
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nanohybrid polymer brushes on silica for bioseparation†
Lingdong
Jiang
a,
Héctor
Bagán
 a,
Tripta
Kamra
ab,
Tongchang
Zhou
a and
Lei
Ye
*a
a,
Tripta
Kamra
ab,
Tongchang
Zhou
a and
Lei
Ye
*a
aDivision of Pure & Applied Biochemistry, Department of Chemistry, Lund University, Box 124, 221 00 Lund, Sweden. E-mail: lei.ye@tbiokem.lth.se; Fax: +46 46 2224611; Tel: +46 462229560
bDivision of Synchrotron Radiation Research, Department of Physics, Lund University, Box 118, 221 00 Lund, Sweden
First published on 7th April 2016
Abstract
Boronic acid based affinity materials are of great importance for effective enrichment of biomolecules containing a cis-diol structure, for example glycoproteins. In this work, we developed a new pH- and temperature-responsive boronate affinity material for effective separation of glycoproteins. A nanohybrid material composed of silica cores and flexible polymer brushes, denoted as Si@poly(NIPAm-co-GMA)@APBA, was prepared via surface-initiated atom transfer radical polymerization (SI-ATRP) in combination with Cu(I)-catalyzed azide–alkyne cycloaddition (CuAAC) click reaction. The size, morphology and composition of the obtained nanohybrid were characterized by dynamic light scattering (DLS), scanning electron microscopy (SEM), Fourier transform infrared spectroscopy (FT-IR), elemental analysis and thermogravimetric analysis (TGA). The density of polymer brushes on the surface of silica nanoparticles was determined to be 0.7 molecules per nm2. The maximum binding capacities of the nanohybrid Si@poly(NIPAm-co-GMA)@APBA for ovalbumin (OVA) and horseradish peroxidase (HRP) were determined to be 87.6 mg g−1 and 22.8 mg g−1, respectively. Glycoprotein binding on the nanohybrid could be controlled by varying the pH of the binding buffer. By increasing the temperature from 20 °C to 35 °C, glycoprotein binding onto the nanohybrid was decreased. This new pH- and temperature-responsive nanohybrid will be useful for a number of biotechnological and biomedical applications, for example, for protein separation and drug delivery.
Introduction
Organic/inorganic hybrid materials have attracted ever increasing attention in recent years due to their unique structural characteristics and potential applications in bioseparation,1 targeted drug/gene delivery,2,3 theranostics and more.4 Typically, hybrid particles are composed of inorganic cores and surrounding organic layers in the form of polymer shells or polymer brushes. The chemical properties of hybrid particles and their responsiveness to external stimuli (e.g., pH, temperature or light) are mainly determined by the organic layer,5 whereas both the size and shape of the inorganic cores and the organic layer determine the physical properties of the particles. Due to their mechanical stability, high surface area, and ease of synthesis, silica/polymer hybrid nanoparticles have been studied most extensively.6Glycosylation is one of the most important and ubiquitous post-translational modifications of proteins, which plays a crucial role in various biological and physiological activities, e.g. molecular recognition, protein folding, cell division and differentiation, signal transduction, immune response, and so on.7,8 Additionally, it has been shown that altered, aberrant or missing glycosylation is correlated with many human diseases.9 Recent advances in mass spectrometry have made direct determination of glycoproteins feasible.10 However, it is still a challenging task to analyze protein glycosylation in complex samples due to the inherently low abundance of glycoproteins in complex biological samples. Therefore, it is necessary to develop efficient methods for selective purification and isolation of glycoproteins in order to obtain a complete mapping of glycoproteomes and to achieve reliable biomarker analysis.
Boronic acid ligand-functionalized materials such as monolithic capillaries11–15 and magnetic nanoparticles16–19 have been well developed and have shown great potential for selective recognition, immobilization and enrichment of glycoproteins from complex biological samples. Boronic acids can covalently bind the cis-diol moiety of biomolecules to generate cyclic esters in mildly basic aqueous media, while under acidic conditions the boronate ester bond can be hydrolyzed, and thus the capture/release process can be easily controlled through a switch of the pH value.20 Since cis-diol binding is based on covalent interactions, it allows compounds adsorbed by non-covalent interactions to be washed away. This unique property of boronic acid makes it an ideal affinity ligand for capture of glycoproteins. Lin et al. prepared boronic acid-functionalized silica nanoparticles by combining surface molecular imprinting with boronate affinity interactions.21 The protein imprinted silica nanoparticles exhibited high adsorption capacity and excellent recognition toward the template glycoprotein. Zhang et al. prepared boronic acid functionalized Fe3O4 composites using distillation–precipitation polymerization in combination with click chemistry and applied the magnetic particles to separate glycoproteins from egg white samples.19 More recently, Ye et al. described an appealing boronate-affinity sandwich assay (BASA) for low abundance glycoproteins by combining protein imprinted polymers with boronate-affinity-based Raman probes.22 The determination of trace glycoproteins in complex samples was enabled by the effective enrichment of glycoproteins and surface enhanced Raman scattering.
Poly(N-isopropylacrylamide) (PNIPAm) is a thermally responsive polymer, which undergoes a phase transition at its lower critical solution temperature (LCST) of 32 °C.23 Below the LCST it is soluble and has a hydrated, extended chain conformation in aqueous solution, while it becomes insoluble above the LCST due to rapid, reversible chain dehydration to form aggregates. To broaden the applications of thermally responsive polymeric systems, it is necessary to introduce suitable comonomers with desired functional characteristics into PNIPAm.24,25 Glycidyl methacrylate (GMA) is a very valuable monomer due to its epoxy group that can undergo ring-opening reactions, and can be used to introduce various functional groups into polymer chains.26 However, using traditional polymerization methods, it is impossible to control the polymer structure, making it difficult to control the stimuli-response properties and the functionalization of polymers. The controlled radical polymerization (CRP) technique has provided a new and powerful avenue towards engineering the structure and properties of synthetic polymers.27 Surface-initiated atom transfer radical polymerization (SI-ATRP) is among the most efficient and robust CRP processes for grafting polymer brushes on a solid support,28,29 as it allows us to regulate not only a predetermined degree of polymerization with a narrow molecular weight distribution, but also the shape and topology of the grafted polymer chains. In the literature, grafting of the PNIPAm brush on the surfaces of various substrates, including silica nanoparticles,30,31 silica beads,32,33 silicon wafers,34,35 polystyrene beads36 and chitosan,37 has been achieved by SI-ATRP. The grafting-from mechanism also guarantees that all polymer chains are synthesized on the surface, and no polymer waste is formed in the reaction solution. In addition, the polymer chains produced by SI-ATRP preserve terminal halogen atoms that can be converted into various functional end groups, which offers different possibilities for further surface functionalization.
In previous studies, we have successfully grafted a homopolymer of boronic acid on silica38 and synthesized a boronic acid-terminated thermo-sensitive polymer for affinity separation of saccharides.39 In this work, we take advantage of SI-ATRP to graft copolymer brushes from NIPAm and GMA on amino-functionalized silica to prepare firstly a generic nanohybrid material, Si@poly(NIPAm-co-GMA), which can be used to derive a number of affinity materials, such as boronate affinity materials, antibody-based affinity materials and immobilized metal-ion affinity chromatography (IMAC) materials through further functionalization of GMA moieties. In this work, we demonstrate that by post-functionalization of the GMA moieties with propargylamine, multiple alkyne groups can be introduced into the grafted polymer brushes, which allow azide-tagged fluorogenic boronic acid to be linked to the polymer brushes via Cu(I)-catalyzed azide–alkyne cycloaddition (CuAAC) click reaction. Using this synthetic approach we obtained a new type of thermo-sensitive and boronic acid-tagged core–shell nanohybrid, Si@poly(NIPAm-co-GMA)@APBA, which provides high efficiency separation for glycoproteins. The nanohybrid particles were characterized using dynamic light scattering (DLS), Fourier transform infrared spectroscopy (FT-IR), spectrofluorometry, scanning electron microscopy (SEM), elemental analysis and thermogravimetric analysis (TGA). Furthermore, we quantified the density of polymer brushes on the silica cores using a novel fluorescence labelling technique. The terminal bromine groups in the polymer brushes of the nanohybrid particles were first converted into azide groups, which were then coupled with an alkyne-tagged fluorescein isothiocyanate (FITC) through the CuAAC click chemistry. By measuring the fluorescence intensity of the derivatized particles we were able to quantify the density of the polymer brushes on the particle surface. We also studied the glycoprotein binding characteristics of the nanohybrid material and demonstrated that the flexible polymer brushes on silica are able to bind glycoproteins (OVA and HRP) through selective boronate affinity interaction. In addition, the feasibility of applying the nanohybrid to enrich glycoproteins in a real complex sample (egg white) was demonstrated. Based on the results obtained, we believe that the synthetic approach and the hybrid material developed in this work could provide new opportunities for the design of more efficient adsorbents for the capture and enrichment of bioactive cis-diols and glycoproteins from complex samples.
Experimental
Materials
Tetraethylorthosilicate (TEOS), (3-aminopropyl)triethoxysilane (APTES), triethylamine (TEA), 2-bromoisobutyryl bromide (BIBB), N-isopropylacrylamide (NIPAm), glycidyl methacrylate (GMA), propargylamine, Alizarin Red S (ARS), CuSO4, sodium ascorbate, sodium azide, tris(2-dimethylaminoethyl)amine (Me6TREN), fluorescein isothiocyanate (FITC), bovine serum albumin (BSA), ovalbumin (OVA), horseradish peroxidase (HRP), 2,2′-azinobis[3-ethylbenzthiazoline-6-sulfonic acid] diammonium salt (ABTS), sodium dodecyl sulfonate (SDS), sulfuric acid, acetonitrile (ACN), methanol, ammonia, toluene, N,N-dimethylformamide (DMF), tetrahydrofuran (THF), isopropanol and acetone were purchased from Sigma-Aldrich and used without further purification. CuBr was stirred overnight in acetic acid, centrifuged, washed with water and methanol and dried in a vacuum desiccator before use. Ultrapure water (18.2 MΩ cm) was obtained from an ELGA LabWater System (Vivendi Water Systems Ltd). 3-(2-Azidoacetylamino)phenylboronic acid (APBA) was synthesized according to our previously published procedure.40 Polyacrylamide gels (4–20%, Mini-protean) used in SDS-PAGE were obtained from Bio-Rad.Preparation of silica nanoparticles
Silica nanoparticles were synthesized using a one-step Stöber procedure.41 Typically, methanol (100 mL), water (33 mL) and ammonia (25%, 22.4 mL) were added to a 1000 mL glass beaker and agitated using a magnetic stirrer. A mixture of 130 mL of methanol containing 13.8 mL of TEOS was quickly added to this solution. The reaction mixture was stirred at ambient temperature for 8 h. The silica nanoparticles were isolated by centrifugation, washed with water and methanol several times, and dried in a vacuum desiccator.Preparation of amino-functionalized silica nanoparticles
The silica nanoparticles were immersed into 1% APTES solution (0.5 mL of APTES dissolved in 49.5 mL of anhydrous toluene) and allowed to react for 24 h at reflux temperature with stirring. The particles were then washed with acetone and ethanol several times to remove residual reagents and dried in a vacuum desiccator. The amino-functionalized silica nanoparticle is denoted as Si@NH2.Preparation of initiator-functionalized silica nanoparticles
Si@NH2 (0.25 g) and triethylamine (0.40 mL, 2.9 mmol) were mixed in THF (12 mL) and cooled in an ice-water bath. To the suspension BIBB (0.575 g, 2.5 mmol) was added slowly. The reaction mixture was warmed to room temperature and stirred overnight. The nanoparticles were isolated and purified following the procedures used for the synthesis of silica nanoparticles. The BIBB-functionalized silica nanoparticle is denoted as Si@initiator.Preparation of Si@poly(NIPAm-co-GMA) by SI-ATRP
Si@initiator (0.10 g), NIPAm (0.17 g, 1.5 mmol), GMA (0.21 g, 1.5 mmol), CuBr (0.0072 g, 0.05 mmol) and 2-propanol (8.0 mL) were added to a 25 mL flask. The mixture was deoxygenated by bubbling with nitrogen gas for 15 min. Me6TREN (0.024 g, 0.1 mmol) was then added to allow the formation of the CuBr/Me6TREN complex. After another 15 min of nitrogen bubbling, the reaction mixture was sealed, heated to 60 °C and magnetically stirred under a nitrogen atmosphere for 24 h. After the reaction, the mixture was exposed to air to terminate polymerization. The nanoparticles were isolated and purified following the procedures as described above. The obtained product is denoted as Si@poly(NIPAm-co-GMA).Preparation of Si@poly(NIPAm-co-GMA)@alkyne particles
To introduce alkyne groups on the Si@poly(NIPAm-co-GMA) particles, Si@poly(NIPAm-co-GMA) (0.05 g) was submerged in a solution of propargylamine in 2-propanol (2% v/v, 30 μL/1.5 mL) and allowed to react for 24 h at 55 °C. During this period the reaction flask was rotated slowly in a DNA hybridization oven. The obtained particles were isolated and purified following the procedures as described above. The product is denoted as Si@poly(NIPAm-co-GMA)@alkyne.Preparation of Si@poly(NIPAm-co-GMA)@APBA by click chemistry
Si@poly(NIPAm-co-GMA)@alkyne (0.09 g) and APBA (0.05 g) were dispersed in a solution of methanol![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O (v/v, 2/1, 6 mL). The mixture was deoxygenated by bubbling with nitrogen gas for 15 min. After that, CuSO4 solution (20 μL, 100 mM) and sodium ascorbate solution (200 μL, 100 mM) were added. After another 15 min of nitrogen bubbling, the reaction mixture was sealed and magnetically stirred at room temperature under a nitrogen atmosphere for 24 h. The obtained particles were isolated and purified following the procedures as described above. The obtained product is denoted as Si@poly(NIPAm-co-GMA)@APBA.
:H2O (v/v, 2/1, 6 mL). The mixture was deoxygenated by bubbling with nitrogen gas for 15 min. After that, CuSO4 solution (20 μL, 100 mM) and sodium ascorbate solution (200 μL, 100 mM) were added. After another 15 min of nitrogen bubbling, the reaction mixture was sealed and magnetically stirred at room temperature under a nitrogen atmosphere for 24 h. The obtained particles were isolated and purified following the procedures as described above. The obtained product is denoted as Si@poly(NIPAm-co-GMA)@APBA.
Preparation of Si@poly(NIPAm-co-GMA)@N3 particles
Si@poly(NIPAm-co-GMA) was first treated with 0.5 M sulfuric acid at 70 °C for 12 h to open the epoxy ring from GMA. The particles were washed with water and methanol, and then dried in a vacuum desiccator. The dried particles (0.05 g) and sodium azide (0.025 g) were dispersed in 6 mL of DMF and allowed to react for 12 h at 60 °C in a DNA hybridization oven. The obtained particles were isolated and purified following the procedures as described above. The product is denoted as Si@poly(NIPAm-co-GMA)@N3.Preparation of alkyne-functionalized fluorescein isothiocyanate (FITC-alkyne)
FITC (0.01 g, 0.0128 mmol) and propargylamine (90 μL, 0.703 mmol) were dissolved in 110 μL of DMF in a microcentrifuge tube and stirred in the dark for 24 h at room temperature.42 DMF and excess of propargylamine were then evaporated in a vacuum chamber, and the remaining product was dissolved in methanol.Quantification of polymer brush density on Si@poly(NIPAm-co-GMA)@N3 with FITC-alkyne
Si@poly(NIPAm-co-GMA)@N3 (0.005 g) were suspended in 880 μL of water. The mixture was sonicated for 5 min to give a homogeneous suspension. After addition of FITC-alkyne (200 μg mL−1, 100 μL), methanol (900 μL), CuSO4 (100 mM, 20 μL) and sodium ascorbate (100 mM, 100 μL), the mixture was stirred in the dark for 24 h at room temperature. After the reaction, the nanoparticles were collected by centrifugation and washed with a mixture of methanol and H2O (1:1, v/v) thoroughly until no florescence signal can be detected in the washing solvent. The remaining unreacted FITC-alkyne in the supernatant and the washing solution was quantified by measuring its maximum fluorescence intensity (Ex: 490 nm, Em: 514 nm) using a spectrofluorometer. The density of the polymer brushes on the particle surface was calculated from the amount of FITC-alkyne consumed during fluorescence labelling.
Protein binding with Si@poly(NIPAm-co-GMA)@APBA
Si@poly(NIPAm-co-GMA)@APBA (0.002 g) were added to a solution (2 mL) of protein sample at different concentrations in phosphate buffered saline (PBS) (20 mM phosphate, pH 9.0 or pH 7.4, containing 0.5 M NaCl). The suspensions were gently shaken at 20 °C or 35 °C overnight and then centrifuged. After centrifugation, the particles were removed, and the UV-vis absorption of the supernatant at 280 nm for OVA and BSA was measured to calculate the protein concentration. To calculate the concentration of HRP, ABTS, a water-soluble ELISA substrate for HRP was used to measure the enzyme activity. Briefly, 0.1 mL of diluted HRP solution (concentration between 0.5 μg mL−1 and 2.5 μg mL−1) were mixed with ABTS solution (0.2 mL) and shaken for 5 min before 0.4 mL of stop solution (1% SDS solution) was added. After 1 h, the UV-vis absorption of the solution at 650 nm was measured. The amount of protein bound by the Si@poly(NIPAm-co-GMA)@APBA particles was calculated from the difference between the initial protein concentration and the protein concentration in the supernatant.The experimental data are presented as the adsorption capacity per unit mass (mg) of the Si@poly(NIPAm-co-GMA)@APBA particles, and the adsorption capacity (Q) is calculated using the equation:

Here C0 (mg mL−1) is the initial concentration of the protein solution, Ct (mg mL−1) is the equilibrium concentration of the protein, V (mL) is the volume of the protein solution, and m (mg) is the mass of the particles.
The selective adsorption of glycoproteins was also investigated by sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) analysis. Typically, 0.006 g of Si@poly(NIPAm-co-GMA)@APBA were added into 1 mL of BSA solution (1 mg mL−1; PBS, 20 mM, pH 9.0), 1 mL of OVA solution (1 mg mL−1; PBS, 20 mM, pH 9.0), and 1 mL of 100-fold diluted egg white (PBS, 20 mM, pH 9.0). The mixture was gently shaken overnight at room temperature. Then, the Si@poly(NIPAm-co-GMA)@APBA particles were separated and washed with the same PBS solution, before the proteins were eluted in HAc–NaAc buffer solution (20 mM, pH 4.0, 0.2 mL). The eluted proteins were lyophilized and dissolved in PBS (20 mM, pH 9.0, 0.1 mL). Finally, the proteins collected in the different supernatants were subjected to the SDS analysis.
Characterization
Attenuated total reflection (ATR) infrared spectra were recorded at room temperature in the range of 4000–525 cm−1 with a resolution of 4 cm−1 and 16 scans using a Perkin-Elmer FT-IR instrument. Thermal gravimetric analysis (TGA) was performed in synthetic air. The samples were heated to 700 °C at 10 °C min−1 and held for 30 min. Elemental analysis (C, H, N) was carried out using an Elemental Analyser, Model 1108 (Carlo-Erba, Milan, Italy). Elemental analysis for Br and B content was performed by Mikroanalytisches Laboratorium Kolbe (Germany). UV-Vis absorption spectra were recorded using a Cary 60 UV/Vis spectrophotometer (Agilent Technologies). Fluorescence emission was measured using a QuantaMaster C-60/2000 spectrofluorometer (Photon Technology International, Lawrenceville, NJ). The surface morphologies of silica particles were observed using a scanning electron microscope (Thermal Field Emission SEM LEO 1560, Zeiss, Oberkochen, Germany) and a transmission electron microscope (Tecnai Spirit BioTWIN, FEI Company, Oregon, USA). The hydrodynamic diameter of the nanoparticles was measured at 25 °C by dynamic light scattering using a Zetasizer Nano ZS instrument (Malvern Instruments, United Kingdom).Results and discussion
In this work, the process for the synthesis of Si@poly(NIPAm-co-GMA)@APBA particles is illustrated in Scheme 1. Firstly, silica nanoparticles were synthesized using a one-step Stöber procedure. The silica particles were modified with APTES to give amino-functionalized Si@NH2 particles, which were used to immobilize the ATRP initiator BIBB, leading to the Si@initiator particles. The copolymer brushes, poly(NIPAm-co-GMA), were grafted on the surface of Si@initiator via SI-ATRP. In order to introduce the azide-functionalized APBA into the polymer brushes, the Si@poly(NIPAm-co-GMA) particles were first reacted with propargylamine to produce multiple alkyne groups, which were then used to conjugate the fluorogenic APBA through the CuAAC click reaction. The CuAAC click reaction between Si@poly(NIPAm-co-GMA)@alkyne and the azide-functionalized APBA was performed in a mixture of methanol and H2O (2:1, v/v) in the presence of CuSO4 and sodium ascorbate, leading to Si@poly(NIPAm-co-GMA)@APBA, a new type of affinity nanohybrid material. The advantage of this synthetic design is that the intermediate particles, Si@poly(NIPAm-co-GMA), can be used as a general reagent to synthesize different types of affinity adsorbents, depending on the types of the organic amines used to react with the epoxy groups.
 | ||
| Scheme 1 Synthesis of Si@poly(NIPAm-co-GMA)@APBA particles by combining SI-ATRP with CuAAC click reaction. | ||
Characterization of particles using SEM, TEM and DLS
The size and shape of the silica core, Si@NH2, Si@initiator and Si@poly(NIPAm-co-GMA)@APBA particles were examined using SEM, TEM and DLS techniques (Fig. 1). SEM analysis revealed that the silica core particles have a uniform and spherical shape with an average diameter of about 260 nm (Fig. 1a). The hydrodynamic diameter of the silica core particles was determined to be 266 nm (PDI: 0.08) (Fig. 1e), which is close to that measured by SEM (Fig. 1a). After introduction of the amino groups and the initiator, the particles were still uniform and spherical (Fig. 1b and c), and the hydrodynamic diameter increased to 299 nm (PDI: 0.218) (Fig. 1h) and 302 nm (PDI: 0.275) (Fig. 1i), respectively, as determined by the DLS measurement. The hydrodynamic diameters increased due to the change in the surface properties after the modification of the silica core with APTES and the initiator molecules. After the grafting of polymer brushes on the Si@initiator, the shape of the particles became a little irregular (Fig. 1d and f). From the TEM images (Fig. 1e and f) it is easy to observe the increase of particle size (by about 40 nm) caused by the polymer brushes. In solution the polymer-grafted particles (Si@poly(NIPAm-co-GMA)@APBA) displayed a significantly larger hydrodynamic size (about 1.2 μm) (Fig. 1h), which can be attributed to the solvation of the surface-grafted polymer brushes. | ||
| Fig. 1 SEM images of silica core particles (a), Si@NH2 (b), Si@initiators (c) and Si@poly(NIPAm-co-GMA)@APBA particles (d). TEM images of Si@initiators (e) and Si@poly(NIPAm-co-GMA)@APBA particles (f). The hydrodynamic particle size of silica core particles (g), Si@NH2 (h), Si@initiators (i) and Si@poly(NIPAm-co-GMA)@APBA particles (j) measured in deionized water. | ||
Characterization of particles by compositional analyses
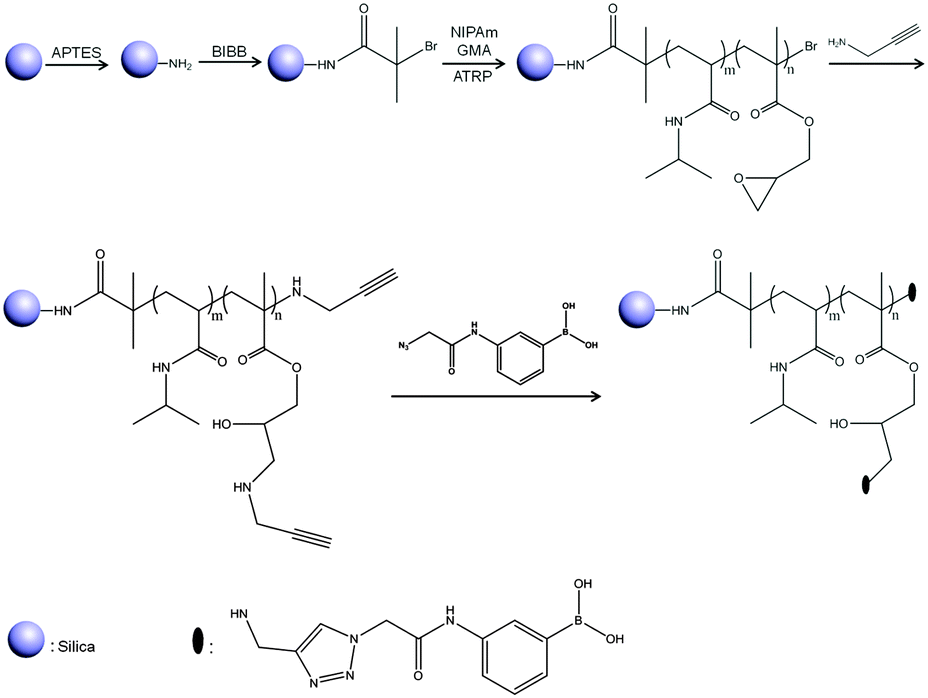
FT-IR spectra were used to characterize the organically modified silica (Fig. 2). The distinct absorption band at 1053 cm−1 is associated with the asymmetric stretching vibrations of Si–O–Si (curve (a)). The bands at about 1637 and 1534 cm−1 in curve (b) are associated with amide I and amide II bands formed after immobilization of the ATRP initiator. The peaks at 1725 cm−1 and 1387 cm−1 in curve (c) are assigned to the stretching vibration of ester carbonyl groups and the anti-symmetric stretching band of carbonyl groups, respectively. The band at 1456 cm−1 is assigned to the methylene scissoring band. All these bands indicate the successful synthesis of the polymer brushes tethered to the particle surface. Moreover, the weak peak at 909 cm−1 is assigned to the symmetrical stretching of the epoxy group from the GMA monomer. After the reaction with propargylamine, the epoxy band in the Si@poly(NIPAm-co-GMA) particles disappeared, suggesting the successful introduction of the alkyne groups into the Si@poly(NIPAm-co-GMA)@alkyne particles (curve (d)). Due to the limited sensitivity of FT-IR, it is difficult to observe spectral variation after conversion of the alkynes into the boronic acid derivatives (curve (e)). To prove the successful introduction of APBA into the particles via the CuAAC reaction, a diol-containing dye, ARS, was employed. It is known that ARS displays a dramatic increase in fluorescence intensity in response to its binding to a boronic acid with a high affinity, and ARS can be used as a general fluorescent reporter to detect boronic acids.43 As can be seen from Fig. S1 (ESI†), after being treated with ARS, the Si@poly(NIPAm-co-GMA)@APBA particles displayed a strong fluorescence emission (Ex: 469 nm, Em: 600 nm), indicating the successful coupling of APBA to the Si@poly(NIPAm-co-GMA)@alkyne particles via the CuAAC reaction.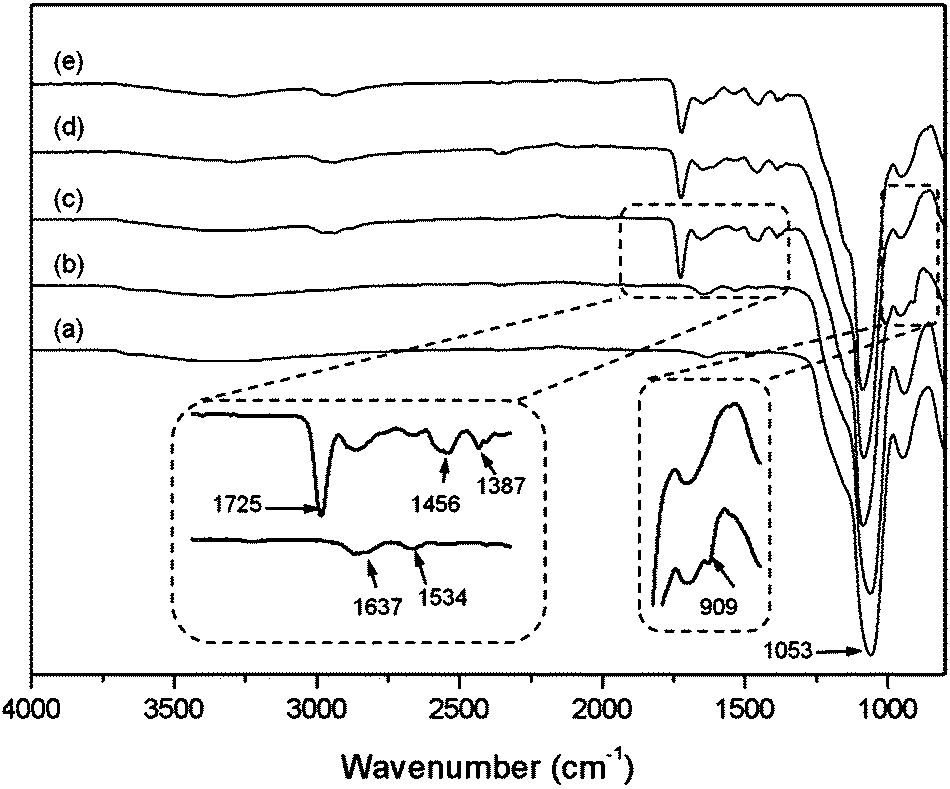 | ||
| Fig. 2 FT-IR spectra of Si@NH2 (a), Si@initiators (b), Si@poly(NIPAm-co-GMA) (c), Si@poly(NIPAm-co-GMA)@alkyne (d) and Si@poly(NIPAm-co-GMA)@APBA (e). | ||
The results of elemental analysis for the different materials are listed in Table 1. On the basis of the nitrogen content in Si@NH2 particles and the bromine content in Si@initiator particles, the density of the aminopropyl group and the initiator on the silica particles was calculated to be 0.51 mmol g−1 and 0.42 mmol g−1, respectively. After the SI-ATRP process, the content of C, H and N increased significantly, which resulted from the grafted polymer brushes made of the organic monomers, NIPAm and GMA.
| Entry | Sample name | %C | %H | %N | %Br | %B |
|---|---|---|---|---|---|---|
| n.d.: not determined. | ||||||
| 1 | Si@NH2 | 3.00 | 1.74 | 0.68 | — | — |
| 2 | Si@initiator | 5.16 | 1.67 | 0.63 | 2.96 | — |
| 3 | Si@poly(NIPAm-co-GMA) | 34.74 | 3.83 | 3.10 | 0.61 | — |
| 4 | Si@poly(NIPAm-co-GMA)@APBA | n.d. | n.d. | n.d. | 0.45 | 0.16 |
| 5 | Acid-treated Si@poly(NIPAm-co-GMA) | n.d. | n.d. | n.d. | 0.60 | — |
| 6 | Si@poly(NIPAm-co-GMA)@N3 | n.d. | n.d. | n.d. | 0.17 | — |
The content of organic materials in the three different particles (Si@NH2, Si@initiator and Si@poly(NIPAm-co-GMA)) was further investigated by TGA analysis. In Fig. 3, the weight loss below 250 °C (approximately 6%) can be attributed to the evaporation of residual organic solvent and water. The difference in organic content (5%) between Si@NH2 and Si@initiator (curves (a) and (b)) is attributed to the immobilized ATRP initiator. If the mass of silica is used as a reference, the amount of the initiator relative to that of silica becomes 6.6%, suggesting that the immobilized initiator on silica is 0.44 mmol g−1. This value is in agreement with the result obtained from the elemental analysis (0.42 mmol g−1), and is too large if the immobilized initiator is considered only to pack into a monolayer on the surface. Obviously, some of the initiator molecules were located underneath and were not able to contribute to the SI-ATRP. For Si@poly(NIPAm-co-GMA), a large weight loss occurred when the temperature increased from 250 °C to 700 °C (curve (c)). This large weight loss is due to the decomposition of the organic polymer on the surface of Si@poly(NIPAm-co-GMA). Based on the mass retention of the TGA curve (curve (c)), the amount of poly(NIPAm-co-GMA) brushes grafted on silica is calculated to be 0.917 g per g silica. For the boronic acid-attached particle Si@poly(NIPAm-co-GMA)@APBA, the boron content was measured to be 0.16% (Table 1, entry 4). Provided that all the alkyne groups in Si@poly(NIPAm-co-GMA)@alkyne have been conjugated to APBA, the density of the boronic acids in the nanocomposite Si@poly(NIPAm-co-GMA)@APBA is estimated to be approximately 0.3 mmol g−1 silica.
 | ||
| Fig. 3 TGA analysis of the Si@NH2 (a), Si@initiator (b) and Si@poly(NIPAm-co-GMA) (c) particles. | ||
Characterization of brush density and molecular weight
To determine the molecular weight of the grafted polymer brushes, we first treated Si@poly(NIPAm-co-GMA) particles with HF to remove the inorganic core. After treatment with HF, the organic polymer precipitate was collected by centrifugation and washed with water and methanol. The polymer was recovered with a yield of 43.5% based on the initial particle mass. This value is in rough agreement with the organic content estimated from the TGA analysis (Fig. 3, curve (c)). Unfortunately, the isolated polymer has very low solubility in the common solvents used for GPC analysis, therefore it was not possible to measure the molecular weight of the grafted polymer brushes by GPC analysis.Considering that the TGA analysis gives an accurate value of the organic content in the Si@poly(NIPAm-co-GMA) particles, we envisaged that an apparent molecular weight of the polymer brush can be calculated if the density of the polymer brushes on the particle surface can be determined. To measure the density of the polymer brushes, we designed a fluorescence labelling method that allows only the terminal bromine groups to be converted into azide, which can be labelled with FITC-alkyne via high efficiency CuAAC reaction. To avoid azidation at the GMA residues in Si@poly(NIPAm-co-GMA), we first treated the particles in 0.5 M sulfuric acid to make sure that all the epoxy rings in the polymer brushes were converted into hydroxyl groups. By comparing the bromine content in the particle Si@poly(NIPAm-co-GMA) and the acid-treated Si@poly(NIPAm-co-GMA) (Table 1, entries 3 and 5), we can conclude that the treatment with 0.5 M sulfuric acid did not affect the bromine in the particle. In the second step, the acid-treated particles were reacted with NaN3 to convert the terminal bromine into azide groups. To measure the efficiency of conversion of the terminal bromine into the azide group, the bromine content in the acid-treated Si@poly(NIPAm-co-GMA) and Si@poly(NIPAm-co-GMA)@N3 was determined by elemental analysis (Table 1, entries 5 and 6). The results show that around 72% bromine disappeared after the conversion. The 28% unreacted bromine is most likely located underneath the particle surface and could not be converted into terminal azide groups.
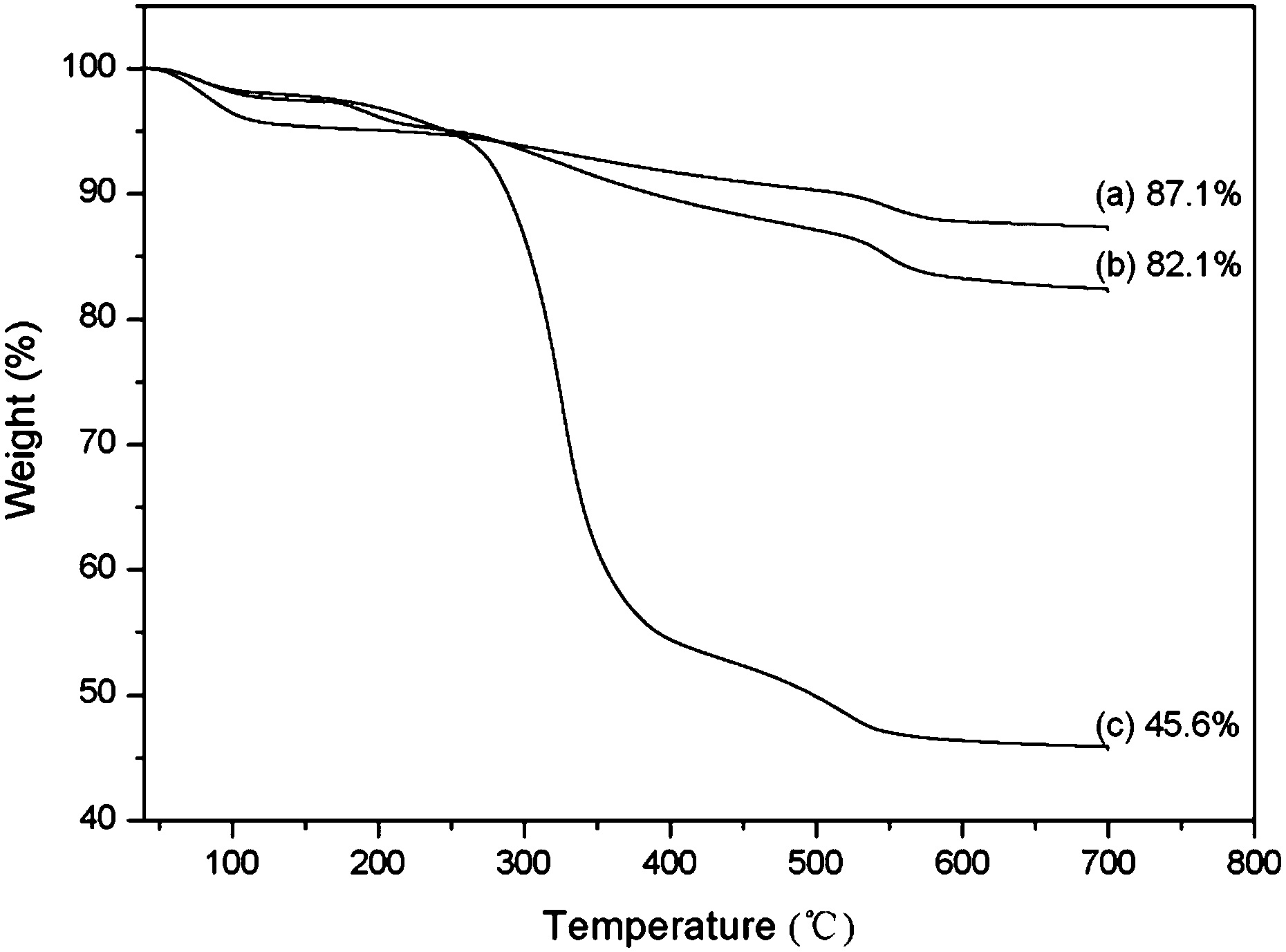
In the FT-IR spectrum in Fig. 4 (curve (b)), an absorption peak located at 2103 cm−1 is clearly observed from Si@poly(NIPAm-co-GMA)@N3 particles, which were obtained by azidation of the acid-treated Si@poly(NIPAm-co-GMA) particles. This absorption peak is ascribed to the asymmetric azide stretching vibration. After the CuAAC click reaction with FITC-alkyne, this absorption peak disappeared completely (Fig. 4, curve (c)), suggesting that the Si@poly(NIPAm-co-GMA)@N3 particles have been successfully conjugated with FITC-alkyne through the click reaction. Assuming that all the azide groups converted from the terminal bromine in the poly(NIPAm-co-GMA) brushes have reacted with FITC-alkyne, the density of the polymer brushes on the Si@poly(NIPAm-co-GMA) particles can be calculated from the consumed FITC-alkyne through fluorescence intensity measurements. Using this method, the density of the polymer brushes on the surface of silica was found to be 13.4 μmol g−1. If we assume the density of silica nanoparticles to be identical to that of bulk silica (2.07 g cm−3), then the density of the polymer chains grafted on the silica surface can be estimated to be ∼0.7 molecules per nm2. Considering the content of polymer brushes in the Si@poly(NIPAm-co-GPA) particles (0.917 g g−1 silica), the molecular weight of the poly(NIPAm-co-GPA) brushes is estimated to be ∼68000.
 | ||
| Fig. 4 FT-IR spectra of acid-treated Si@poly(NIPAm-co-GMA) (a), acid-treated Si@poly(NIPAm-co-GMA)@N3 (b) and acid-treated Si@poly(NIPAm-co-GMA)@FITC (c) particles. | ||
The fluorescence labelling by CuAAC is demonstrated as a versatile method for quantification of chemically accessible, surface-attached bromine atoms from the ATRP initiator. The obtained density value is considered reliable, especially to account for the surface-attached living initiators because the designed labelling reaction works only for the surface-exposed ATRP initiators. We envisage that the method for quantification of terminal halides developed here will be applicable to many other particle/initiator systems to deliver reliable and fast results.
Protein binding with Si@poly(NIPAm-co-GMA)@APBA particles
To determine the selectivity and efficiency of Si@poly(NIPAm-co-GMA)@APBA particles for separation of glycoproteins, we tested the protein binding with the particles using two glycoproteins, OVA and HRP, and a non-glycoprotein BSA as models.The equilibrium binding isotherms of Si@poly(NIPAm-co-GMA)@APBA for the three model proteins are shown in Fig. 5. A series of protein solutions of varying concentrations (0.2–1.0 mg mL−1 for BSA and OVA, 0.01–0.1 mg mL−1 for HRP) in phosphate buffer (PBS, 0.02 M; pH 9.0) containing 0.5 M NaCl were incubated with Si@poly(NIPAm-co-GMA)@APBA particles. NaCl was added to minimize non-specific adsorptions caused by electrostatic interactions. Under these conditions the biomolecules with cis-diol groups such as glycoproteins (OVA and HRP) were specifically captured by the Si@poly(NIPAm-co-GMA)@APBA particles. As can be seen from Fig. 5a and b, the Si@poly(NIPAm-co-GMA)@APBA particles showed significantly higher specific binding for the glycoproteins OVA and HRP than the non-glycoprotein BSA.
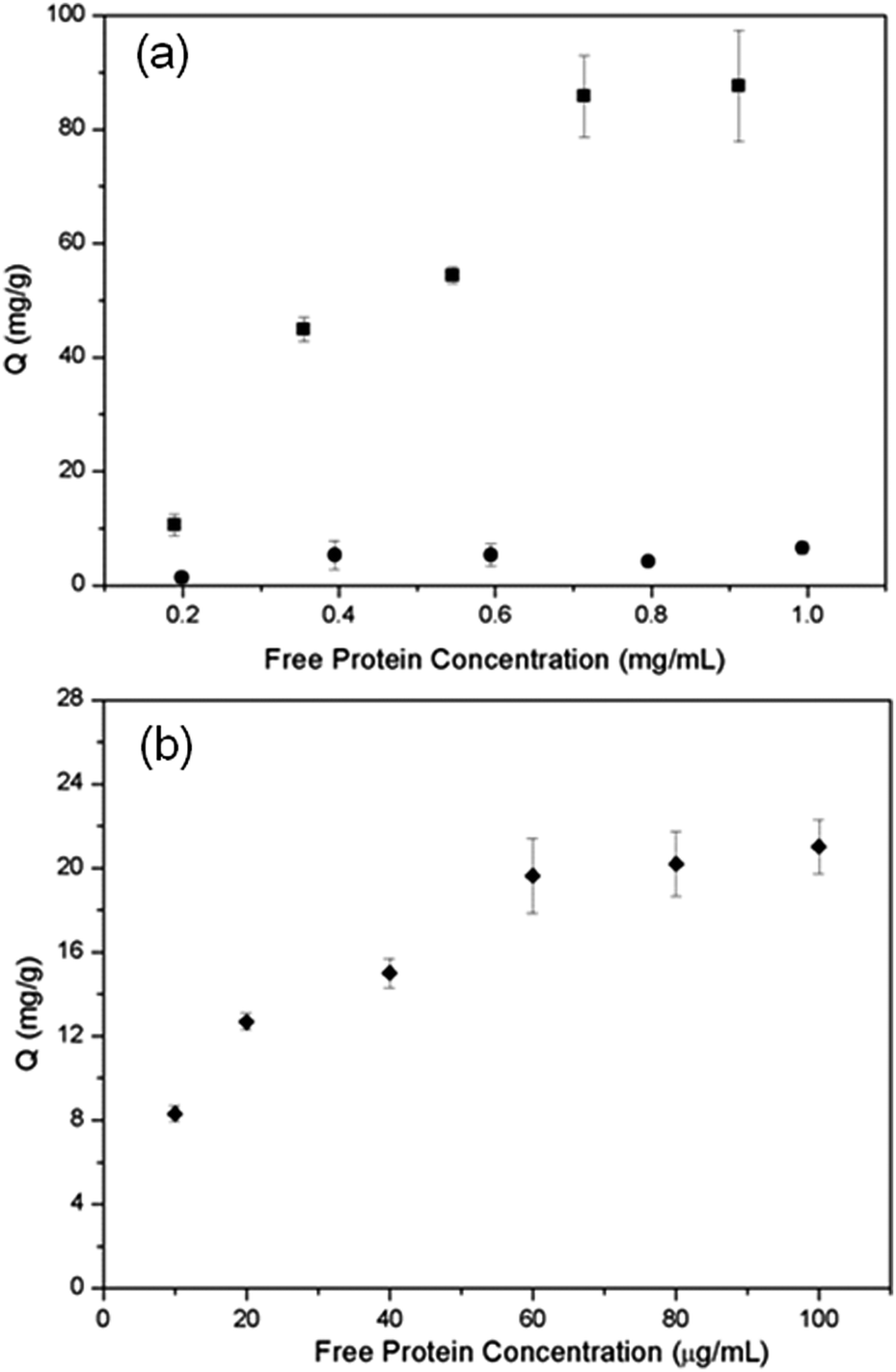 | ||
| Fig. 5 (a) Protein binding isotherm measured at pH 9.0 for OVA (■) and BSA (●). (b) Protein binding isotherm measured at pH 9.0 for HRP (◆). The concentration of the composite particles was 1 mg mL−1. | ||
The binding capacity of Si@poly(NIPAm-co-GMA)@APBA towards the two glycoproteins is 87.6 and 22.3 mg g−1 for OVA and HRP, respectively. In contrast, the maximum binding of the non-glycoprotein (BSA) by the particles is less than 6.6 mg g−1.
These results indicate that the specific affinity interactions between the boronic acid polymer brushes and the glycoproteins are dominant. Therefore, the multiple boronic acid ligands in the polymer brushes on the Si@poly(NIPAm-co-GMA)@APBA particles indeed contribute to the efficient glycoprotein binding.
Conventional boronate affinity separation requires the use of high pH solvent for covalent binding. Due to the risk of degradation of labile biomolecules under basic pH conditions, much attention has been paid to developing boronic acid-functionalized materials that can be used under neutral or physiological pH conditions. In our previous work we have shown that the clickable boronic acid APBA can be immobilized on a solid support to enable cis-diol binding at physiological pH.38,39 In this work, the protein binding offered by Si@poly(NIPAm-co-GMA)@APBA towards the glycoproteins and the non-glycoprotein at two different pH values (pH 7.4 and 9.0) was also investigated. As shown in Fig. 6, the binding capacity of Si@poly(NIPAm-co-GMA)@APBA at pH 7.4 was lower than that measured at pH 9.0. Nevertheless, the particles were still able to bind OVA and HRP with a capacity of 44.2 and 16.5 mg g−1, respectively, which are significantly higher than the non-glycoprotein BSA.
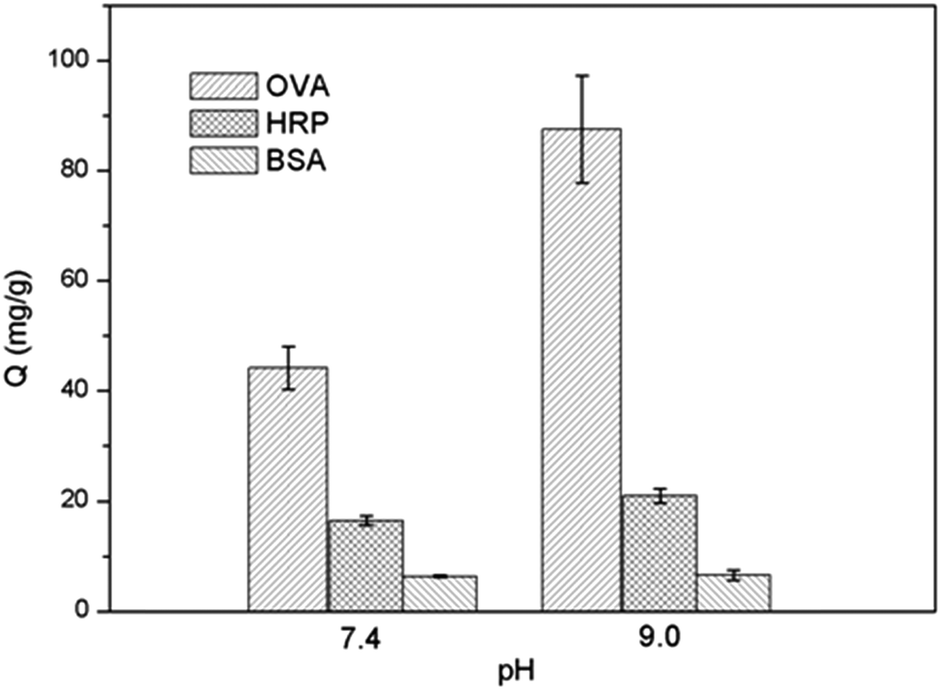 | ||
| Fig. 6 Effect of pH on protein binding to Si@poly(NIPAm-co-GMA)@APBA particles. | ||
The pH-dependent behavior of glycoprotein binding can be explained by the structural variation of the appended boronic acids. At high pH (pH 9.0), boronic acid exists in the form of a tetragonal anion that can react with cis-diols to form cyclic esters easily.44 At low pH boronic acid tends to have a trigonal configuration, which makes it more difficult to bind cis-diols. In this work, the phenylboronic acid was linked to the polymer brushes through a 1,2,3-triazole group. The triazole group can help keep the B atom in a sp3 configuration even under neutral or physiological pH conditions.38–40 As a result, the Si@poly(NIPAm-co-GMA)@APBA particles are able to retain sufficient binding for glycoproteins at neutral pH. Compared with previously reported boronate affinity materials such as stimuli-responsive core–shell nanospheres (binding capacity for OVA below 10 mg g−1),24 macroporous cryogels (binding capacity for OVA below 25 mg g−1)45 and magnetic composite microspheres (binding capacity for HRP below 25 mg g−1)46 that require an alkaline environment for glycoprotein binding, the new boronate affinity particles developed in this work display higher glycoprotein binding under physiological pH conditions, making the new affinity materials more suitable for separation of glycoproteins from biological samples.
Modulation of protein binding by temperature
Temperature responsive materials have also drawn increasing attention due to their wide range of applications in separation science and biomedicine.47 The incorporation of polyNIPAm in polymer brushes offers a possibility to modulate protein binding via temperature control. The effect of temperature on the protein binding to Si@poly(NIPAm-co-GMA)@APBA was further investigated. As shown in Fig. 7, at 35 °C, the binding capacity of Si@poly(NIPAm-co-GMA)@APBA for OVA and HRP decreased to 53.5 and 13.7 mg mL−1, respectively. Although glycoprotein binding was not completely diminished, the altered temperature indeed showed an obvious effect. The effect of temperature on glycoprotein binding can be explained by the structural change of the polymer brushes. At 35 °C (above the LCST of polyNIPAm), the polymer brushes became dehydrated and collapsed, making the appended boronic acids less accessible to bind the glycoproteins. As a comparison, binding of the non-glycoprotein BSA by the same particles was low and was not affected by the temperature variation, because BSA adsorption did not involve the appended boronic acids. Due to their temperature responsive properties, it may be possible to tune the binding capacity of the boronate affinity particles by simply varying the environmental temperature, which may bring in additional options for bioseparation.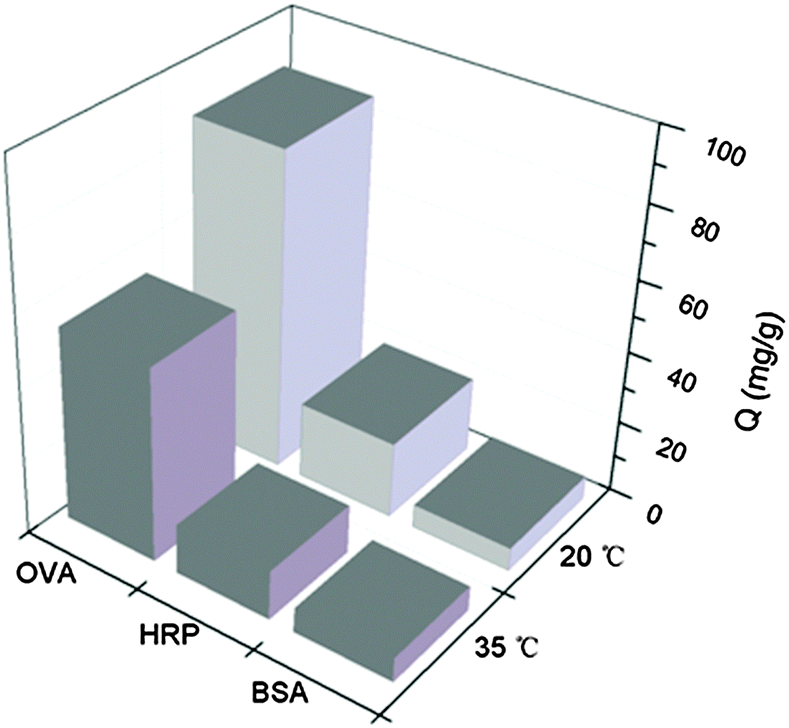 | ||
| Fig. 7 Effect of temperature on protein binding to Si@poly(NIPAm-co-GMA)@APBA particles at pH 9.0. The concentrations of OVA, BSA and HRP are 1 mg mL−1, 1 mg mL−1 and 100 μg mL−1, respectively. | ||
Separation of glycoproteins from complex samples
The efficiency of Si@poly(NIPAm-co-GMA)@APBA for separating glycoproteins from complex samples was evaluated by SDS-PAGE analysis. OVA (≥98%) and egg white glycoproteins were selected as the target proteins, and non-glycoprotein BSA (≥98%) was used as a control. The different protein samples were first incubated with the composite particles. After equilibrium binding, the particles were separated and transferred into an acidic buffer to release the bound proteins. The proteins in all the liquid phases were then collected and subjected to the SDS-PAGE analysis. In Fig. 8, the strong band in lane 2 corresponds to the BSA standard (65 kDa). After being treated with Si@poly(NIPAm-co-GMA)@APBA, the BSA band maintained the same intensity (lane 3), and no BSA band could be found in the eluted fraction (lane 4). This result supports that BSA binding to the affinity particles is insignificant. In contrast, compared to the OVA standard (lane 5), after treatment with the affinity particles the OVA band (46 kDa) became much weaker (lane 6), and a clear OVA band appeared in the eluted fraction (lane 7). Obviously, this was because the glycoprotein was removed by the affinity particles during incubation, and the bound glycoprotein could be released by acid elution. | ||
| Fig. 8 SDS-PAGE analysis of proteins. Lane 1, protein marker; lane 2, BSA standard; lane 3, unbound BSA; lane 4, BSA eluted from the particles; lane 5, OVA standard; lane 6, unbound OVA; lane 7, OVA eluted from the particles; lane 8, diluted egg white; lane 9, unbound egg white proteins; lane 10, eluted egg white proteins. | ||
When egg white was diluted and treated with the same affinity particles, similar glycoprotein binding results were observed. In the diluted egg white, there are three abundant protein bands corresponding to the glycoproteins ovotransferrin (OVT, 76.7 kDa) and OVA (46 kDa), and the non-glycoprotein lysozyme (Lyz, 14.4 kDa) (lane 8). After treatment with Si@poly(NIPAm-co-GMA)@APBA particles, the two bands at 76 kDa and 46 kDa became weaker, while the band corresponding to Lyz (14.4 kDa) remained unchanged (lane 9). In lane 10 for the acid-eluted proteins, only two bands corresponding to the two glycoproteins were observed. This result indicates again that the Si@poly(NIPAm-co-GMA)@APBA particles are efficient affinity adsorbents for separation of glycoproteins from complex samples.
Conclusions
In this work, we have synthesized a new type of thermo-sensitive and boronic acid-tagged core–shell nanohybrid, Si@poly(NIPAm-co-GMA)@APBA, via SI-ATRP in combination with CuAAC click reaction. The SI-ATRP allowed a facile control over the structure and homogeneity of the polymer brushes on the surface, and the click reaction enabled a higher efficiency of ligand immobilization. The intermediate polymer brushes, poly(NIPAm-co-GMA), were grafted from an amino-functionalized silica core via the SI-ATRP process followed by post-functionalization with propargylamine, and then conjugated to an azide-tagged fluorogenic boronic acid via click reaction under mild conditions. Due to a high density of boronic acid ligands appended on the long flexible polymer brushes, the as-prepared nanohybrid, Si@poly(NIPAm-co-GMA)@APBA, exhibited excellent specificity and affinity towards glycoproteins. The boronate affinity nanohybrid allowed facile capture/release of target glycoproteins in a pH-switchable fashion. In addition, the feasibility of application to real complex samples was demonstrated by selective enrichment of glycoproteins from egg white samples. Furthermore, a practical method to quantify surface-attached ATRP initiators on particles has been developed, where an alkyne-tagged FITC derivative was used as a probe to titrate the terminal azide groups on acid-treated Si@poly(NIPAm-co-GMA) through CuAAC click chemistry. The synthetic method developed in this work provides a versatile approach for preparing polymer brush-based affinity adsorbents not only for glycoproteins, but also for other bioactive molecules using specific interactions that can be provided by antibodies, aptamers and immobilized metal affinity chromatography (IMAC).Acknowledgements
This work was supported by the Swedish Research Council FORMAS (Grant No. 212-2013-1350) and the Danish Council for Strategic Research (project FENAMI, DSF-10-93456). L. D. Jiang thanks the China Scholarship Council (CSC) for a PhD fellowship.References
- T. Barroso, T. Casimiro, A. M. Ferraria, F. Mattioli, A. AguiarRicardo and A. C. Roque, Adv. Funct. Mater., 2014, 24, 4528–4541 CrossRef CAS.
- M. Safari, M. Ghiaci, M. Jafari-Asl and A. A. Ensafi, Appl. Surf. Sci., 2015, 342, 26–33 CrossRef CAS.
- L. Sun, T. Liu, H. Li, L. Yang, L. Meng, Q. Lu and J. Long, ACS Appl. Mater. Interfaces, 2015, 7, 4990–4997 CAS.
- S. Kango, S. Kalia, A. Celli, J. Njuguna, Y. Habibi and R. Kumar, Prog. Polym. Sci., 2013, 38, 1232–1261 CrossRef CAS.
- I. Cobo, M. Li, B. S. Sumerlin and S. Perrier, Nat. Mater., 2015, 14, 143–159 CrossRef CAS PubMed.
- I. A. Rahman and V. Padavettan, J. Nanomater., 2012, 2012, 1–15 CrossRef.
- J. D. Marth and P. K. Grewal, Nat. Rev. Immunol., 2008, 8, 874–887 CrossRef CAS PubMed.
- K. Ohtsubo and J. D. Marth, Cell, 2006, 126, 855–867 CrossRef CAS PubMed.
- H. Narimatsu, H. Sawaki, A. Kuno, H. Kaji, H. Ito and Y. Ikehara, FEBS J., 2010, 277, 95–105 CrossRef CAS PubMed.
- J. H. Lee, Y. Kim, M. Y. Ha, E. K. Lee and J. Choo, J. Am. Soc. Mass Spectrom., 2005, 16, 1456–1460 CrossRef CAS PubMed.
- H. Li, H. Wang, Y. Liu and Z. Liu, Chem. Commun., 2012, 48, 4115–4117 RSC.
- Z. A. Lin, J. L. Pang, Y. Lin, H. Huang, Z. W. Cai, L. Zhang and G. N. Chen, Analyst, 2011, 136, 3281–3288 RSC.
- F. Yang, J. Mao, X. He, L. Chen and Y. Zhang, Anal. Bioanal. Chem., 2013, 405, 5321–5331 CrossRef CAS PubMed.
- M. H. Levy, J. Plawsky and S. M. Cramer, J. Sep. Sci., 2013, 36, 2358–2365 CrossRef CAS PubMed.
- Z. Lin, J. Wang, X. Tan, L. Sun, R. Yu, H. Yang and G. Chen, J. Chromatogr. A, 2013, 1319, 141–147 CrossRef CAS PubMed.
- M. Zhao, Y. Xie, C. Deng and X. Zhang, J. Chromatogr. A, 2014, 1357, 182–193 CrossRef CAS PubMed.
- L. Zhang, Y. Xu, H. Yao, L. Xie, J. Yao, H. Lu and P. Yang, Chem. – Eur. J., 2009, 15, 10158–10166 CrossRef CAS PubMed.
- W. Zhou, N. Yao, G. Yao, C. Deng, X. Zhang and P. Yang, Chem. Commun., 2008, 5577–5579 RSC.
- X. Zhang, X. He, L. Chen and Y. Zhang, J. Mater. Chem. B, 2014, 2, 3254–3262 RSC.
- T. D. James, K. Sandanayake and S. Shinkai, Angew. Chem., Int. Ed. Engl., 1996, 35, 1910–1922 CrossRef.
- Z. Lin, L. Sun, W. Liu, Z. Xia, H. Yang and G. Chen, J. Mater. Chem. B, 2014, 2, 637–643 RSC.
- J. Ye, Y. Chen and Z. Liu, Angew. Chem., Int. Ed., 2014, 53, 10386–10389 CrossRef CAS PubMed.
- H. G. Schild, Prog. Polym. Sci., 1992, 17, 163–249 CrossRef CAS.
- F. Gao, X. Ma, X. He, W. Li and Y. Zhang, Colloids Surf., A, 2013, 433, 191–199 CrossRef CAS.
- A. E. Ivanov, I. Y. Galaev and B. Mattiasson, J. Mol. Recognit., 2006, 19, 322–331 CrossRef CAS PubMed.
- S. Edmondson and W. T. Huck, J. Mater. Chem., 2004, 14, 730–734 RSC.
- H. Roghani-Mamaqani, V. Haddadi-Asl and M. Salami-Kalajahi, Polym. Rev., 2012, 52, 142–188 CrossRef CAS.
- K. Matyjaszewski and N. V. Tsarevsky, Nat. Chem., 2009, 1, 276–288 CrossRef CAS PubMed.
- C. M. Hui, J. Pietrasik, M. Schmitt, C. Mahoney, J. Choi, M. R. Bockstaller and K. Matyjaszewski, Chem. Mater., 2013, 26, 745–762 CrossRef.
- Y. Wang, J. Chen, J. Xiang, H. Li, Y. Shen, X. Gao and Y. Liang, React. Funct. Polym., 2009, 69, 393–399 CrossRef CAS.
- T. Wu, Y. Zhang, X. Wang and S. Liu, Chem. Mater., 2008, 20, 101–109 CrossRef CAS.
- K. Nagase, J. Kobayashi, A. Kikuchi, Y. Akiyama, H. Kanazawa and T. Okano, Langmuir, 2007, 23, 9409–9415 CrossRef CAS PubMed.
- K. Nagase, M. Kumazaki, H. Kanazawa, J. Kobayashi, A. Kikuci, Y. Akiyama, M. Annaka and T. Okano, ACS Appl. Mater. Interfaces, 2010, 2, 1247–1253 CAS.
- E. Turan, S. Demirci and T. Caykara, Thin Solid Films, 2010, 518, 5950–5954 CrossRef CAS.
- N. Idota, K. Nagase, K. Tanaka, T. Okano and M. Annaka, Langmuir, 2010, 26, 17781–17784 CrossRef CAS PubMed.
- A. Mizutani, K. Nagase, A. Kikuchi, H. Kanazawa, Y. Akiyama, J. Kobayashi, M. Annaka and T. Okano, J. Chromatogr. A, 2010, 1217, 522–529 CrossRef CAS PubMed.
- H. Bao, L. Li, L. H. Gan, Y. Ping, J. Li and P. Ravi, Macromolecules, 2010, 43, 5679–5687 CrossRef CAS.
- Z. Xu, K. M. A. Uddin, T. Kamra, J. Schnadt and L. Ye, ACS Appl. Mater. Interfaces, 2014, 6, 1406–1414 CAS.
- Z. Xu, K. M. A. Uddin and L. Ye, Macromolecules, 2012, 45, 6464–6470 CrossRef CAS.
- K. M. A. Uddin and L. Ye, J. Appl. Polym. Sci., 2013, 128, 1527–1533 Search PubMed.
- W. Stöber, A. Fink and E. Bohn, J. Colloid Interface Sci., 1968, 26, 62–69 CrossRef.
- C. Xu and L. Ye, Chem. Commun., 2011, 47, 6096–6098 RSC.
- G. Springsteen and B. Wang, Chem. Commun., 2001, 1608–1609 RSC.
- D. Li, Y. Chen and Z. Liu, Chem. Soc. Rev., 2015, 44, 8097–8123 RSC.
- F. Gao, X. Zhao, X. He, W. Li and Y. Zhang, Anal. Methods, 2013, 5, 6700–6708 RSC.
- M. Pan, Y. Sun, J. Zheng and W. Yang, ACS Appl. Mater. Interfaces, 2013, 5, 8351–8358 CAS.
- D. Roy, J. N. Cambre and B. S. Sumerlin, Prog. Polym. Sci., 2010, 35, 278–301 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6tb00241b |
| This journal is © The Royal Society of Chemistry 2016 |