
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Conjugated polymers for the optical control of the electrical activity of living cells†
Susana
Vaquero
a,
Caterina
Bossio
a,
Sebastiano
Bellani‡
ab,
Nicola
Martino§
ab,
Elena
Zucchetti
ab,
Guglielmo
Lanzani
*ab and
Maria Rosa
Antognazza
*a
aCenter for Nano Science and Technology, IIT@PoliMi, Via Pascoli 70/3, 20133 Milano, Italy. E-mail: guglielmo.lanzani@iit.it; mariarosa.antognazza@iit.it
bPolitecnico di Milano, Dip.to di Fisica, P.zza L. da Vinci 32, 20133 Milano, Italy
First published on 13th July 2016
Abstract
The possibility to optically excite the electrical activity of living cells by using exogenous absorbers is gaining more and more interest in the neuroscience and biotechnology community. Conjugated polymers, inherently sensitive to visible light, were recently proposed as candidates to this goal. To date, however, only one polymer type, namely regio-regular poly-3-hexylthiophene, has been tested as the active material. In this work four different conjugated polymers, regarded as prototypes of their category, are investigated as photoactive bio-interfaces. The selected materials have different absorption spectra, morphology, light emission efficiency and charge transport properties. We analyze their key-enabling properties, such as electrochemical stability, surface morphology, wettability, sterilization compatibility, interaction with protein adhesion layers and toxicity, throughout all the necessary steps for the realization of an efficient bio-optical interface. We demonstrate that all considered polymers are characterized by good biocompatibility and cell seeding properties, and can optimally sustain thermal sterilization. Conversely, electrochemical stability and cell photostimulation efficacy can vary a lot among different materials, and should be carefully evaluated case by case. Reported results represent the starting point for the implementation of bio-polymer interfaces sensitive to different colors and, in perspective, for the realization of a three-chromatic artificial visual prosthesis.
1. Introduction
In recent years the possibility to exogenously modulate the electrical activity of living cells by using optical tools, both in vitro and in vivo, has attracted considerable attention.1 Several photoactive materials, capable of converting the light stimulus into electrical, chemical or thermal stimuli, have been reported2,3 including photoconductive silicon,4–6 semiconducting,7,8 metallic micro- and nanoparticles9–11 and carbon microparticles.12 In this field, conjugated polymers recently emerged as ideal candidates for the optically-driven control of electrical activity in several types of in vitro cultures, including primary neurons, astrocytes, and non-excitable cells.13–15 Similar protocols have also been reported in the study of excised tissues, in particular in explanted retinas in different configurations and different animal models.16,17 Interestingly, the possibility not only to excite, but also to inhibit neuronal activity has been recently demonstrated in neural networks, acute brain slices and retinal tissues, opening up interesting opportunities for the use of conjugated polymers in neuroscience.18So far only poly-3-hexylthiophene (rr-P3HT) has been tested as a photoactive layer, either in the pristine form or in blend with different electron acceptors. This choice was motivated by the widely assessed environmental stability of rr-P3HT, its well-known optoelectronic properties, its easy processability and biocompatibility.19 Yet, many other organic semiconductors are nowadays available, characterized by different optical, electronic and morphological properties. A comparative study among them, considering key enabling properties such as electrochemical stability, surface morphology, wettability, sterilization compatibility and interaction with protein adhesion layers, eventually used for cell seeding, is thus in demand for a number of different reasons. Firstly, the study of different material properties in combination with cell cultures is expected to contribute to the understanding of the polymer/electrolyte/cell interface, which is almost completely missing in the literature at the moment. Secondly, for practical in vitro uses, possible interferences with specific light responsive biomarkers of common use in neuroscience investigations should be avoided, posing constraints on the absorption spectral range of the exogenous photoactive materials. Thirdly, within the broad scope of this emerging technology platform, the realization of an artificial retinal prosthesis is an outstanding goal. An implant based on conjugated polymers presents a number of potential advantages over other technologies currently available, which are mainly silicon-based. A prominent one is certainly the intrinsic sensitivity to colors. The realization of photodiodes mimicking the spectral response of natural photoreceptors has been already demonstrated.20 In order to transfer this result to the artificial retinal application, it is necessary to assess the actual tuneability of the organic semiconductors also in terms of cell interaction.
In this work, we considered four different polymers as study-cases: two polythiophene derivatives with different optical band gaps (poly[2,1,3-benzothiadiazole-4,7-diyl[4,4-bis(2-ethylhexyl)-4H-cyclopenta[2,1-b:3,4-b′]dithiophene-2,6-diyl]], PCPDTBT, and regio-regular poly(3-hexylthiophene), rr-P3HT), a poly-phenylene vinylene derivative (poly[2-methoxy-5-(2-ethylhexyloxy)-1,4-phenylene-vinylene], MEH-PPV) and a polyfluorene derivative (poly[9,9-dioctylfluorenyl-2,7-diyl], PFO). These materials are characterized by different absorption spectra, morphology, light emission efficiency and charge transport properties. We analyzed them by a combination of morphological, electrochemical, biochemical (toxicity) and electrophysiological measurements, throughout all the necessary steps for the realization of an efficient and reliable bio-optical interface. These include sterilization, deposition of adhesion proteins, cell seeding and cell culturing. For the biological counterpart, we chose the human embryonic kidney cells (HEK-293) as a valuable model, since they can be easily cultured on different materials and they are one of the best known models for the study of the cell membrane and ion channels.
Our results demonstrate that all considered polymers can sustain thermal sterilization protocols and protein adsorption, but not all of them are suitable for optical stimulation. In particular, the high band gap PFO showed the poorest biocompatibility and failed in establishing a functional coupling with living cells. Conversely, tested polymers absorbing in the green and in the red part of the visible spectrum were all effective in sustaining living cell optical stimulation, but showed different stability properties.
2. Experimental
2.1 Materials and preparation of cell/polymer interfaces
Rr-P3HT was purchased from Sigma Aldrich; PCPDTBT from 1-Material, Inc.; MEH-PPV and PFO from American Dye Source, Inc. All polymers were used without any further purification. Dulbecco's modified Eagle's medium (DMEM), trypsin–EDTA, penicillin, streptomycin, phosphate buffer saline (PBS) tablets, tetrazolium salt (MTT) and fibronectin (from bovine plasma) were purchased from Sigma Aldrich. Fetal bovine serum (FBS) was purchased from Euroclone. All chemicals were used as received.Polymers were deposited from organic solvent solutions on square 18 × 18 mm2 glass (VWR) substrates by spin-coating. Solutions were prepared in chlorobenzene at 50 °C for rr-P3HT, PCPDTBT and MEH-PPV at a final concentration of 20 g l−1, 30 g l−1 and 7 g l−1, respectively. For PFO, a chloroform solution of 10 g l−1 was prepared. Thicknesses of thin film polymers were in the range of 100 nm (PCPDTBT: 105 ± 14; P3HT: 103 ± 16; MEHPPV: 89 ± 9; PFO: 120 ± 9). Prior to polymer deposition, glass substrates were subsequently cleaned in cycles of 10 minutes with ultrapure water (Milli-Q water), acetone and isopropanol in an ultrasonic bath. After that, all substrates were dried with a N2 gun. Polymers were then spin-coated on the cleaned glass substrates with a two step procedure: (i) 3 s at 800 rpm, (ii) 60 s at 1600 rpm. The polymer thin films were thermally sterilized at 120 °C for 2 h, exception made for PFO that was sterilized at 80 °C, in order to avoid peculiar topography changes provoked by annealing temperature, as previously reported in the literature.21 To promote cell adhesion, a layer of fibronectin (2 μg ml−1 in PBS) was deposited on the surface of the polymers and incubated for 1 hour at 37 °C. After rinsing the fibronectin with PBS, the HEK-293 cells were grown in the presence of their culture medium on the different selected polymers.
The same protocol described for the preparation of the polymer–cell bio-interphases was applied for electrochemical measurements, with the exception that square 18 × 18 mm2 ITO-glass substrates were used for polymer deposition. In this case no cells were seeded after fibronectin deposition.
2.2 Surface characterization (contact angle and AFM)
The wettability of the different polymer films was characterized by using an OCA-15 Optical Contact Angle Measuring Instrument (Data physics). Static water contact angles, defined as the angles formed by the water drop at the three-phase boundary where water, the polymer surface and air intersect, were determined using the sessile drop method (4 μl, Milli-Q water). For each sample an average of nine measurements were made.The morphology of the different polymers was investigated using an Agilent 5500 Atomic Force Microscope (AFM) in tapping mode at 25 °C. Scans were performed over three length scales (5 × 5, 1 × 1 and 0.250 × 0.250 μm) at a scan speed of 0.5 Hz, ranging 0.2–0.5 lines second−1, optimizing it for each material and each length scale. Root mean square (RMS) roughness values were calculated from AFM topography images using Gwyddion software.
Contact angles as well as AFM images were determined for the polymers directly after spin-coating, after sterilization and after fibronectin incubation for 1 hour at 37 °C.
2.3 Fibronectin (Fn) absorption: QCM-D measurements
Protein absorption measurements were carried out using a four-channel Quartz Crystal Microbalance with a Dissipation monitoring (QCM-D) Q-sense E4 system (Biolin Scientific, Sweden). QCM sensors (AT-cut crystals with gold electrodes QSX301 with a 5 MHz fundamental resonance frequency) were coated with the different polymers and thermally sterilized for 2 h. The QCM sensors supporting the polymer films were then transferred to the Q-sense flow modules. A PBS solution was first flown in order to evaluate the hydration of the films. Saturation was achieved when values of frequency and dissipation were constant with time. Following PBS saturation, a 2 μg ml−1 fibronectin solution in PBS was introduced to the flow modules (10 μg min−1 flow rate) until the protein saturation was obtained. The experiments were carried out at a constant temperature of 22 ± 0.02 °C and all solutions were degassed before running the experiment. Each experiment was run in triplicate. Student's t-test was performed in order to evaluate the statistical significance.The Q-tools software package v.3.0.10.286 (Biolin Scientific) was used to fit the data. The Voigt model was applied to determine the mass of the viscoelastic protein layer deposited. The best fit was obtained by using the following input parameters: layer density (1150 kg m−3), fluid density (1000 kg m−3), layer viscosity (10−4–10−2 kg m−1 s−1), shear modulus (105–107 Pa) and mass (1–4000 ng cm−2). The 9th and 11th harmonic overtones were used for all modeling calculations.
2.4 Electrochemical impedance spectroscopy and cyclic voltammetry
Electrochemical stability in water of the different polymers was evaluated by means of electrochemical impedance spectroscopy (EIS) and cyclic voltammetry (CV). The effective capacitance was extracted from the complex impedance by assuming an equivalent circuit including a capacitor connected in parallel to a resistor, accordingly to a widely accepted modeling of the polymer/electrolyte interface.22 EIS analysis was performed in 0.2 M NaCl at room temperature using a three electrode cell comprising the ITO/polymer as the working electrode, a platinum wire as the counter electrode and saturated Ag/AgCl as the reference electrode. Impedance measurements were recorded between 1 Hz and 100 kHz with an AC amplitude of 10 mV using a potentiostat (PGSTAT302N, Metrohm Autolab). CV experiments were carried out using the same three electrode configuration at a scan rate of 100 mV s−1. The different polymers were characterized under three conditions: directly after spin-coating (pristine polymer), after sterilization and after fibronectin incubation for 1 hour at 37 °C. All experiments were carried out in dark.2.5 Cell culture and cytotoxicity analysis (MTT)
HEK-293 cells were cultured in cell culture flasks containing Dulbecco's modified Eagle's medium (DMEM) with 10% fetal bovine serum (FBS), 100 U ml−1 penicillin, 100 μg ml−1 streptomycin and 100 U ml−1L-glutamine. Culture flasks were maintained in a humidified incubator at 37 °C with 5% CO2. When at confluence, HEK-293 cells were enzymatically dispersed using trypsin–EDTA and then plated on the different polymer substrates at a concentration of 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells cm−2.
000 cells cm−2.
HEK-293 proliferation was evaluated after 2 h and after 1, 2 and 3 days in vitro with the MTT assay (thiazolyl blue tetrazolium bromide) for all polymers. For every time point the culture medium was removed and replaced with DMEM without phenol red, supplemented with 0.1 mg ml−1 of the MTT reagent. Cells were further re-incubated at 37 °C for 2 h. The MTT reagent was then removed and, after drying, 1 ml of ethanol was added to dissolve formazan crystals. The absorbance of the obtained solution was finally measured at 560 nm using a spectrophotometer (Cary 50, Agilent Technologies). For each case, MTT assay was carried out four times. Student's t-test was performed in order to evaluate the statistical significance.
2.6 Electrophysiology
Intracellular recordings were performed using a patch-clamp setup (Axopatch 200B, Axon Instruments) coupled to an inverted microscope (Nikon Eclipse Ti-U). The light source for excitation of the polymer was provided by a LED system (Lumencor Spectra X) fibre-coupled to the fluorescence port of the microscope; the illuminated spot on the sample had an area of 0.23 mm2. HEK-293 cells were measured at 1–2 DIV in whole-cell configuration with freshly pulled glass pipettes (3–6 MΩ), filled with the following intracellular solution [mM]: 12 KCl, 125 K-gluconate, 1 MgCl2, 0.1 CaCl2, 10 EGTA, 10 HEPES, 10 ATP-Na2. The extracellular solution contained [mM]: 135 NaCl, 5.4 KCl, 5 HEPES, 10 glucose, 1.8 CaCl2, 1 MgCl2. Only single HEK-293 cells were selected for recordings. All measurements were performed at room temperature. Acquisition was performed using the pClamp 10 software suite (Axon Instruments) and all data were elaborated with Origin 8.0.3. Results and discussion
3.1 Surface characterization of the different polymers
The first necessary condition to obtain an effective bio-organic interface is the attainment of a well-adherent cell culture on top of the polymer surface. Cells are extremely sensitive to stimuli provided by their surroundings; in particular, two crucial factors control cellular adhesion, i.e., the wettability and the roughness of the substrate surface. The preferential adhesion of cells to rough surfaces is a widely-known phenomenon, where the specific or non-specific adsorption of adhesion proteins, usually employed for in vitro cultures, plays a crucial role.23–25 In this work, we chose fibronectin as a model serum protein, since it has been shown to be a good mediator in the initial adhesion and spreading of various cell types on different materials,24,25 and to control critical surface properties, such as wettability.26 In addition, it previously succeeded in promoting the adhesion of HEK-293 cells on rr-P3HT thin films.26 Based on these premises, we first evaluated the wettability of polymers, their surface morphology and the dynamics of the fibronectin adsorption to the polymer. In principle, these properties can intrinsically vary a lot among the considered materials, and they may also undergo different changes upon thermal sterilization. | ||
| Fig. 1 Static water contact angles for the different polymers investigated. | ||
 | ||
| Fig. 2 AFM topography images of pristine (left) and sterilized polymers (right) (sample area, 5 × 5 μm2). From top to the bottom: PCPDTBT, rr-P3HT, MEH-PPV and PFO. | ||
The subsequent step in the preparation of a bio-polymer interface concerns the deposition of the protein adhesion layer. The morphology of fibronectin has been previously investigated in the case of several biomaterials,32,33 including conducting polymers like polypyrrole,34 but to the best of our knowledge it has never been evaluated on top of light-sensitive organic semiconductors. Fig. 3 shows the phase images of the sterilized polymers before and after fibronectin deposition (0.25 × 0.25 μm2 scale). Phase imaging is a powerful tool, sensitive to surface stiffness/softness and adhesion between the tip and the surface. It is particularly useful in analyzing surface coatings, presenting different viscoelastic properties, which do not strictly affect the topography images. The differences observed among the considered polymers might be attributed to a different conformation of the protein on materials of different hydrophobicity35,36 or also to a different interaction of the fibronectin with the different surfaces.37 A dedicated study, beyond the scope of the present work, would be actually needed to exactly identify their origin.38 Here, more interestingly, we just notice substantial differences among the phase images of sterilized polymers before and after protein deposition, thus confirming the absorption of fibronectin on top of all the considered polymers, which is crucial for the subsequent cell seeding and it is a factor conditioning the overall biocompatibility of the device.
 | ||
| Fig. 3 AFM phase images of sterilized polymers before (left) and after (right) fibronectin deposition. From top to bottom: PCPDTBT, rr-P3HT, MEHPPV and PFO. Sample area, 0.250 × 0.250 μm2. | ||
By analysis of the AFM images, we quantitatively evaluated the polymer roughness factors in the pristine case, after sterilization and after fibronectin deposition. The roughness root mean square (RMS) was calculated at 1 μm length scales (Fig. 4). As predicted from the topography images (Fig. 2), PCPDTBT and rr-P3HT pristine polymers were found to be relatively rough, with an RMS of 4.5 and 5.5 nm at 1 μm, respectively. On the other hand, MEH-PPV and PFO pristine polymers revealed smoother surfaces, with an RMS of 0.4 and 0.25 nm at 1 μm respectively. The thermal treatment, necessary for sample sterilization, did not significantly affect the RMS values of the polymers, which only slightly increased. Conversely, fibronectin deposition led in all cases to a significant enhancement in the RMS. This rise was much more pronounced for flat polymers such as MEH-PPV and PFO, which increased their RMS by a factor of 1.8 and 2, respectively, considerably higher than the 1.1 and 1.4 increases obtained for the rougher polymers, PCPDTBT and rr-P3HT, respectively.
 | ||
| Fig. 4 RMS measured at 1 μm length scales. (a) PCPDTBT, (b) rr-P3HT, (c) MEH-PPV and (d) PFO. | ||
The characterization of the polymer surfaces in terms of morphology and wettability, upon the subsequent steps of polymer sterilization and deposition of an adhesion protein layer, allowed us to conclude that, despite the inherent differences in the morphology and the roughness of the pristine materials, the major role in the final morphology of the bio-polymer device is played by the fibronectin layer itself. The comparable hydrophilic character of the considered polymers, obtained in all cases upon contact with aqueous solutions, makes possible to deposit an uniform protein layer, all over the polymer surface, which in all cases is expected to have beneficial effects on subsequent cell seeding. However, given the different surface topographies, the dynamics of the protein specific adsorption to the polymer surface might still vary from case to case, and need to be quantitatively assessed.
3.2 Quantitative evaluation of protein adsorption
The Quartz Crystal Microbalance with Dissipation monitoring (QCM-D) is a versatile technique, able not only to provide a quantitative measurement of the mass adsorbed to a substrate via changes in the crystal oscillation frequency, but also the evaluation of the viscoelastic properties of the adhered layer by the dissipation factor.34 Frequency and dissipation curve dynamics for the four investigated polymers are shown in Fig. 5 (Fig. 5a and b, respectively).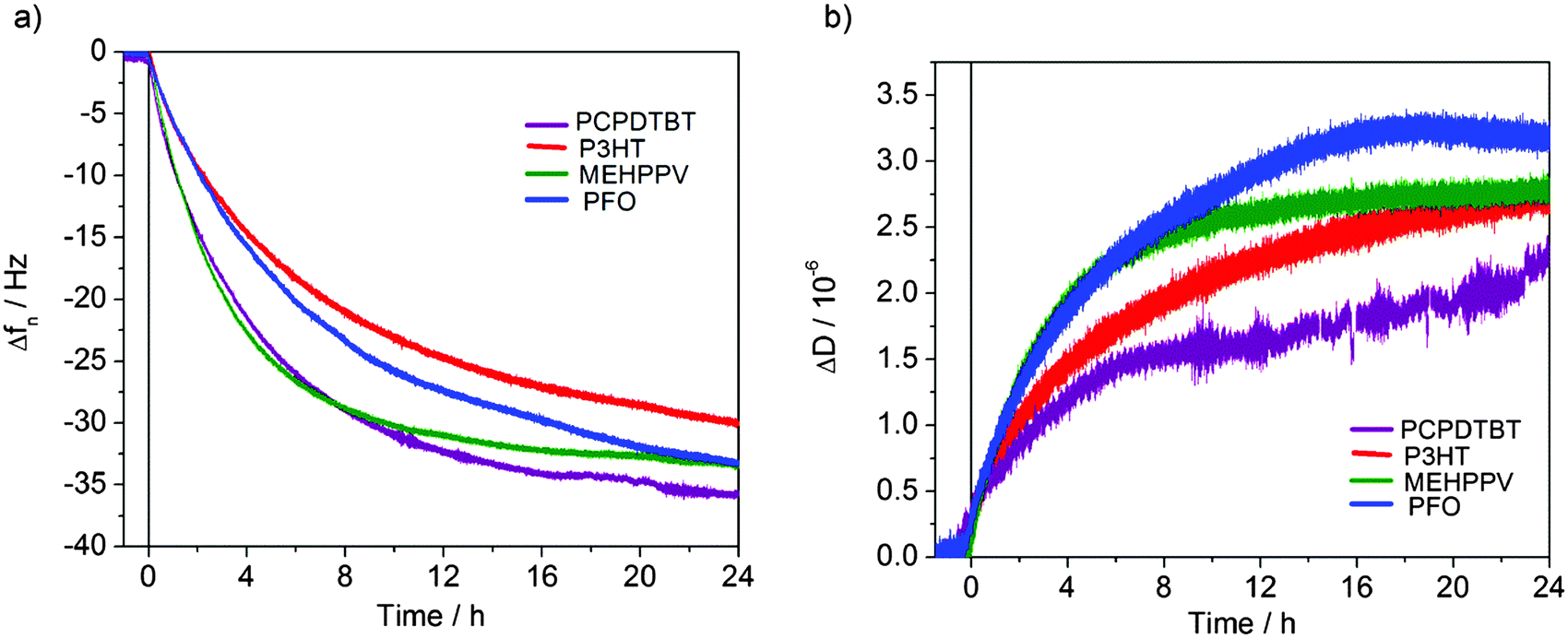 | ||
| Fig. 5 Representative normalized frequency (a) and dissipation (b) shifts upon fibronectin adsorption for the polymer surface (overtone n = 9). | ||
Saturation was achieved for all polymers after 24 h, as frequency and dissipation shifts were almost stable with time.
The total frequency as well as the dissipation shifts showed slight differences among polymers, with values after 24 hours in the range of 30–35 Hz for frequency and 2–3 × 10−6 for dissipation. The mass of adsorbed protein on each surface was examined at different times of fibronectin exposure. The Sauerbrey model,39 commonly used to determine surface bound mass, utilizes the QCM frequency shift data (Δf) to model the properties of the adsorbed layer. However, this model is only applicable when the mass is rigidly coupled to the sensor surface. When the adsorbed layer is not rigid enough, it does not oscillate in phase with the crystal and a dissipation shift (ΔD) consequently rises. In our measurements, ΔD > 0 and therefore the Voigt model was employed to estimate the mass of adsorbed protein.34,40
Fig. 6a shows the mass values of fibronectin for each polymer at 1 h (i.e., the time of protein incubation before cell seeding in our culturing protocol), 12 h and 24 h. After 1 h, rr-P3HT, MEH-PPV and PFO showed similar mass values, around 150 ng cm−2, whereas PCPDTBT displayed a higher value of 225 ng cm−2. All polymers reached saturation after 24 h, displaying values around 1000 ng cm−2. No statistically significant difference was found among the considered polymers, for each considered time point.
 | ||
| Fig. 6 (a) QCM-D modelling results for the mass of fibronectin adsorbed on the different polymers at 1 h, 12 h and 24 h. (b) ΔD(−Δfn)−1 data for fibronectin adsorption onto the different polymers at 1 h, 12 h and 24 h (overtone n = 9). Data are reported as average values ± SD (n = 3 samples for each polymer type). No statistically significant difference was observed among the different polymers (Student's t-test, P value > 0.1 in all cases). | ||
There are several research papers devoted to investigate fibronectin adsorption to a range of materials.41–43 However, the exact influence of surface roughness on fibronectin binding is currently a matter of controversy in the literature. While some authors postulate that an increase in surface roughness leads to increase in fibronectin adsorption,42,44,45 the opposite has also been reported.46–48 Generalizations on the influence of surface topography are thus complex. Also other factors like hydrophobicity have been considered, and it was reported that hydrophilic materials have a positive effect on fibronectin binding.36,49 In our case, the considered polymers did not show major differences in contact angle values, which could explain why the values of adsorbed fibronectin among different polymers were not significantly different.
To gain some insight into the viscoelastic properties of the adsorbed layer,50 the ratio ΔD(−Δfn)−1 was also evaluated at 1 h, 12 h and 24 h for all polymers (Fig. 6b). A lower ΔD(−Δfn)−1 is representative of a more rigid protein layer.51 At 1 h, evaluated values range from 0.08 to 0.11 × 10−6 Hz−1, in agreement with previous results reported for more concentrated fibronectin solutions with polypyrrole polymers.34 At longer times, slightly lower values are obtained ranging from 0.065 to 0.09 Hz−1. However, Student's t-test revealed no significant differences among the considered polymers.
3.3 Effect of sterilization and protein deposition on the capacitance of the polymers
A crucial parameter which must be taken into account in the realization of a functional bio-polymer interface is the electrochemical stability of the active layer in an aqueous environment, for the pristine material but also after the necessary steps of sterilization and treatment with adhesion proteins. Impedance spectroscopy (Fig. 7a–c) and cyclic voltammetry (Fig. 7b–d) measurements were carried out on the four considered polymers at the different steps of substrate preparation. Fig. 7 shows the results obtained for PCPDTBT and rr-P3HT. | ||
| Fig. 7 Electrochemical measurements of PCPDTBT (top) and rr-P3HT (bottom), as deposited (black line), after sterilization (red line), and after protein incubation (blue line), in 0.2 M NaCl. (a) and (c) Bode plot for PCPDTBT and rr-P3HT, respectively. (b) and (d) Cyclic voltammetry at 100 mV s−1 for PCPDTBT and rr-P3HT, respectively. | ||
In this case fully comparable results among considered conditions were obtained. In particular, cyclic voltammetry measurements showed similar current densities, thus indicating that the interface capacitance of the two polymers exposed to the electrolyte remains the same upon sterilization and even after fibronectin deposition. Conversely, in the case of MEH-PPV and PFO it was not possible to obtain repeatable measurements among different replicas, even under the same conditions, thus indicating a lower degree of electrochemical stability, when directly exposed to water. Moreover, we observed that MEH-PPV and PFO thin films in contact with water tended to delaminate more frequently and faster than polythiophene derivatives.
Capacitance values were then calculated by fitting the impedance spectroscopy data to the corresponding equivalent circuit, according to a widely accepted model.22 Capacitance vs. frequency curves for rr-P3HT and PCPDTBT are shown in Fig. 8, for the three conditions. PCPDTBT (Fig. 8a) and rr-P3HT (Fig. 8b) polymers showed similar capacitances, on the order of 1–2 μF cm−2 (in agreement with previous reports for sole exposure to saline water52) under all the conditions investigated, thus demonstrating that the steps necessary for subsequent cell seeding do not seriously affect the capacitance properties.
 | ||
| Fig. 8 Capacitance values versus frequency extrapolated from the corresponding equivalent circuit derived from impedance measurements for pristine polymers, sterilized polymers and polymers after protein incubation. (a) PCPDTBT; (b) rr-P3HT. | ||
This outcome might find a possible explanation in the peculiar molecular orientation of the thiophene backbone at the interface with the electrolyte, with a preferential edge-on orientation, as recently reported in the case of rr-P3HT and other thiophene derivatives.53
3.4 Polymer effect on cell viability
We evaluated the cell viability on the different polymers by the tetrazolium salt (MTT) assay. Images of cells cultured on top of the different organic semiconducting polymers can be found in the ESI† (Fig. S2). Fig. 9a shows the absorption of formazan at 4 different time points, up to 3 days in vitro (DIV). | ||
| Fig. 9 MTT data plot for cell proliferation up to 3 days in vitro for different semiconducting polymers in terms of (a) absorbance and (b) percentage viability, as compared to the corresponding control at each time point. Data are reported as average (over n = 4 samples per each polymer) ± SE. Statistical significance is also reported (Student's t-test, *P < 0.1; **P < 0.05). | ||
Higher absorption values are related to a higher cell proliferation, as only mitochondrial dehydrogenases of living cells are able to transform MTT in formazan. As expected, cell proliferation increased with incubation time reaching the maximum absorbance at day 3 in all cases.
In particular, it can be observed that after 3 DIV the cell proliferation in PCPDTBT, rr-P3HT and MEH-PPV was as high as in the glass positive control, confirming the low degree of cytotoxicity of these polymers as substrates for cell culturing. However, it must be noticed that some samples based on MEH-PPV were discarded, due to partial delamination during cell culture. PFO showed the lower viability data, scarcely repeatable, and after 2 DIV all samples started to partially detach from the glass coverslip. As complementary information, Fig. 9b shows the percentage viability of cells as compared to the corresponding glass control substrates, at each time point.
3.5 Polymer-mediated cellular photostimulation
The effect of polymer photoexcitation on the electrical activity of in vitro cell cultures was finally assessed by electrophysiological measurements. In particular, we measured the variation in the HEK-293 cell membrane potential upon illumination of the photoactive substrate with pulses of visible light. Recently, we used these cells to characterize in detail the polymer-mediated cell photoexcitation process, in the case of rr-P3HT thin films on glass or on ITO substrates.15 We found two different mechanisms, predominant on different time scales, leading respectively to depolarization and hyperpolarization of the cell membrane. A thorough characterization suggested a thermal origin for both.Whole-cell recordings were carried out in current-clamp (I = 0) configuration in response to 200 ms pulses of light after 1–2 DIV. LED light sources of different wavelengths were employed for excitation, depending on the polymers' absorption spectra (Fig. S3, ESI†). The film thickness was controlled in order to have suitable optical density in all samples.
Representative cellular responses (n = 10 cells per type of polymer) recorded on PCPDTBT, rr-P3HT and MEH-PPV substrates together with control glass substrates (black curves) are reported in Fig. 10a, b and c, respectively, for a fixed light intensity, on the order of a few tens of mW mm−2. Membrane potential values of HEK-293 cells were in the range of −30 mV for both cells cultured on glass as well as on semiconducting polymers and were stable during the illumination protocol. No change in cellular shape was observed after illumination in glass control, PCPDTBT, P3HT and MEHPPV samples. In line with the data previously reported for rr-P3HT,15 and confirmed also here (Fig. 10b), in the cases of PCPDTBT and MEH-PPV (Fig. 10a and c, respectively) we observed an initial depolarization signal, followed by a hyperpolarization effect. No signal was observed in glass control samples. Both the depolarization and hyperpolarization maximum amplitudes scaled with the photoexcitation density, which was varied from 5 to 44 mW mm−2 for excitation in the red (used for PCPDTBT) and from 8 to 61 mW mm−2 for excitation in the cyan (used for MEH-PPV and rr-P3HT) (Fig. 10d).
 | ||
| Fig. 10 (a–c) Representative membrane potential variation measured in HEK-293 cells (n = 10 per each type of polymer) cultured on different photoactive polymers under illumination (grey area in the plot indicates light on). PCPDTBT excitation wavelength peak, λ = 635 nm; P3HT and MEH-PPV, λ = 475 nm. Black lines refer to control samples, with cells cultured on top of glass substrates and under the same photoexcitation density used for the corresponding polymer (d) variation of depolarization and hyperpolarization signals of conjugated polymers with increasing photoexcitation density. PCPDTBT (violet symbols) was excited at λ = 635 nm, in a range between 5 and 44 mW mm−2; rr-P3HT (red symbols) and MEH-PPV (green symbols) were excited at λ = 475 nm, in a range between 8 and 61 mW mm−2. Depolarization data correspond to the maximum depolarization peak; hyperpolarization data were obtained as the average value over the last 15 ms illumination period. | ||
Given the close resemblance of the signal recorded in rr-P3HT, PCPDTBT and MEH-PPV we infer that the origin of the variation in the cell membrane potential upon photostimulation is the same in all three cases, i.e. the increase in temperature mediated by polymer absorption. These polymers are characterized by different electronic levels (PCPDTBT: HOMO = 5.3 eV, LUMO = 3.8 eV; P3HT: HOMO = 4.9 eV, LUMO = 3.2 eV; MEHPPV: HOMO = 5.1 eV, LUMO = 2.7 eV; PFO: HOMO = 6. 65 eV, LUMO = 3.55 eV),54–56 as well as by different charge generation efficiency and charge transport, which further supports this conclusion, by excluding the occurrence of direct photoelectrical excitation under the considered conditions. The case of PFO (Fig. 11a), excited by blue LED light peaking at 435 nm, is fundamentally different. To begin with, control samples, at the highest available photoexcitation density, showed a hyperpolarization effect for this excitation. This could be due to an endogenous response of the cell, which was previously reported in many different situations under blue light excitation and related to the production of reactive oxygen species.57–60 In the presence of PFO, blue illumination rapidly leads to a variation in the membrane equilibrium potential and to detrimental cellular effects, clearly visible by alterations in the cell morphology (ESI,† Fig. S4, panels a and b), up to cell death effects, as demonstrated by fluorescence staining with propidium iodide (panel c). Moreover, we noticed that blue light shined onto PCPDTBT samples, closely matching the secondary absorption peak of this polymer (Fig. S3, ESI†), did not lead to cell death, even despite the fact that optical absorbance in the blue region of PCPDTBT is 5-times bigger than in PFO, and showed photoexcited signals with dynamics similar to those observed in Fig. 10 (Fig. 11b). Overall, these results indicate that PFO itself induces cellular damage upon blue light illumination.
 | ||
| Fig. 11 Membrane potential variation measured in HEK-293 cells cultured on (a) PFO and (b) PCPDTBT under illumination in the blue (λ = 435 nm). In both cases, black lines refer to control samples, with cells cultured on top of glass substrates and illuminated at the same photoexcitation density used for the corresponding polymer. | ||
We speculate this could be due to the energy position of the HOMO and LUMO levels in PFO,56,61 that can support a substantial rate of reactive oxygen species production, causing the death of HEK cells. Further studies are currently ongoing to prove this hypothesis.
4. Conclusions
We explored the possibility to extend the polymer-based interface, recently demonstrated for cell optical modulation in the green to the whole visible spectral range. We investigated four polymer semiconductor thin films in a biological environment and in contact with living cells, as candidates for optically-mediated control of the electrical activity of living cells. The selected materials are representative of different organic semiconductor classes, including rr-P3HT as a reference standard. The polymers have band gap energies across the visible range, from red to blue, and different yields of radiative recombination following photoexcitation. We characterized hydrophobicity, surface morphology, electrochemical stability, interface capacitance, biocompatibility, and finally the specific photoexcitation functionality. The low band gap material PCPDTBT showed excellent properties, including stability and cell photoexcitation capability. The phenylene-vinylene derivative, MEH-PPV, showed good properties in terms of biocompatibility and protein adsorption, but displayed poor electrochemical stability. Both polymers were able to optically modulate the membrane potential according to the thermal effect previously demonstrated in rr-P3HT under similar conditions. These results suggest that PPV-derivatives may be useful for in vitro studies, which do not require prolonged exposure to an aqueous environment, but should be replaced with more stable materials for in vivo, long-term applications. Thiophene-based materials confirmed their superior biocompatibility and stability. Finally PFO, with poor stability and limited biocompatibility, totally failed in cell photoexcitation, becoming indeed phototoxic. This work demonstrates that conjugated polymers can support thermal sterilization procedures and suggests that effective cell seeding and proliferation can be easily obtained. It shows however that electrochemical stability and cell photostimulation efficacy are not granted, and it points out a specific hurdle when using large band gap materials that can induce photo-chemical reactions.Overall, this work represents the necessary starting point for the implementation of polymer-based interfaces for optical modulation of cellular electrical activity all over the visible spectral window, and for the realization of a trichromatic sensor for artificial visual prosthesis.
Acknowledgements
This work was supported by EU through project FP7-PEOPLE-212-ITN 316832-OLIMPIA, Telethon – Italy (grants GGP12033 and GGP14022), Fondazione Cariplo (grant ID 2013-0738).References
- M. Scanziani and M. Häusser, Nature, 2009, 461, 930–939 CrossRef CAS PubMed.
- Y. Bareket-Keren and L. Hanein, Int. J. Nanomed., 2014, 9, 65–83 CrossRef PubMed.
- M. R. Antognazza, N. Martino, D. Ghezzi, P. Feyen, E. Colombo, D. Edeman, F. Benfenati and G. Lanzani, Adv. Mater. Interfaces, 2015, 27, 7662–7669 CrossRef CAS PubMed.
- M. A. Colicos, B. E. Collins and M. J. Sailor, Cell, 2001, 107, 605–616 CrossRef CAS PubMed.
- J. Suzurikawa, M. Nakao, Y. Jimbo, R. Kanzaki and H. Takahashi, Sens. Actuators, B, 2014, 192, 393–398 CrossRef CAS.
- J. Campbell, D. Singh, G. Hollett, S. M. Dravid, M. J. Sailor and J. Arikkath, Front. Cell Neurosci., 2014, 8, 142–151 Search PubMed.
- T. C. Pappas, W. M. S. Wickramanyake, E. Jan, M. Motamedi, M. Brodwick and N. a. Kotov, Nano Lett., 2007, 7, 513–519 CrossRef CAS PubMed.
- K. Lugo, X. Miao, F. Rieke and L. Y. Lin, Biomed. Opt. Express, 2012, 3, 447–454 CrossRef CAS PubMed.
- H. Huang, S. Delikanli, H. Zeng and D. M. Ferkey, Nat. Nanotechnol., 2010, 5, 602–606 CrossRef CAS PubMed.
- N. Farah, A. Zoubi, S. Matar, L. Golan, A. Marom, C. R. Butson and I. Brosh, J. Neural Eng., 2013, 10, 056004 CrossRef PubMed.
- S. A. Stanley, J. E. Gagner, S. Damanpour, M. Yoshida and J. S. Dordick, Sci. Signaling, 2012, 336, 604–608 CAS.
- B. Migliori, M. Di Ventra and W. Kristan, AIP Adv., 2012, 2, 032154 CrossRef PubMed.
- D. Ghezzi, M. R. Antognazza, M. Dal Maschio, E. Lanzarini, F. Benfenati and G. Lanzani, Nat. Commun., 2011, 2, 166 CrossRef PubMed.
- V. Benfenati, N. Martino, M. R. Antognazza, A. Pistone, S. Toffanin, S. Ferroni, G. Lanzani and M. Muccini, Adv. Healthcare Mater., 2014, 3, 392–399 CrossRef CAS PubMed.
- N. Martino, P. Feyen, M. Porro, C. Bossio, E. Zucchetti, D. Ghezzi, F. Benfenati, G. Lanzani and M. R. Antognazza, Sci. Rep., 2015, 5, 8911 CrossRef CAS PubMed.
- D. Ghezzi, M. R. Antognazza, R. Maccarone, S. Bellani, E. Lanzarini, N. Martino, M. Mete, G. Pertile, S. Bisti, G. Lanzani and F. Benfenati, Nat. Photonics, 2013, 7, 400–406 CrossRef CAS PubMed.
- V. Gautam, D. Rand, Y. Hanein and K. S. Narayan, Adv. Mater., 2014, 26, 1751–1756 CrossRef CAS PubMed.
- P. Feyen, E. Colombo, D. Endeman, M. Nova, L. Laudato, N. Martino, M. R. Antognazza, G. Lanzani, F. Benfenati and D. Ghezzi, Sci. Rep., 2016, 1–14 Search PubMed.
- N. Mzoughi, A. Abdellah, Q. Gong, H. Grothe, P. Lugli, B. Wolf and G. Scarpa, Sens. Actuators, B, 2012, 171–172, 537–543 CrossRef CAS.
- V. Gautam, M. Bag and K. S. Narayan, J. Am. Chem. Soc., 2011, 133, 17942–17949 CrossRef CAS PubMed.
- X. Chen, H. Wan, H. Li, F. Cheng, J. Ding, B. Yao, Z. Xie, L. Wang and J. Zhang, Polymer, 2012, 53, 3827–3832 CrossRef CAS.
- R. Porrazzo, S. Bellani, A. Luzio, E. Lanzarini, M. Caironi and M. R. Antognazza, Org. Electron., 2014, 15, 2126–2134 CrossRef CAS.
- A. Curtis and C. Wilkinson, Biomaterials, 1998, 18, 1573–1583 CrossRef.
- T. J. Webster, C. Ergun, R. H. Doremus, R. W. Siegel and R. Bizios, J. Biomed. Mater. Res., 2000, 51, 475–483 CrossRef CAS PubMed.
- M. M. Stevens and J. H. George, Science, 2005, 310, 1135–1139 CrossRef CAS PubMed.
- G. Scarpa, A. L. Idzko, S. Götz and S. Thalhammer, Macromol. Biosci., 2010, 10, 378–383 CrossRef CAS PubMed.
- T. Nguyen, I. B. Martini, J. Liu and B. J. Schwartz, J. Phys. Chem. B, 2000, 104, 237–255 CrossRef CAS.
- R. J. Kline, M. D. McGehee, E. N. Kadnikova, J. Liu and J. M. J. Fréchet, Adv. Mater., 2003, 15, 1519–1522 CrossRef CAS.
- E. S. Hellerich, J. J. Intemann, M. Cai, R. Liu, M. D. Ewan, B. C. Tlach, M. Jeffries-EL, R. Shinar and J. Shinar, J. Mater. Chem. C, 2013, 1, 5191–5199 RSC.
- N. Prasad, I. Singh, A. Kumari, D. Madhwal, S. Madan, S. K. Dixit, P. K. Bhatnagar and P. C. Mathur, J. Lumin., 2015, 159, 166–170 CrossRef CAS.
- F. Deschler, D. Riedel, A. Deák, B. Ecker, E. von Hauff and E. Da Como, Synth. Met., 2015, 199, 381–387 CrossRef CAS.
- G. K. Toworfe, R. J. Composto, C. S. Adams, I. M. Shapiro and P. Ducheyne, J. Biomed. Mater. Res., Part A, 2004, 71, 449–461 CrossRef PubMed.
- S. Ahn, L. F. Deravi, S. J. Park, B. E. Dabiri, J. S. Kim, K. K. Parker and K. Shin, Adv. Mater., 2015, 2838–2845 CrossRef CAS PubMed.
- P. J. Molino, M. J. Higgins, P. C. Innis, R. M. I. Kapsa and G. G. Wallace, Langmuir, 2012, 28, 8433–8445 CrossRef CAS PubMed.
- M. Bergkvist, J. Carlsson and S. Oscarsson, J. Biomed. Mater. Res., 2003, 64, 349–356 CrossRef PubMed.
- H. M. Kowalczyńska, M. Nowak-Wyrzykowska, R. Kołos, J. Dobkowski and J. Kamiński, J. Biomed. Mater. Res., Part A, 2005, 72, 228–236 CrossRef PubMed.
- J. J. Gray, Curr. Opin. Struct. Biol., 2004, 14, 110–115 CrossRef CAS PubMed.
- M. E. Brennan-Fournet, M. Huerta, Y. Zhang, G. Malliaras and R. M. Owens, J. Mater. Chem. B, 2015, 3, 9140–9147 RSC.
- F. Hook, M. Rodahl, P. Brzezinski and B. Kasemo, Langmuir, 1998, 7463, 729–734 CrossRef.
- I. Reviakine, D. Johannsmann and R. P. Richter, Anal. Chem., 2011, 22, 8838–8848 CrossRef PubMed.
- J. G. Steelea, C. McFarlandb, B. A. Daltonc, G. Johnsond, D. M. Evanse and C. R. Howlett, J. Biomater. Sci., Polym. Ed., 1994, 5, 245–257 CrossRef.
- C. González-García, S. R. Sousa, D. Moratal, P. Rico and M. Salmerón-Sánchez, Colloids Surf., B, 2010, 77, 181–190 CrossRef PubMed.
- A. G. Hemmersam, K. Rechendorff, M. Foss, D. S. Sutherland and F. Besenbacher, J. Colloid Interface Sci., 2008, 320, 110–116 CrossRef CAS PubMed.
- H. L. Khor, Y. Kuan, H. Kukula, K. Tamada and W. Knoll, Biomacromolecules, 2007, 8, 1530–1540 CrossRef CAS PubMed.
- M. S. Lord, B. G. Cousins, P. J. Doherty, J. M. Whitelock, A. Simmons and R. L. Williams, Biomaterials, 2006, 27, 4856–4862 CrossRef CAS PubMed.
- K. Cai, J. Bossert and K. D. Jandt, Colloids Surf., B, 2006, 49, 136–144 CrossRef CAS PubMed.
- M. N. Sela, L. Badihi, G. Rosen and D. Steinberg, Clin. Oral Implant Res., 2007, 18, 630–638 CrossRef PubMed.
- M. B. Hovgaard, K. Rechendorff and J. Chevallier, J. Phys. Chem. B, 2008, 112, 8241–8249 CrossRef CAS PubMed.
- M. Dargahi, V. Nelea, A. Mousa, S. Omanovic and M. T. Kaartinen, RSC Adv., 2014, 4, 47769–47780 RSC.
- N. B. Eisele, I. Fredrik and S. F. Andersson, Biomacromolecules, 2012, 13, 2322–2332 CrossRef CAS PubMed.
- P. J. Molino, Z. Yue, B. Zhang, A. Tibbens, X. Liu, R. M. I. Kapsa, M. J. Higgins and G. G. Wallace, Adv. Mater. Interfaces, 2014, 1, 1300122 CrossRef.
- S. Bellani, D. Fazzi, P. Bruno, E. Giussani, E. V. Canesi, G. Lanzani and M. R. Antognazza, J. Phys. Chem. C, 2014, 118, 6291–6299 CAS.
- R. Porrazzo, S. Bellani, a. Luzio, C. Bertarelli, G. Lanzani, M. Caironi and M. R. Antognazza, APL Mater., 2015, 3, 014905 CrossRef.
- V. D. Mihailetchi, H. X. Xie, B. de Boer, L. J. A. Koster and P. W. M. Blom, Adv. Funct. Mater., 2006, 16, 699–708 CrossRef CAS.
- P. B. Miranda and D. Moses, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 64, 081201 CrossRef.
- C. Soci, I. W. Hwang, D. Moses, Z. Zhu, D. Waller, R. Gaudiana, C. J. Brabec and A. J. Heeger, Adv. Funct. Mater., 2007, 17, 632–636 CrossRef CAS.
- P. Hockberger, T. A. Skimina, V. E. Centone, C. Lavin, S. Chu and S. Dadras, Cell Biol., 1999, 96, 6255–6260 CAS.
- O. M. Pereira, J. R. Smith and L. Packer, Photochem. Photobiol., 1976, 24, 237–242 CrossRef CAS PubMed.
- Y. Kuse, K. Ogawa, K. Tsuruma, M. Shimazawa and H. Hara, Sci. Rep., 2014, 4, 5223 CAS.
- B. F. Godley, F. A. Shamsi, F. Q. Liang, S. G. Jarrett, S. Davies and M. Boulton, J. Biol. Chem., 2005, 280, 21061–21066 CrossRef CAS PubMed.
- L. S. Liao, M. K. Fung, C. S. Lee, S. T. Lee, M. Inbasekaran, E. P. Woo and W. W. Wu, Appl. Phys. Lett., 2000, 76, 3582 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6tb01129b |
| ‡ Istituto Italiano di Tecnologia, Via Morego, 30, 16163 Genova, Italy. |
| § Wellman Center for Photomedicine, Massachusetts General Hospital & Harvard Medical School. |
| This journal is © The Royal Society of Chemistry 2016 |