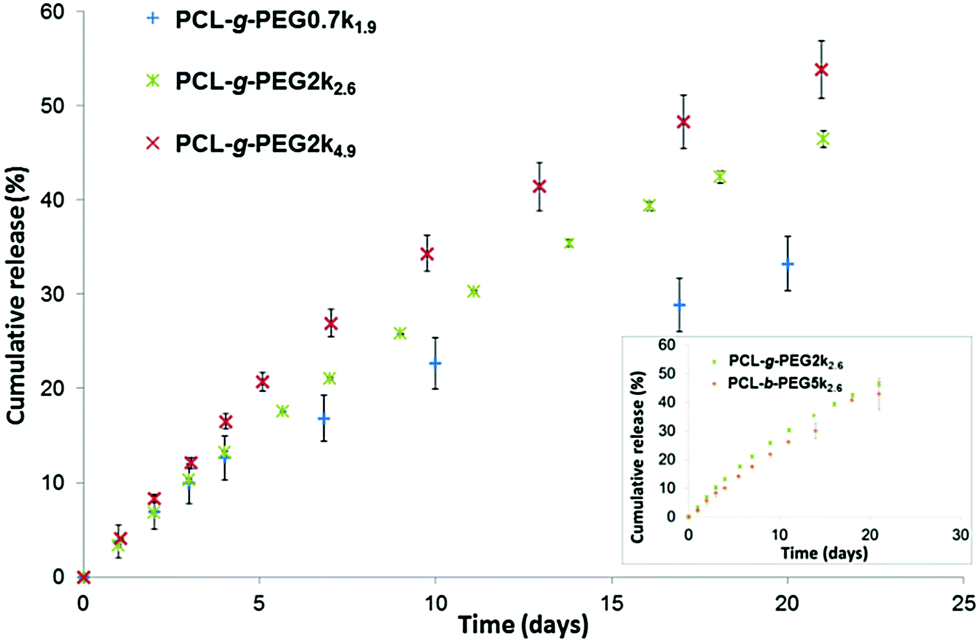 | ||
| Fig. 3 Cumulative release of curcumin from PCL-g-PEG nanoaggregates as a function of EG/CL ratio. The inset shows the comparison between PCL-g-PEG2k2.6 and PCL-b-PEG5k2.6 copolymers. (Data are expressed as means ± SD and correspond to measurements in triplicate.) | ||
Similar release profiles were obtained with PCL-g-PEG2k2.6 and PCL-b-PEG5k2.6 copolymers (Fig. 3, inset). The two architectures show the same drug loading and release capacities, their main difference is therefore the smaller size of the nanoaggregates in the graft copolymer (DH ≈ 26 nm) compared to the ones obtained from the linear block copolymer (DH ≈ 55 nm). This is not the case for the block copolymers with the same EG/CL = 2.6 ratios but with a lower molecular weight. A 21% release was reported after 24 h for PEG2k–PCL2k diblocks, and ca. 50% release after 7 to 9 days.38,51 For comparison, similar ratios were obtained after 5 days and ca. 20 days with the graft copolymer PCL-g-PEG2k2.6.
Based on the results obtained during our study, we can conclude that our nanoaggregates are able to release their payload in a controlled manner, which depends on the hydrophilicity of the used PCL-g-PEG. It is worth mentioning that although the reported release kinetics may appear slow, the situation in the body is slightly different. First, as nanoaggregates circulate in the body the released drug is immediately redistributed, thus the release kinetics will probably be significantly higher. Second, in the frame of cancer therapy, such nanoaggregates are expected to rapidly accumulate in the tumor tissue due to the EPR effect, where their payload will be more rapidly released in vitro due to the faster in vivo degradation of copolyesters in the more acidic tumor environment.
Toxicity of curcumin loaded nanoaggregates
Curcumin loaded PCL-g-PEG nanoaggregates were tested against the breast cancer cell line MCF-7. All tests were performed with associated controls to check the non-toxicity of various culture media used in cytotoxicity tests. In more detail, the effect of a few percents of PBS in the culture medium has been assessed as the buffer is used to prepare the nanoaggregate suspensions prior to cell seeding. The same protocol was used with less than 0.1% of DMSO as a cosolvent to ensure the solubility of free curcumin in the culture medium in accordance with previous studies.52 All media showed no toxicity (Fig. S3, ESI†). In a first assay, the dose dependent viability of MCF-7 cells after 24 h of treatment was tested with the hydrophobic PCL-g-PEG0.7k1.2 to confirm the non-toxicity of unloaded nanoaggregates, and to evaluate the toxicity of curcumin-loaded micelles with a range of curcumin concentration between 0.18 and 18 μg mL−1. These concentrations were selected from the literature.53Fig. 4a confirms the absence of cytotoxicity of the unloaded nanoaggregates. The pertinence of the range of drug concentration was also confirmed with significant differences being obtained in terms of cell viability at low (0.18 μg mL−1) and high (18 μg mL−1) curcumin doses (Fig. 4b). In addition, equivalent cytotoxicities towards the MCF-7 cells were observed for the same dose of curcumin, free or entrapped in the nanoaggregates, which confirmed the activity of the drug released from the PCL-g-PEG nanoaggregates. In more detail, the calculated IC50 values for free curcumin and curcumin-loaded nanoaggregates were very close to the values of 3 μg mL−1 and 5.5 μg mL−1, respectively.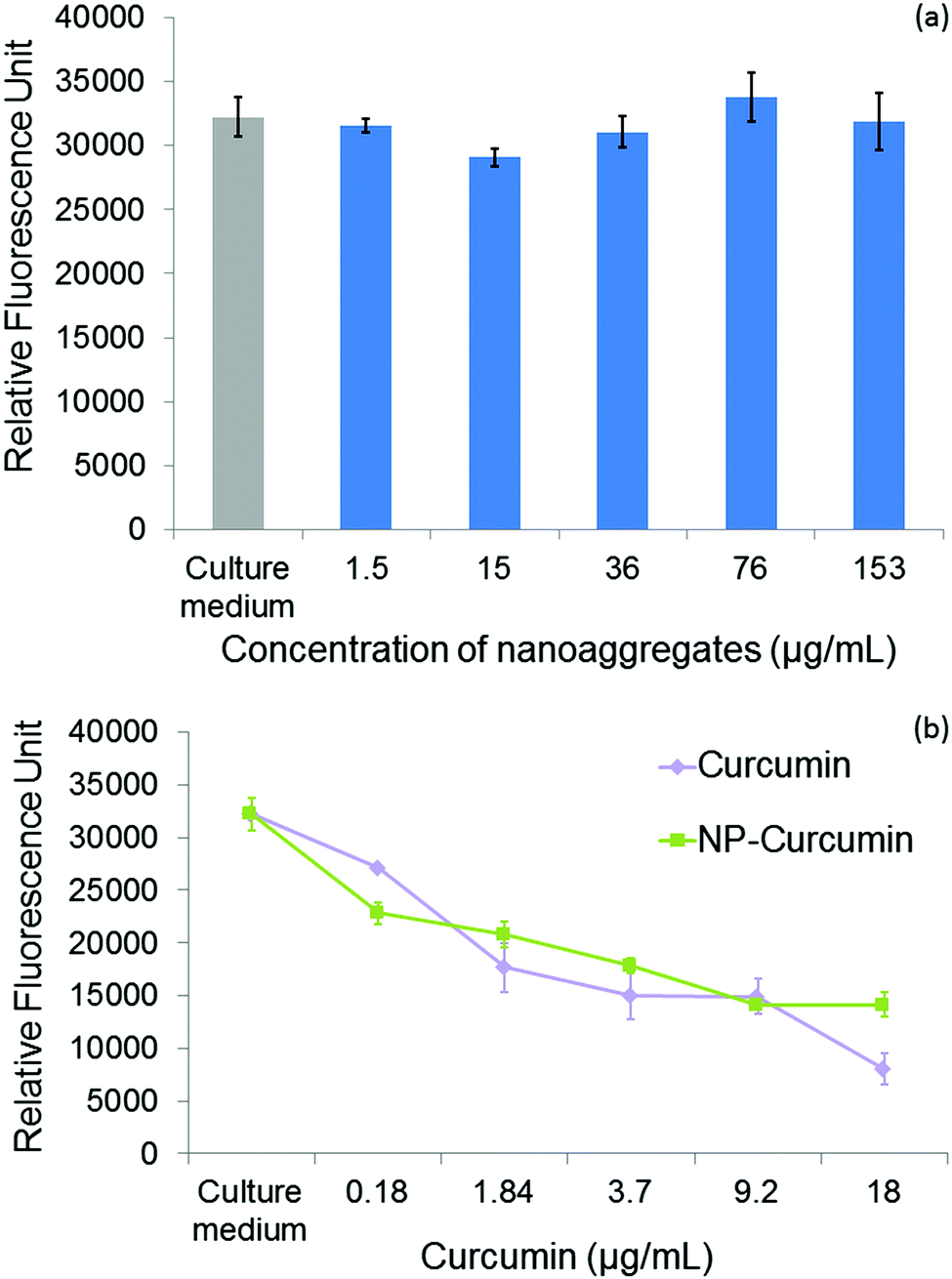 | ||
| Fig. 4 Dose dependent viability of MCF-7 cells after 24 h of treatment (a) with blank nanoaggregates (concentration of nanoaggregates corresponds to the quantity required to encapsulate the determined dose of curcumin in the range 0.18 to 18 μg mL−1) and (b) with free curcumin (squares) or curcumin loaded PCL-g-PEG0.7k1.2 nanoaggregates (diamonds). (Concentrations correspond to μg mL−1 curcumin in the culture medium and data are expressed as means ± SD and correspond to measurements in triplicate.) | ||
Based on these results, curcumin concentrations of 0.18 μg mL−1 (medium viability) and 18 μg mL−1 (low viability) were selected to evaluate the impact of the copolymer amphiphilicity on their cytotoxicity towards MCF-7 cells. Four copolymers with EG/CL ratios ranging from 1.9 to 5.4 were tested. The amount of loaded nanoaggregates added to the culture medium was adjusted based on the calculated drug loadings (Table 4) to provide the selected concentrations of curcumin. Control experiments with blank nanoaggregates were run in parallel. After treatment with blank micelles for 24 h and 48 h, the cell viability was above 90% for the whole range of concentrations examined during the treatment, which confirmed the non-toxicity of various copolymers for all EG/CL ratios (Fig. S4, ESI†). In other words, copolymers had no activity and their effect on the cytotoxicity determination of curcumin loaded in nanoaggregates. MCF-7 cell viability was then evaluated in the presence of curcumin-loaded nanoaggregates as shown in Fig. 5.
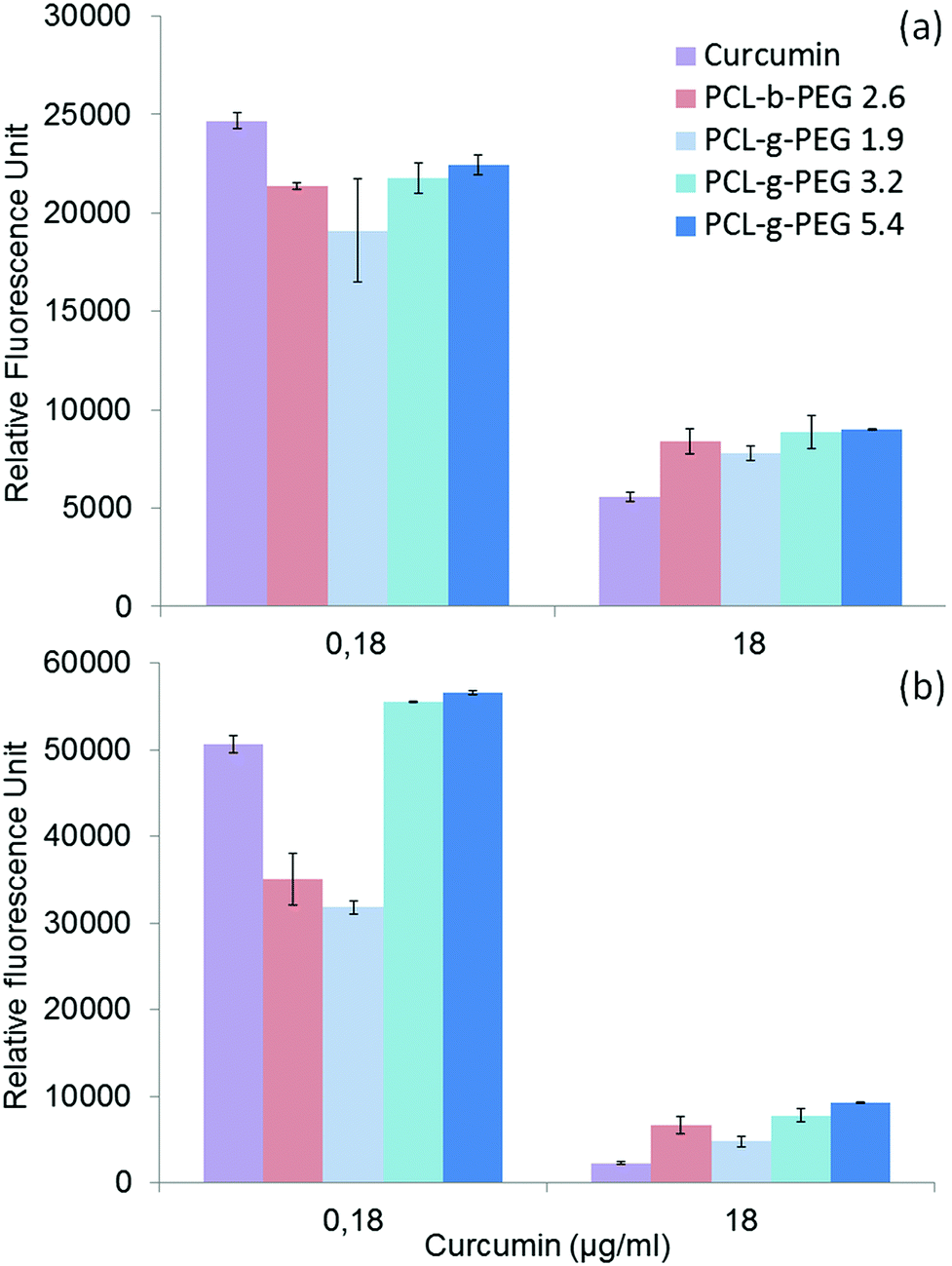 | ||
| Fig. 5 In vitro cell cytotoxicity of curcumin and curcumin-loaded nanoaggregates as a function of the PCL-g-PEG copolymer amphiphilicity after (a) 24 h and (b) 48 h. (Data are expressed as means ± SD and correspond to measurements in triplicate.) | ||
After 24 h cell viabilities were in the range 30 to 40% for all formulations, including free curcumin. However, at the lower dose of 0.18 μg mL−1 the most hydrophobic copolymer PCL-g-PEG0.7k1.9 yielded the highest cytotoxicity. After 48 h of treatment the cell viability was only 60% for PCL-g-PEG0.7k1.9 whereas all other formulations were not cytotoxic. These results show a higher cytotoxicity towards MCF-7 cancer cells for the most hydrophobic copolymer. At low curcumin doses, a strong increase of activity is gained from the use of the PCL-g-PEG0.7k1.9 copolymer compared to free curcumin. Considering the drug release profiles (Fig. 3), the observed differences cannot be ascribed to higher curcumin content in the culture medium as all copolymers would release the same amount of their payload after 48 h (∼7%). The difference in activity is therefore based on differences in the PCL-g-PEG composition, architecture, and consequently on different amphiphilicities of various nanoaggregates. Cellular uptake dependence on these factors is believed to be involved here. It was previously reported that a higher cellular uptake was observed for hydrophobic PEG2k– or PEG5k–PLA block and miktoarm copolymers with EG/LA >2.5,17 whereas some others reported the inverse effect for PEG2k–PCL block copolymers with EG/CL < 2.5.19 This discrepancy in interpretations demonstrates the difficulty in linking the cytotoxic effect of drug loaded vectors with a single parameter. Besides the pure release parameters, the resulting cytotoxicity depends on many factors that can influence the endocytosis pathways, such as the particle size, morphology, surface charge, and of course, the amphiphilicity of the copolymers.
In the present study, the higher efficiency of PCL-g-PEG0.7k1.9 over its grafted counterparts is mainly related to the PEG chain length rather than the pure amphiphilic balance, the nanoaggregate sizes or the grafting number as supported by the following points. First, despite very different EG/CL ratios, all copolymers lead to loaded nanoaggregates of similar sizes (ca. 40 nm in diameter), which implies that size is not a critical factor. Second, considering the average grafting number, one should note that the grafting number of PCL-g-PEG0.7k1.9 and PCL-g-PEG2k5.4 is ∼20 instead of 10 in PCL-g-PEG2k3.2. Despite the twice as low grafting number and lower hydrophilicity, curcumin-loaded PCL-g-PEG2k3.2 nanoaggregates behave like PCL-g-PEG2k5.4 in terms of cell cytotoxicity. In the studied EG/CL range, nanoaggregates with shorter PEG chains allow improved cell membrane interactions which result in a better cellular uptake, as illustrated in Scheme 1.
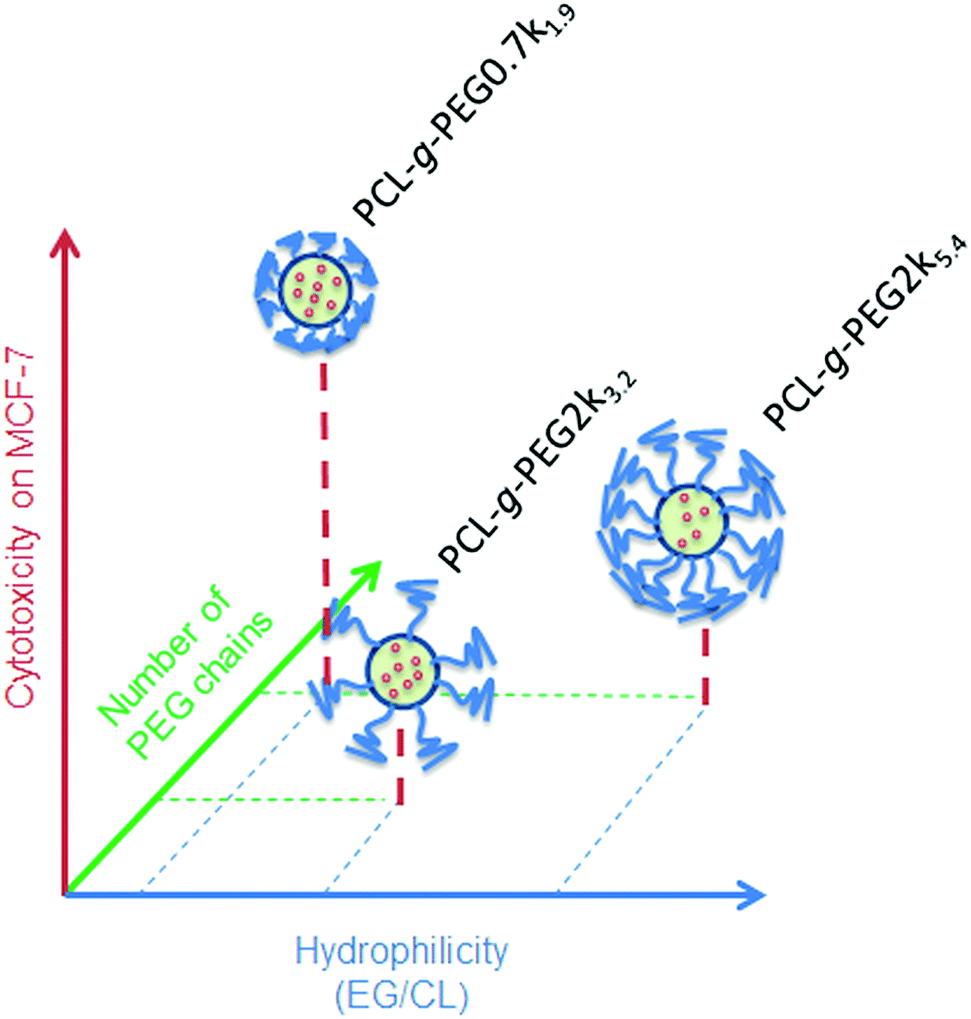 | ||
| Scheme 1 Relative cytotoxicity of curcumin loaded nanoaggregates as a function of the EG/CL ratio, grafting number and PEG molecular weight. | ||
This is confirmed by the dissipative dynamics simulation study carried out by Zhang et al.18 They investigated how nanoparticles coated with different amphiphilic copolymers penetrate cells as a function of the entry force and the penetration force needed to cross the cell membrane. Hydrophobicity, hydrophilic/hydrophobic block repartition and also the rigidity of the polymer presented to the membrane had a strong impact. Rigid copolymers showed better capability and needed less time to pass through the cell membrane. This is in total accordance with our findings, where shorter, more rigid and less hydrophilic PEG0.7k segments are expected to require less energy to pass through the cell membrane compared to PEG2k ones. An in-depth study based on fluorescence microscopy will be undertaken in a future work to elucidate this particular point.
Finally, the importance of the architecture is clearly visible when comparing block and graft copolymers. Based on the PEG molecular weight and the EG/CL ratio, one might expect a lower efficiency for the PCL-b-PEG5k2.6 diblock copolymer but it is comparable to the PCL-g-PEG0.7k1.9 graft copolymer. The polymer rigidity might again be a critical factor here.
In summary, the modification provided by our synthetic approach for fabricating PCL-g-PEG copolymers offers great opportunities and high potential for efficient DDSs with high cytotoxicity against the selected MCF-7 cancer cell line.
Of course, to prove the applicability of our nanoaggregates as DDS, further investigations are needed and underway both in terms of formulation and freeze-drying of the DDS and in terms of in vivo applicability. However, considering the obtained results, it is our belief that the proposed PCL–PEG graft copolymers represent a good alternative to the classical diblock copolymers. To date, although the latter might appear as more viable economically, PCL–PEG graft copolymers have various advantages compared to their linear counterparts. First, their CACs are about 10 times lower, which is of importance for formulation and economic reasons. Second, although minor populations of larger aggregates are obtained with the graft copolymers, they present more compact structures with twice lower hydrodynamic diameters compared to their linear counterparts, which may be of benefit for tissue penetration. Last but not least, the proposed PCL–PEG graft copolymers present the main advantage of being tunable, depending on the target application. When diblock copolymers can only be functionalized on their chain ends, graft copolymers obtained by the thiol–yne approach, thanks to the presence of unreacted alkyne groups (cf. grafting efficiency), could be further modified with higher substitution degrees or multifunctionalities. This particular point should be the matter of future work.
Conclusion
In this work, the easy modification offered by the photo-thiol–yne approach to generate amphiphilic copolymers was shown. PCL-g-PEG copolymers whose amphiphilicity was easily tuned from hydrophobic (EG/CL = 1.9) to hydrophilic (EG/CL = 5.4) have been synthesized. Their CAC is low, in the range 1 to 5 mg L−1, and it increased with the increase of hydrophilicity. The grafting number was found to be critical for the CAC as PCL-g-PEGs with the same grafting number present similar CAC values independent of the EG/CL ratio. Nanoaggregates were easily loaded with various drugs with the DL being dependent on the APIs. Best results were obtained with curcumin that could be loaded at 12 wt%. Curcumin loaded nanoaggregates present sizes in the range 23 nm to 55 nm for all copolymers. All curcumin release curves present the no burst effect although a faster release is obtained from nanoaggregates with higher hydrophilicity. Finally, in terms of cytotoxicity against MCF-7 cells, PCL-g-PEG0.7k1.9 is a better DDS candidate due to a combination of higher hydrophobicity and shorter PEG chains that may lead to a better cellular uptake in vitro. Of course, the selection of suitable candidates for treatment of selected diseases has to be based on in vivo evaluation and we believe that the results summarized in the present paper could be usable for the selection of polymer nanoaggregates for future preliminary in vivo experiments.Acknowledgements
The authors wish to thank the Azm & Saade Association for Assala Al Samad's PhD fellowship, Prof. Rachid Makhloufi from the Institut Français Prague as well as Campus France for Eva Koziolová's internship (Prisme Project 185CZEB160040), Stéphane Dejean for 1H NMR analyses and Aurélien Lebrun of the Laboratoire de mesures physiques (LMP)-University Montpellier 2 for DOSY NMR analyses. The work was also supported by the Ministry of Education, Youth and Sports of CR within the National Sustainability Program I (NPU I), Project POLYMAT LO1507 and Grant Agency of the Czech Republic (grant 15-02986S).Notes and references
- G. S. Kwon and K. Kataoka, Adv. Drug Delivery Rev., 2012, 64, 237–245 CrossRef.
- H. Otsuka, Y. Nagasaki and K. Kataoka, Adv. Drug Delivery Rev., 2012, 64, 246–255 CrossRef.
- G. Gaucher, R. H. Marchessault and J. C. Leroux, J. Controlled Release, 2010, 143, 2–12 CrossRef CAS PubMed.
- J. Nicolas, S. Mura, D. Brambilla, N. Mackiewicz and P. Couvreur, Chem. Soc. Rev., 2013, 42, 1147–1235 RSC.
- V. Weissig, T. K. Pettinger and N. Murdock, Int. J. Nanomed., 2014, 9, 4357–4373 CrossRef CAS PubMed.
- A. L. Sisson, D. Ekinci and A. Lendlein, Polymer, 2013, 54, 4333–4350 CrossRef CAS.
- M. A. Woodruff and D. W. Hutmacher, Prog. Polym. Sci., 2010, 35, 1217–1256 CrossRef CAS.
- T. K. Dash and V. B. Konkimalla, J. Controlled Release, 2012, 158, 15–33 CrossRef CAS PubMed.
- F. Bahadori, A. Dag, H. Durmaz, N. Cakir, H. Onyuksel, U. Tunca, G. Topcu and G. Hizal, Polymers, 2014, 6, 214–242 CrossRef.
- X. Gao, B. L. Wang, X. W. Wei, W. Rao, F. Ai, F. Zhao, K. Men, B. W. Yang, X. Y. Liu, M. J. Huang, M. L. Gou, Z. Y. Qian, N. Huang and Y. Q. Wei, Int. J. Nanomed., 2013, 8, 971–982 CrossRef PubMed.
- S. A. Senevirathne, K. E. Washington, M. C. Biewer and M. C. Stefan, J. Mater. Chem. B, 2016, 4, 360–370 RSC.
- M. Ukawala, T. Rajyaguru, K. Chaudhari, A. S. Manjappa, S. Pimple, A. K. Babbar, R. Mathur, A. K. Mishra and R. S. R. Murthy, Drug Delivery, 2012, 19, 155–167 CrossRef CAS PubMed.
- K. Yoon, H. C. Kang, L. Li, H. Cho, M. K. Park, E. Lee, Y. H. Bae and K. M. Huh, Polym. Chem., 2015, 6, 531–542 RSC.
- Z. X. Du, J. T. Xu and Z. Q. Fan, Macromol. Rapid Commun., 2008, 29, 467–471 CrossRef CAS.
- E. A. Simone, T. D. Dziubla, F. Colon-Gonzalez, D. E. Discher and V. R. Muzykantov, Biomacromolecules, 2007, 8, 3914–3921 CrossRef CAS PubMed.
- R. S. Lee and Y. T. Huang, Polym. J., 2010, 42, 304–312 CrossRef CAS.
- C. Garofalo, G. Capuano, R. Sottile, R. Tallerico, R. Adami, E. Reverchon, E. Carbone, L. Izzo and D. Pappalardo, Biomacromolecules, 2014, 15, 403–415 CrossRef CAS PubMed.
- L. Y. Zhang, M. Becton and X. Q. Wang, J. Phys. Chem. B, 2015, 119, 3786–3794 CrossRef CAS PubMed.
- Z. Zhang, Q. Q. Qu, J. R. Li and S. B. Zhou, Macromol. Biosci., 2013, 13, 789–798 CrossRef CAS PubMed.
- P. Elamanchili, C. Mceachern and H. Burt, J. Pharm. Sci., 2009, 98, 945–958 CrossRef CAS PubMed.
- C. Hoskins, P. K. Thoo-Lin and W. P. Cheng, Ther. Delivery, 2012, 3, 59–79 CrossRef CAS.
- J. Logie, S. C. Owen, C. K. McLaughlin and M. S. Shoichet, Chem. Mater., 2014, 26, 2847–2855 CrossRef CAS.
- Z. S. Gao and A. Eisenberg, Macromolecules, 1993, 26, 7353–7360 CrossRef CAS.
- C. L. Lo, C. K. Huang, K. M. Lin and G. H. Hsiue, Biomaterials, 2007, 28, 1225–1235 CrossRef CAS PubMed.
- F. Coumes, V. Darcos, D. Domurado, S. M. Li and J. Coudane, Polym. Chem., 2013, 4, 3705–3713 RSC.
- X. Jiang, E. B. Vogel, M. R. Smith and G. L. Baker, Macromolecules, 2008, 41, 1937–1944 CrossRef CAS.
- X. W. Jiang, M. R. Smith and G. L. Baker, Macromolecules, 2008, 41, 318–324 CrossRef CAS.
- S. Onbulak, S. Tempelaar, R. J. Pounder, O. Gok, R. Sanyal, A. P. Dove and A. Sanyal, Macromolecules, 2012, 45, 1715–1722 CrossRef CAS.
- H. Y. Li, R. Riva, R. Jérôme and P. Lecomte, Macromolecules, 2007, 40, 824–831 CrossRef CAS.
- R. Riva, P. Schmeits, F. Stoffelbach, C. Jérôme, R. Jérôme and P. Lecomte, Chem. Commun., 2005, 5334–5336, 10.1039/B5102182k.
- K. Zhang, Y. Wang, W. P. Zhu, X. D. Li and Z. Q. Shen, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 2045–2052 CrossRef CAS.
- B. Parrish, R. B. Breitenkamp and T. Emrick, J. Am. Chem. Soc., 2005, 127, 7404–7410 CrossRef CAS PubMed.
- J. Rieger, P. Dubois, R. Jérôme and C. Jérôme, Langmuir, 2006, 22, 7471–7479 CrossRef CAS PubMed.
- J. Coudane, E. Laurent and M. Vert, Macromol. Rapid Commun., 2004, 25, 1865–1869 CrossRef CAS.
- S. Ponsart, J. Coudane, J. McGrath and M. Vert, J. Bioact. Compat. Polym., 2002, 17, 417–432 CrossRef CAS.
- A. Al Samad, Y. Bakkour, C. Fanny, F. El Omar, J. Coudane and B. Nottelet, Polym. Chem., 2015, 6, 5093–5102 RSC.
- X. Gao, F. J. Zheng, G. Guo, X. X. Liu, R. R. Fan, Z. Y. Qian, N. Huang and Y. Q. Wei, J. Mater. Chem. B, 2013, 1, 5778–5790 RSC.
- L. Liu, L. Sun, Q. Wu, W. Guo, L. Li, Y. Chen, Y. Li, C. Y. Gong, Z. Qian and Y. Q. Wei, Int. J. Pharm., 2013, 443, 175–182 CrossRef CAS PubMed.
- H. Danafar, K. Rostamizadeh, S. Davaran and M. Hamidi, Drug Dev. Ind. Pharm., 2014, 40, 1411–1420 CrossRef CAS PubMed.
- Y. J. Wang, M. H. Pan, A. L. Cheng, L. I. Lin, Y. S. Ho, C. Y. Hsieh and J. K. Lin, J. Pharm. Biomed. Anal., 1997, 15, 1867–1876 CrossRef CAS PubMed.
- M. Wilhelm, C. L. Zhao, Y. C. Wang, R. L. Xu, M. A. Winnik, J. L. Mura, G. Riess and M. D. Croucher, Macromolecules, 1991, 24, 1033–1040 CrossRef CAS.
- Y. Cao, J. Q. Zhao, Y. M. Zhang, J. F. Liu, J. J. Liu, A. J. Dong and L. D. Deng, RSC Adv., 2015, 5, 28060–28069 RSC.
- X. W. Wei, C. Y. Gong, M. Y. Gou, S. Z. Fu, Q. F. Guo, S. Shi, F. Luo, G. Guo, L. Y. Qiu and Z. Y. Qian, Int. J. Pharm., 2009, 381, 1–18 CrossRef CAS PubMed.
- P. J. W. Schuijl, H. J. T. Bos and L. Brandsma, Recl. Trav. Chim. Pays-Bas, 1969, 88, 597–608 CrossRef CAS.
- M. Lachia, F. Denes, F. Beaufils and P. Renaud, Org. Lett., 2005, 7, 4103–4106 CrossRef CAS PubMed.
- E. F. Choo, J. Boggs, C. Q. Zhu, J. W. Lubach, N. D. Catron, G. Jenkins, A. J. Souers and R. Voorman, Drug Metab. Dispos., 2014, 42, 207–212 CrossRef CAS PubMed.
- F. Danhier, N. Lecouturier, B. Vroman, C. Jérôme, J. Marchand-Brynaert, O. Feron and V. Preat, J. Controlled Release, 2009, 133, 11–17 CrossRef CAS PubMed.
- R. K. Maheshwari, A. K. Singh, J. Gaddipati and R. C. Srimal, Life Sci., 2006, 78, 2081–2087 CrossRef CAS PubMed.
- M. M. Yallapu, M. Jaggi and S. C. Chauhan, Drug Discovery Today, 2012, 17, 71–80 CrossRef CAS PubMed.
- Z. M. Song, W. X. Zhu, N. Liu, F. Y. Yang and R. L. Feng, Int. J. Pharm., 2014, 471, 312–321 CrossRef CAS PubMed.
- M. L. Gou, K. Men, H. S. Shi, M. L. Xiang, J. A. Zhang, J. Song, J. L. Long, Y. Wan, F. Luo, X. Zhao and Z. Y. Qian, Nanoscale, 2011, 3, 1558–1567 RSC.
- L. Y. Zhao, J. C. Du, Y. W. Duan, Y. N. Zang, H. S. Zhang, C. F. Yang, F. L. Cao and G. X. Zhai, Colloids Surf., B, 2012, 97, 101–108 CrossRef CAS PubMed.
- M. Lopez-Lazaro, Mol. Nutr. Food Res., 2008, 52, S103–S127 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Scheme S1. Schematic representation of the preparation of the PCL-g-PEG copolymers by a thiol–yne approach. Fig. S1. Excitation spectra of pyrene as a function of the concentration of PCL-g-PEG2k3.2; Fig. S2. Fit curves for the curcumin release from PCL-g-PEG copolymers; Fig. S3. Viability of MCF-7 cells in different media; Fig. S4. Viability of MCF-7 cells in the presence of non-loaded nanoaggregates; Table S1. SEC analysis of PCL-g-PEG during degradation; and Table S2. The chemical structure and solubility parameters of tested APIs. See DOI: 10.1039/c6tb01841f |
| This journal is © The Royal Society of Chemistry 2016 |