
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAgglutination of bacteria using polyvalent nanoparticles of aggregation-induced emissive thiophthalonitrile dyes†
Bettina
Schmidt
ab,
Shrikrishnan
Sankaran
ab,
Linda
Stegemann
c,
Cristian A.
Strassert
c,
Pascal
Jonkheijm
*ab and
Jens
Voskuhl
*ad
aBioinspired Molecular Engineering Laboratory of the MIRA Institute for Biomedical Technology and Technical Medicine, University of Twente, P.O. Box 217, 7500 AE Enschede, Netherlands
bMolecular Nanofabrication Group of the MESA+ Institute for Nanotechnology, University of Twente, P.O. Box 217, 7500 AE Enschede, Netherlands. E-mail: p.jonkheijn@utwente.nl
cPhysikalisches Institut – Center for Nanotechnology, Westfälische Wilhelms-Universität Münster, Heisenbergstrasse 11, 48149 Münster, Germany
dInstitute of Organic Chemistry, University of Duisburg-Essen, 45117 Essen, Germany. E-mail: jens.voskuhl@uni-due.de
First published on 13th June 2016
Abstract
A novel class of aggregation-induced emissive bis(phenylthio)phthalonitrile dyes were synthesized. These dyes assembled into nanoparticles that were equipped with mannose units. The nanoparticles underwent selective interactions with lectins and bacteria. The bright fluorescent aggregates aid in the visualization of the agglutination of bacteria.
Introduction
Since the appearance of first examples of molecules with aggregation-induced emission (AIE) properties,1 this phenomenon has attracted a lot of interest.2 These compounds bear, contrary to other organic dyes, the ability to show strong fluorescence upon aggregation. Typical molecules that are prone to exhibit AIE have a highly dynamic, propeller-like structure bearing phenyl rings that are able to rotate against the central unit and annihilate excitons in solution.3–6 However, in the solid or in the aggregated state, this rotation is hindered leading to stacking of the molecules and with this π–π-stacking charge transfer can occur leading to emission. This phenomenon was used for diverse purposes such as organic light emitting diodes,7 sensors for metal ions8 and explosives,9 mapping of cell viscosity10 and cell targeting11 as well as interactions with bacteria12 and proteins.13 When molecules exhibit AIE also in buffered media, these molecules can be readily applied in existing bioanalytical systems, which are often based on fluorescent read-out systems.Here we report a facile synthesis route towards a new class of compounds, bis(phenylthio)phthalonitriles (SPn), that show strong aggregation-induced emission in aqueous media (Fig. 1). The synthesized molecules were easily equipped with bioactive moieties such as carbohydrates via Cu(I) catalysed click chemistry.
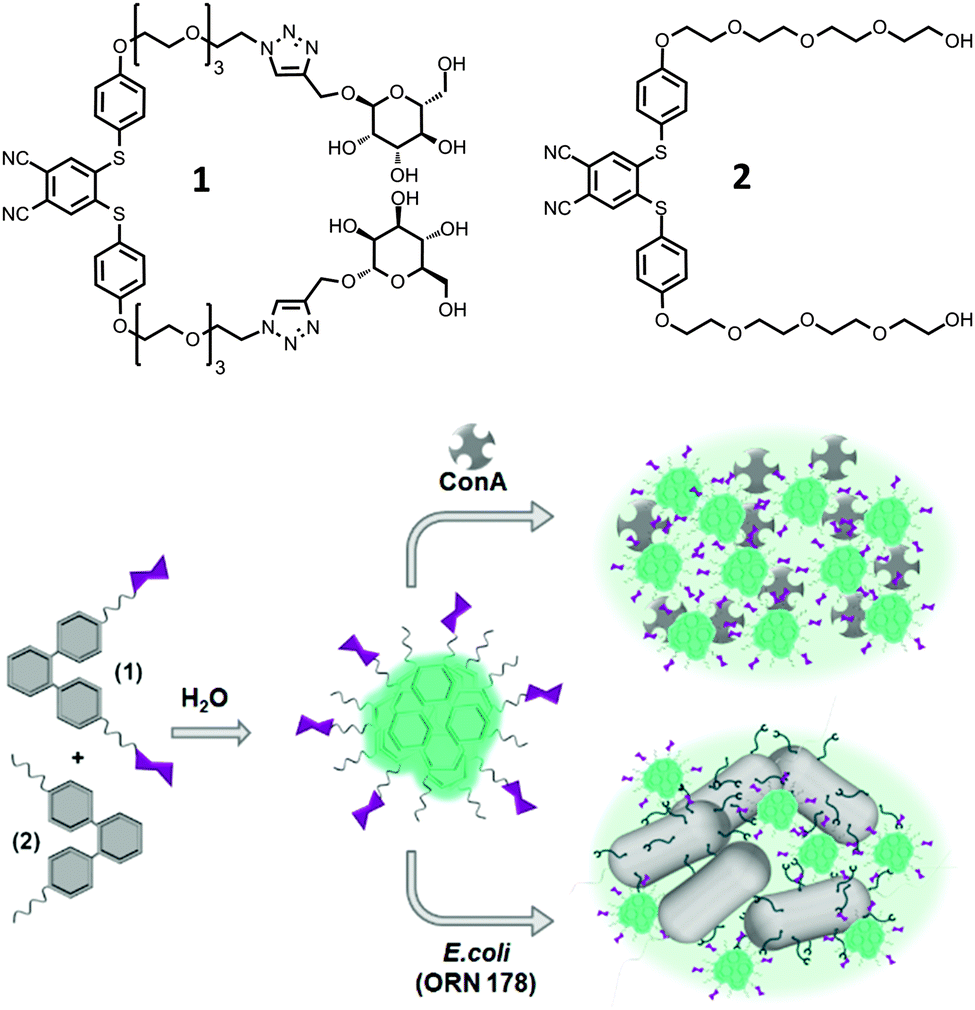 | ||
| Fig. 1 Structures of molecules used in this study and a schematic representation of the aggregation of lectins and bacteria in the presence of particles composed of SPn 1 and 2. | ||
Results and discussion
Synthesis
The synthesis started from commercially available 4,5-dichlorophthalonitrile that was coupled to 4-mercaptophenol in a thioetherification reaction under basic conditions in good yield (see Scheme S1, ESI†). The thioether diol was then coupled in a substitution reaction to different tetraethylene glycol derivatives. SPn 2 was obtained in 54% yield, while the azide-terminated SPn analogue was obtained in 50% yield, which was able to undergo Cu(I)-assisted azide–alkyne click reaction with an alkyne-terminated carbohydrate in its protected form. Here we selected α-D-mannose since it can bind actively to FimH receptors of E. coli strains and to carbohydrate selective proteins, the so-called lectins. After deprotection by sodium methoxide and purification by column chromatography the final compound SPn 1 was isolated in good yield (79%). The structures of the final compound were confirmed by mass spectrometry and 1H and 13C NMR spectroscopy. Details of the synthesis and characterization can be found in the ESI.†AIE properties and self-assembly
Dissolving SPn compound 1 or 2 or mixtures of both in water or buffer led to dispersed aggregates that exhibited strong emission when excited at a wavelength (λ) of 330 nm. In contrast, when the SPn compounds were dissolved in THF or methanol faint emission was detected. When minute amounts of 100 μM solutions of 1 or 2 in water were deposited onto copper grids and negatively stained (1 wt% uranyl acetate), inspection of the samples using a TEM revealed spherical objects of ca. 16 nm in diameter (Fig. 2 and Fig. S1, ESI†). Also SEM samples were prepared from 100 μM water solutions on silicon wafers (Fig. S2, ESI†). The density of the particles was lower in comparison with the TEM images, but the size of the objects was similar, as can be seen in Fig. 2. In solution, larger nanoparticles were detected using dynamic light scattering probably due to hydration of the nanoparticles (Fig. S22, ESI†). These results are indicative of aggregated species that were highly fluorescent in water, whereas in a good solvent, such as THF, the aggregates were dissolved and the molecules were non-fluorescent. | ||
| Fig. 2 TEM image of deposits of 100 μM aqueous solution of 1, negatively stained with 1wt% uranyl acetate, and size distribution of TEM (blue-green) and SEM (black-grey) samples of 1. | ||
The transition between fluorescent aggregates and non-fluorescent molecules was studied in more detail by measuring the fluorescence spectra of a series of solutions of SPn 1 in a varying ratio of THF and water at constant SPn concentration (100 μM, Fig. 3 and Fig. S3, ESI†). A red shift of the λmax in the fluorescence spectra was observed to λ = 488 nm when going from THF to water solutions. When the change in emission intensity at λmax = 488 nm was plotted against the water fraction (Fig. 3C), a sudden rise in emission intensity occurs when the fraction of good solvent THF declined below 20%. This change is also clearly visible in the series of photographs of the solutions in vials shown in Fig. 3A. In the recorded UV/Vis spectra of those THF–water solutions a decrease in absorption was observed (Fig. S4, ESI†). While in THF–water mixtures the emission appeared beyond a water fraction of 80%, in methanol–water mixtures the emission appeared at a water fraction of 60% (Fig. 3C) verifying that the polarity of the solvent is driving the aggregation of SPns.
 | ||
| Fig. 3 (A) Photographs of vials consisting of 100 μM solutions of SPn 1 in water–THF mixtures with increasing water fraction; (B) fluorescence spectra of 100 μM SPn 1 in water–THF mixtures; (C) shift in the fluorescence emission intensity at λ = 488 nm of 1 depending on the water fraction in water–THF and water–methanol mixtures. | ||
In the case of SPn 2 similar spectral changes in emission were observed upon aggregation (Fig. S3, ESI†) indicating that peripheral substitution of the SPn core has little effect on the photophysical properties of the SPn aggregates.
To evaluate the critical aggregation concentration of SPn 1, fluorescence spectra of a series of aqueous solutions of SPn 1 in the range of 0.05–100 μM were measured (Fig. S5, ESI†). The maximum wavelength remained constant in water when diluting from 100 μM to 2.5 μM while the emission intensity decreased linearly. These observations show that aggregates were even present at low concentrations. In contrast, upon diluting further from 2.5 μM to 50 nM, the emission maximum was blue shifted while the emission intensity remained nearly constant. The critical aggregation concentration of SPn 1 in water was determined to be around 1 μM (Fig. S6, ESI†). Compound 2 aggregated more gradually without a clear critical concentration but in the same concentration window as SPn 1 (Fig. S6, ESI†). Furthermore the fluorescence quantum yields in water were measured for 2 (6.4%) and 1 (2.2%). Both the fluorescence quantum yield and the fluorescence lifetime were found to decrease when the fraction of THF in the solution was increased above 10–15% confirming that aggregation was lost (Table S1 and Fig. S10 and S11, ESI†).
Upon addition of increasing amounts of surfactant Triton X-100, the fluorescence emission intensity dropped significantly while the position of λmax did not change (Fig. S7, ESI†). This is presumably based on the expansion and rupture of the aggregates by the interference of Triton X-100 with supramolecular structures acting as a potent detergent in agreement with previous observations.14
Next, we performed an experiment to verify that restricted intramolecular rotation can be the mechanism of the AIE observed for our SPns by titrating highly viscous glycerol into a methanolic solution up to a volume fraction of 90% glycerol.3,15Fig. 4 shows a strong increase in the fluorescence intensity beyond 60 vol% glycerol for SPn 1. This observation is in agreement with AIEgens based on a slower intramolecular rotation of periplanar aligned substituents on the central core of the dye as a result of the addition of a viscous component in the solution.3,15
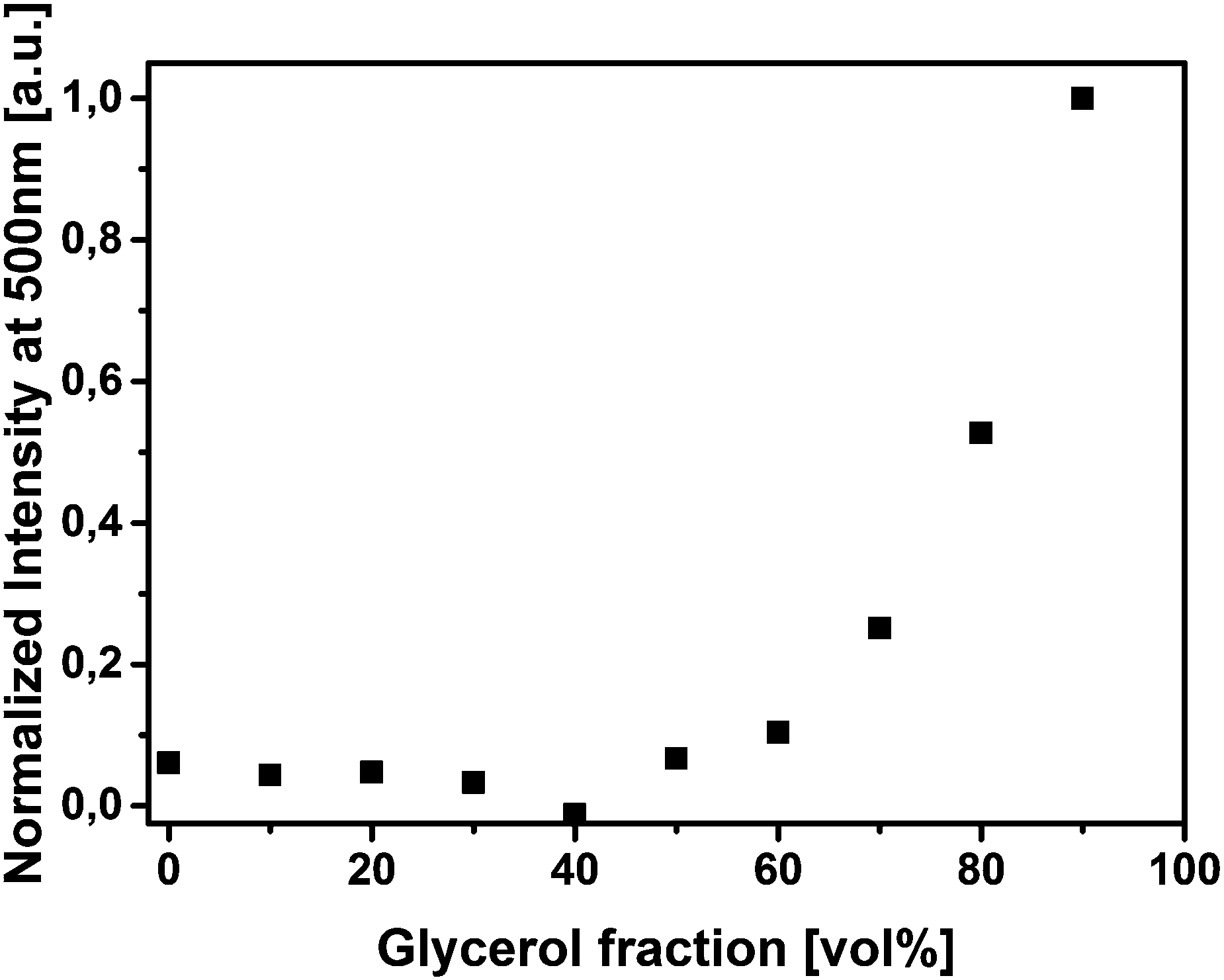 | ||
| Fig. 4 Normalized fluorescence emission intensity at λ = 500 nm as a function of the glycerol volume fraction in methanol–glycerol mixtures of SPn 1 (cSPn = 100 μM). | ||
Agglutination studies using polyvalent SPn assemblies
With these strong emissive nanoparticles in hand, in the next experiment agglutination studies between lectins and the bioactive mannose ligand in SPn aggregates were performed. To this end, Concanavalin A (ConA) was added after 5 min to mixtures of SPn 1 and 2 in different ratios (overall concentration 20 μM, Fig. 5 and Fig. S9, ESI†). The agglutination of SPn assemblies and ConA occurred instantly upon addition and was dependent on the presence of mannose ligands in the aggregates. When SPn 1 was absent, no agglutination was optically detected, while at 10% of 1 a small change in optical density was measured. Agglutination visible to the naked eye started only when 25% of 1 was present in the SPn assemblies.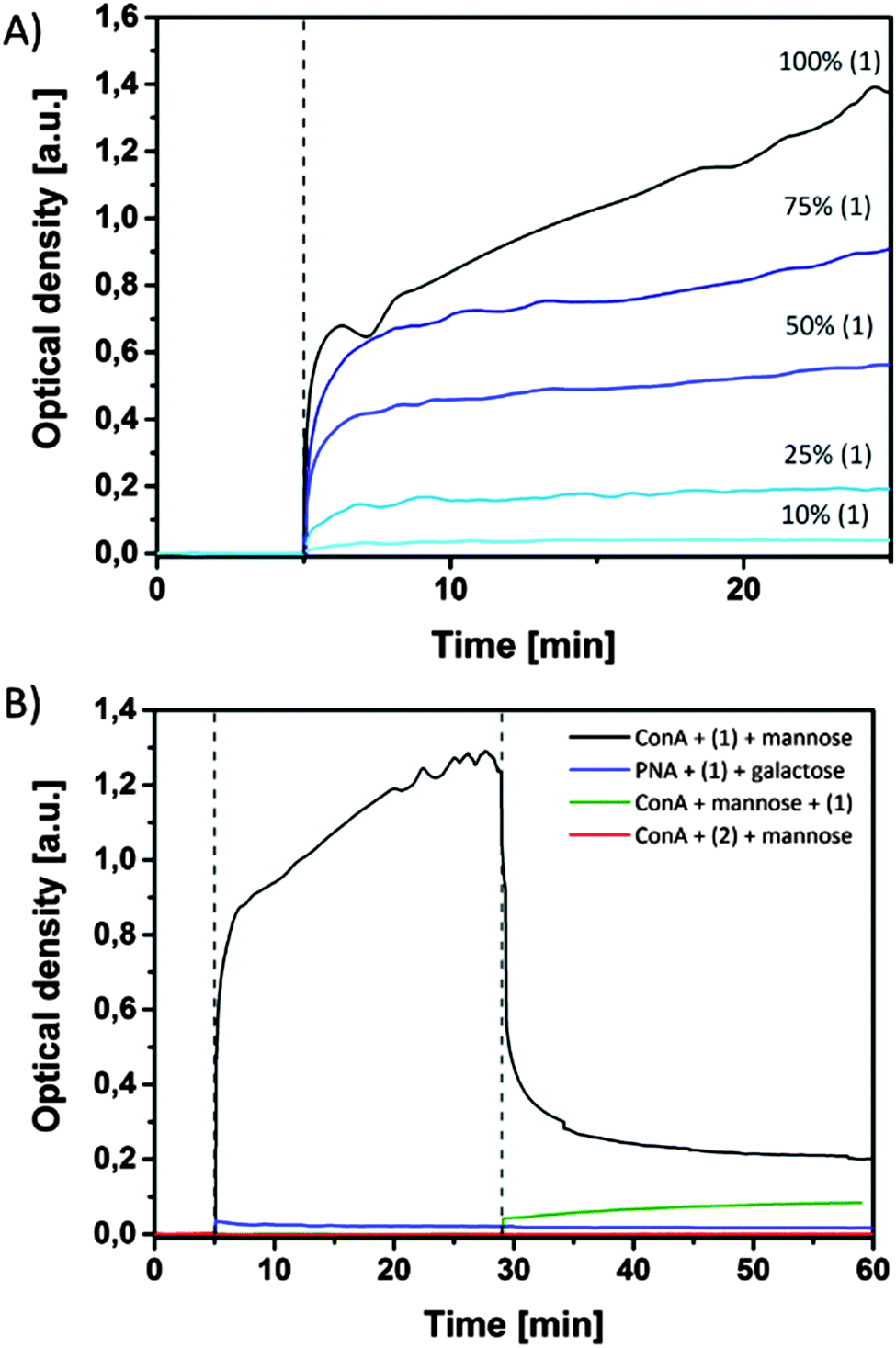 | ||
| Fig. 5 Temporal evolution of the optical density (A) upon adding ConA to solutions of 1 and 2 in different ratios as indicated and (B) upon adding ConA and mannose (αMMP, 10 mM) after 30 min, PNA and galactose after 30 min, ConA and mannose together to a solution of 1 and ConA and mannose together to a solution of 2. As buffer (pH = 7.2) a solution of 10 mM HEPES, 137 mM NaCl, 1 mM CaCl2, and 1 mM MnCl2 was used. | ||
These results show that specific interactions between the mannose moieties and the four binding pockets of the ConA occur. It is known that at least two interactions per protein are needed to induce aggregation.14,16 When the competitor α-methyl-D-mannoside (αMMP, 10 mM) was added to the solution containing the aggregates of assemblies of 1 and ConA, the aggregates re-dissolved within 10 min signified by a significant strong decrease in optical density. This observation indicates that the binding between ConA and mannose-functionalized aggregates is reversible and that an equilibrium was reached between the polyvalent SPn assemblies, ConA and monovalent αMMP. Increasing the concentration of αMMP to 30 mM further prevents the SPn assemblies from aggregating. In another experiment αMMP was added to ConA before adding SPn nanoparticles. The observed increase in optical density was much lower in this case when compared to the experiments described above (Fig. 5, green). This experiment shows that although an excess of αMMP was present, a certain amount of SPn particles was still binding to ConA, resulting in an equilibrium between the three components. When galactose-binding lectin peanut agglutinin (PNA) was used to interact with SPn 1 particles, no agglutination was observed (Fig. 5B), confirming the specific binding of the polyvalent mannose particles to ConA. Importantly, upon agglutination of ConA and SPn particles, the emission properties of the SPn particles were unchanged, which simplifies the use of these particles as fluorescent read-out modality for biological interactions.
In the final experiment the agglutination of bacteria by the polyvalent mannose SPn particles was evaluated. To this end two strains of E. coli were tested.17 ORN 208 bears mutated FimH receptors which should not be able to interact with the mannose SPn nanoparticles. ORN 178 expresses wild-type FimH receptors and should be able to recognize the mannose moieties in the SPn nanoparticles containing 1. Since each aggregate bears hundreds of mannose moieties a polyvalent recognition is expected and bacterial aggregation should occur. We detected no cytotoxic response upon adding the SPn assemblies to the bacterial strains used in this study (Fig. S8, ESI†). From Fig. 6A–C it is apparent that ORN 208 showed neither aggregation nor fluorescence upon incubation with 50 μM SPn 1 aggregates for 2 h, indicating that non-specific interactions, if present, do not lead to bacterial aggregation. In strong contrast, in the case of adding 50 μM SPn 1 nanoparticles to ORN 178, large bacterial aggregates that spanned sizes of around 40 μm in length were observed (Fig. 6D–F). Furthermore fluorescence microscopy images were recorded at the same location. The bacterial aggregates co-localized with the fluorescence confirming the presence of SPn nanoparticles between single bacteria. Aggregation of bacteria started immediately upon addition of the nanoparticles. When the images of samples containing SPn assemblies (50 μM) in the absence of bacteria were recorded, no fluorescent assemblies were detected. When SPn particles of 2 (50 μM), lacking the mannose binding moieties, were incubated with ORN 178, no agglutination was observed confirming the specificity of the interaction between FimH receptors and polyvalent mannose SPn nanoparticles (Fig. S12A, ESI†). Upon dilution of the solution of the SPn 1 particles to 25 μM, the bacterial cluster size significantly decreased (Fig. S13, ESI†). Just above the critical concentration of the SPn assemblies at 5 μM, nearly no clusters, in which more than 10 bacteria agglutinated, were detected, while below the critical concentration at 0.5 μM no clusters were detected. These observations confirm that polyvalent mannose nanoparticles of SPn are required for bacterial agglomeration. After adding αMMP (50 mM, 1 h) to solutions containing ORN 178 and SPn 1 nanoparticles, the large fluorescent bacterial clusters disappeared and only a few, small aggregates remained (Fig. S14, ESI†) confirming the reversibility of the interactions between mannose and FimH receptors.
 | ||
| Fig. 6 Bacterial aggregation induced by 50 μM SPn 1 in medium and imaged by (A) and (D) bright field and (B) and (E) fluorescence microscopy images. Images in (C) and (F) are overlay of these. The upper row (A–C) features strain ORN 208 and the lower row (D–F) features strain ORN 178. Scale bars are 20 μm. | ||
Next, a series of ORN 178 bacterial samples were incubated with SPn assemblies containing different ratios of SPn 1 and 2 (Fig. S12, ESI†). From the bright field images the sizes of bacterial aggregates were analysed. Upon increasing the content of mannose carrying SPn 1, and thereby the mannose density on the SPn nanoparticles, the bacterial aggregation size changed. Interestingly, the maximum bacterial aggregate size was estimated using mixed assemblies of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 SPn 1:SPn 2 and not when mannose-only SPn 1 was used, indicating that crowding of carbohydrates is an important parameter when optimizing the interaction between bacteria and supramolecular particles. The assemblies of SPn containing below 25% 1 showed no bacterial agglutination when defined as clusters of at least 10 bacteria.
:1 SPn 1:SPn 2 and not when mannose-only SPn 1 was used, indicating that crowding of carbohydrates is an important parameter when optimizing the interaction between bacteria and supramolecular particles. The assemblies of SPn containing below 25% 1 showed no bacterial agglutination when defined as clusters of at least 10 bacteria.
Conclusions
Easily accessible thiophthalonitriles form nanoparticles featuring aggregation-induced emission. AIE properties were not changed by attaching mannose moieties to the thiophthalonitriles. The interaction of the polyvalent nanoparticles with ConA and wild-type FimH expressing bacteria was specific and reversible. Moreover, the agglutination of bacteria was dependent on the amount of mannose-functionalized thiophthalonitriles that were mixed into the nanoparticles while maximum agglutination was observed with only 50% of the mannose modified thiophthalonitrile mixed in the nanoparticles. This agglutination process was suitably visualized by monitoring the fluorescent nanoparticles. This new class of AIE dyes bear the opportunity to be used in several biomedical applications since the modification of these compounds in the periphery with bioactive residues is facile, which will be investigated in detail in the future.Experimental
General
Chemicals were purchased from Sigma Aldrich or from Acros Organics and were used without further purification. Reactions were carried out using dried solvent and under an atmosphere of argon. Reactions were monitored by thin-layer chromatography (TLC), which was performed on 0.2 mm Merck precoated silica gel 60 F254 aluminium sheets. Spots were visualized by treatment with basic KMnO4 solution. Column chromatography was carried out on silica gel 60 (0.063–0.2 mm, Merck). NMR spectra were recorded on a Bruker spectrometer (AV400). Chemical shifts are given in units of parts per million (ppm) and expressed relative to the signals of deuterated solvents. Coupling constants (J) are reported in Hertz (Hz). Mass spectra were recorded using a MicroToF spectrometer (Bruker).The TEM measurements were carried out on a Philips CM300ST-FEG TEM machine. The SEM measurements were carried out on a Zeiss Merlin scanning electron microscope.
UV/vis absorption spectra were measured on a Perkin Elmer Lambda 850 spectrophotometer. The UV/vis absorption spectra were corrected for the solvent. Fluorescence spectra were recorded on a Perkin Elmer LS 55 fluorescence spectrometer using an excitation wavelength of 330 nm.
Synthetic procedures of SPn 1 and 2
4,5-Bis((4-(2-(2-(2-(2-hydroxyethoxy)ethoxy)ethoxy)ethoxy)-phenyl)thio)phthalonitrile 2
To a stirred solution of 4,5-bis((4-hydroxyphenyl)thio)phthalonitrile18 (300 mg, 0.8 mmol) in 10 ml of acetonitrile was added K2CO3 (700 mg, 4.8 mmol). The reaction mixture was heated to 90 °C for 30 min followed by the addition of 2-(2-(2-(2-hydroxyethoxy)ethoxy)ethoxy)ethyl 4-methylbenzenesulfonate19 (900 mg, 2.4 mmol) in 10 ml acetonitrile. Stirring was continued at 90 °C for 24 h. The crude mixture was evaporated, re-dissolved in 30 ml of CHCl3 and extracted twice with brine. The organic layer was dried over MgSO4 and evaporated. The residue was subjected to silica column chromatography using CH2Cl2/MeOH 90/10 as eluent. The product was obtained as a colourless waxy solid. Yield 324 mg, 54%. ESI-MS (m/z): calculated for [C36H44N2O10S2Na]+: 751.2317, found: 751.2330. 1H NMR (400 MHz, CDCl3, 298 K): δ = 7.45 (d, J = 8.7 Hz, 4H, 4CH), 7.01 (d, J = 8.8 Hz, 4H, 4CH), 6.84 (s, 2H, 2CH), 4.19 (t, J = 5.0 Hz, 4H, 2CH2), 3.89 (t, J = 5.1 Hz, 4H, 2CH2), 3.74–3.66 (m, 20H, 10CH2), 3.59 (t, J = 5.1 Hz, 4H, 2CH2), 2.49 (s, 2H, 2OH). 13C NMR (100 MHz, CDCl3, 298 K): δ = 161.08, 144.76, 137.49, 129.02, 118.55, 117.02, 115.75, 111.29, 72.64, 71.02, 70.84, 70.76, 70.49, 69.71, 67.87, 61.93.Absorption and fluorescence spectroscopy
Samples were prepared from a 1 mM DCM/MeOH stock solution in the following way: the necessary volume of the stock solution was transferred into a glass vial. The solvent was evaporated at 60 °C. Subsequently, the required amount of water was added and these solutions were sonicated at 50 °C for at least 0.5 h. Solvent mixtures were prepared after this step.Electron microscopy
For the TEM images the solutions of the dinitriles (3 μl of 100 μM in water) were deposited on 200 mesh copper grids for 5 min. After drying the samples, they were stained for 1 min with 5 μl of a uranyl acetate solution (1 wt% in water). After carefully drying the samples the grids were subjected to TEM.For the SEM images the solutions of dinitriles (3 μl of 100 μM in water) were deposited on silicon wafers for 6 min (Fig. S2B–D, ESI†) or 10 min (Fig. S2A, ESI†) before gently removing the remaining solution.
Quantum yield and fluorescence lifetime measurements
Fluorescence lifetimes were recorded on a FluoTime300 spectrometer from PicoQuant equipped with a 300 W ozone-free Xe lamp (250–900 nm), a 10 W Xe flash-lamp (250–900 nm, pulse width <10 μs) with repetition rates of 0.1–300 Hz, an excitation monochromator (Czerny–Turner, 2.7 nm mm−1 dispersion, 1200 grooves per mm, blazed at 300 nm), diode lasers (pulse width <80 ps) operated by a computer-controlled laser driver PDL-820 (repetition rate up to 80 MHz, burst mode for slow and weak decays), two emission monochromators (Czerny–Turner, selectable gratings blazed at 500 nm with 2.7 nm mm−1 dispersion and 1200 grooves per mm, or blazed at 1250 nm with 5.4 nm mm−1 dispersion and 600 grooves per mm), Glan–Thompson polarizers for excitation (Xe-lamps) and emission, a Peltier-thermostatted sample holder from Quantum Northwest (−40–105 °C), and two detectors, namely a PMA Hybrid 40 (transit time spread FWHM <120 ps, 300–720 nm) and a R5509-42 NIR-photomultiplier tube (transit time spread FWHM 1.5 ns, 300–1400 nm) with external cooling (−80 °C) from Hamamatsu. Fluorescence lifetimes were recorded in TCSPC mode using a PicoHarp 300 (minimum base resolution 4 ps). Lifetime analysis was performed using the commercial FluoFit software. The quality of the fit was assessed by minimizing the reduced chi squared function (χ2) and visual inspection of the weighted residuals and their autocorrelation. Luminescence quantum yields were measured using a Hamamatsu Photonics absolute PL quantum yield measurement system (C9920-02) equipped with a L9799-01 CW Xenon light source (150 W), a monochromator, a C7473 photonic multi-channel analyzer, an integrating sphere and by employing U6039-05 PLQY measurement software (Hamamatsu Photonics, Ltd, Shizuoka, Japan). All solvents used were of spectrometric grade.Binding to lectins and bacteria
Five cuvettes were filled with 0.5 mg ml−1 ConA, 1 mM MnCl2 and 1 mM CaCl2. As solvent a solution of 10 mM HEPES and 137 mM NaCl, pH 7, was used. Solutions with different ratios of compound 1 to compound 2 were prepared: 1/2 = 0, 0.25, 0.5, 0.75, 1. Total concentration was always 200 μM. From these solutions 40 μl was added to each cuvette (total volume 400 μl). The agglutination was recorded as video. The data were processed by taking pictures: first 5 min every second one picture, afterwards every 2 minutes.The measurements of optical density depending on time were recorded on a Perkin Elmer Lambda 850 spectrophotometer. The samples were measured in absorption mode at 450 nm for 1 h. Every 2 seconds a value was recorded. The total volume in the cuvette was 1 ml which consisted of 250 μl of 2 mg ml−1 ConA, 1 μl of 1 M MnCl2/CaCl2 and 649 μl of HEPES buffer (10 mM HEPES, 137 mM NaCl, pH 7). 100 μl of 200 μM thiophthalonitrile was added after 5 min of recording. After 29 min 10 μl of 1 M sugar (α-methyl-D-mannoside or D(+)-galactose) was added.
The bacterial strains ORN178 and ORN208 (kind gift from Prof. Luc Brunsveld, TU/e) were grown overnight in LB media using tetracycline as a selective antibiotic. After measuring the OD600 value, samples containing 0.1 OD bacteria, 1 mM MnCl2, 1 mM CaCl2 and 50 μM compound 1 in HEPES buffer were prepared and incubated for 60 min. A drop of the solution was placed between cover slips.
The microscopy pictures were recorded using an Olympus IX71 microscope. The bright field pictures were recorded in monochrome mode with 50 ms exposure time. The fluorescence images were recorded using the following filter: excitation wavelength 350 nm, emission wavelength 420 nm with a long pass filter. As exposure time 500 ms was used. The overlay was created in ImageJ.
To test the bacterial viability E. coli bacterial strains ORN 178 and 208 that were grown overnight were incubated for 1 h at 37 °C with the thiophthalonitriles and cultivated on LB medium-agar plates (tetracycline added as a selective antibiotic). The incubated solutions contained 109 cells per ml (OD600 1) and 50 μM compound 1. The control solution contained no thiophthalonitrile (only water). After incubation the samples were diluted to 105, 104 and 103 cells per ml. From these solutions 10 μl was placed as drops on top of LB agar plates before the plates were tilted to form 3 streams which quickly dried. The plates were kept at 37 °C overnight before the colonies were counted the next day.
Acknowledgements
The authors thank the St-ERC 259183 (P. J.) and ERC PoC 31015 (P. J.) for funding. Mark Smithers and Dr Rico Keim (University of Twente) are acknowledged for their help with SEM and TEM imaging. ORN strains were kindly provided by Prof. L. Brunsveld.Notes and references
- J. Luo, Z. Xie, J. W. Y. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhu and B. Z. Tang, Chem. Commun., 2001, 1740–1741 RSC.
- J. Liang, B. Z. Tang and B. Liu, Chem. Soc. Rev., 2015, 44, 2798–2811 RSC.
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Commun., 2009, 4332–4653 RSC.
- J. W. Chen, B. Xu, X. Y. Ouyang, B. Z. Tang and Y. Cao, J. Phys. Chem. A, 2004, 108, 7522–7526 CrossRef CAS.
- H. Tong, Y. Hong, Y. Dong, Y. Ren, M. Häussler, J. W. Y. Lam, K. S. Wong and B. Z. Tang, J. Phys. Chem. B, 2007, 111, 2000–2007 CrossRef CAS PubMed.
- H. Tong, Y. Dong, Y. Hong, M. Häussler, J. W. Y. Lam, H. H. Y. Sung, X. Yu, J. Sun, I. D. Williams, H. S. Kwok and B. Z. Tang, J. Phys. Chem. C, 2007, 111, 2287–2294 CrossRef CAS.
- J. Huang, R. Tang, T. Zhang, Q. Li, G. Yu, S. Xie, Y. Liu, S. Ye, J. Qin and Z. Li, Chem. – Eur. J., 2014, 20, 5317–5326 CrossRef CAS PubMed.
- L. Zhang, W. Hu, L. Yua and Y. Wang, Chem. Commun., 2015, 51, 4298–4301 RSC.
- A. Qin, J. W. Y. Lam, L. Tang, C. K. W. Jim, H. Zhao, J. Sun and B. Zhong Tang, Macromolecules, 2009, 42, 1421–1424 CrossRef CAS.
- S. Chen, Y. Hong, Y. Zeng, Q. Sun, Y. Liu, E. Zhao, G. Bai, J. Qu, J. Hao and B. Zhong Tang, Chem. – Eur. J., 2015, 21, 4315–4320 CrossRef CAS PubMed.
- Y. Zhang, Y. Chen, X. Li, J. Zhang, J. Chen, B. Xu, X. Fub and W. Tian, Polym. Chem., 2014, 5, 3824–3830 RSC.
- M. K. Müller and L. Brunsveld, Angew. Chem., Int. Ed., 2009, 48, 2921–2924 CrossRef PubMed.
- T. Sanji, K. Shiraishi and M. Tanaka, ACS Appl. Mater. Interfaces, 2009, 1(2), 270–273 Search PubMed.
- (a) J. Voskuhl, M. C. A. Stuart and B. J. Ravoo, Chem. – Eur. J., 2010, 16, 2790–2796 CrossRef CAS PubMed; (b) J. Voskuhl, T. Fenske, M. C. A. Stuart, B. Wibbeling, C. Schmuck and B. J. Ravoo, Chem. – Eur. J., 2010, 16, 8300–8306 CrossRef CAS PubMed.
- J. Chen, C. C. W. Law, J. W. Y. Lam, Y. Dong, S. M. F. Lo, I. D. Williams, D. Zhu and B. Z. Tang, Chem. Mater., 2003, 15(7), 1535–1546 CrossRef CAS.
- R. V. Vico, J. Voskuhl and B. J. Ravoo, Langmuir, 2011, 27, 1391–1397 CrossRef CAS PubMed.
- (a) J. Voskuhl, S. Sankaran and P. Jonkheijm, Chem. Commun., 2014, 50, 15144–15147 RSC; (b) S. L. Harris, P. A. Spears, E. A. Havell, T. S. Hamrick, J. R. Horton and P. E. Orndorff, J. Bacteriol., 2001, 183, 4099–4102 CrossRef CAS PubMed.
- M. Mayukh, C.-W. Lu, E. Hernandez and D. V. McGrath, Chem. – Eur. J., 2011, 17, 8472–8478 CrossRef CAS PubMed.
- K. Brunner, J. Harder, T. Halbach, J. Willibald, S. Spada, F. Gnerlich, K. Sparrer, A. Beil, L. Möckl, C. Bräuchle, K.-K. Conzelmann and T. Carell, Angew. Chem., Int. Ed., 2015, 54, 1946–1949 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of the synthesis and methodology and Fig. S1–S18. See DOI: 10.1039/c6tb01210h |
| This journal is © The Royal Society of Chemistry 2016 |