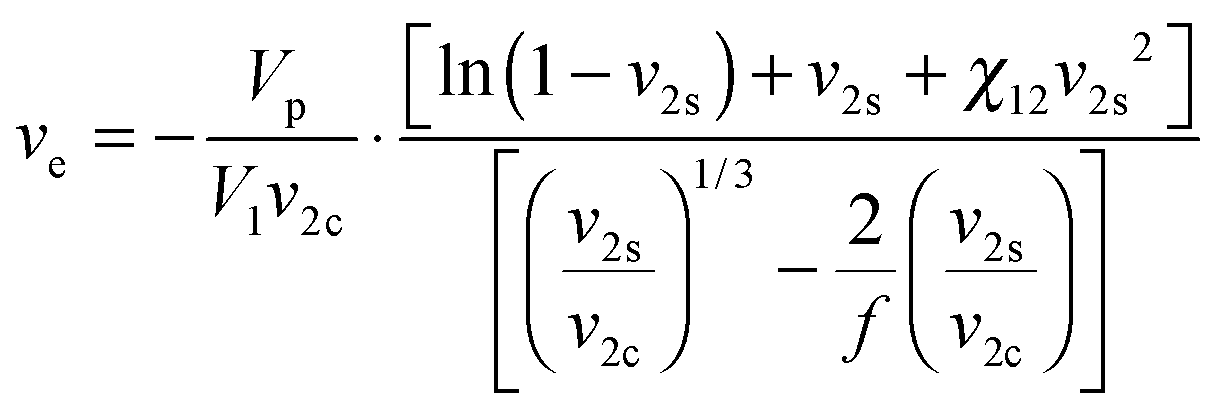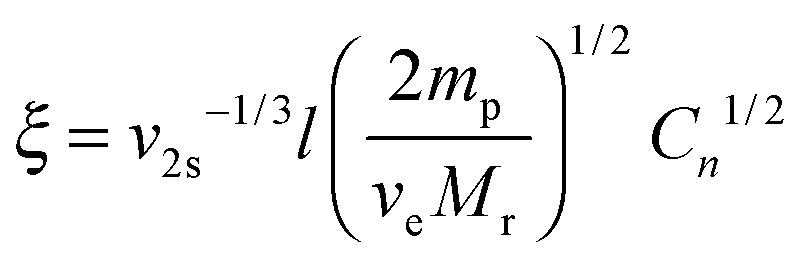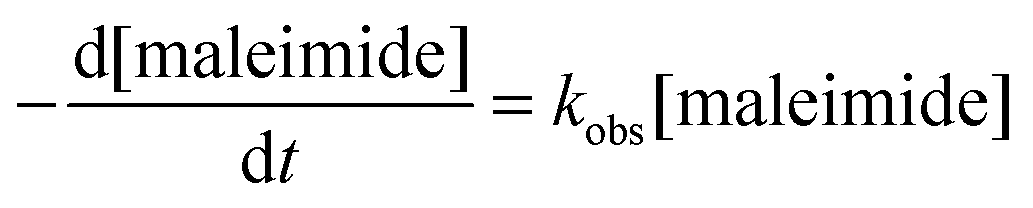Design of hydrogels for delayed antibody release utilizing hydrophobic association and Diels–Alder chemistry in tandem†
Received
25th January 2016
, Accepted 22nd April 2016
First published on 25th April 2016
Abstract
Biodegradable hydrogels were prepared from furan- and maleimide-functionalized eight-armed poly(ethylene glycol) with an average molecular mass of 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da (8armPEG40k-furan and 8armPEG40k-maleimide) using the Diels–Alder (DA) reaction as a cross-linking mechanism. Hydrophobic 6-aminohexanoic acid (C6) and 12-aminododecanoic acid (C12) spacers were introduced between the polymer backbone and the functional end-groups; the influence on the gel properties was studied. Modification with C6 and C12 spacers induced hydrophobic interactions between the macromonomers leading to association and increased viscosity of the polymer solutions; both effects were influenced by the spacer length. In combination with DA cross-linking, hydrophobic derivatives of 8armPEG40k-furan and 8armPEG40k-maleimide led to hydrogels with improved properties. Upon introduction of C12 spacers, gelation of 8armPEG40k hydrogels occurred twice as fast. Interestingly, no effect was observed when only one of the two components had been modified. Our experiments suggest that the association of macromonomers by hydrophobic interactions facilitates chemical cross-linking via DA chemistry. This hypothesis is supported by calculations of the network mesh size and the Young's modulus of compression, which showed an increased cross-linking density of hydrophobically modified hydrogels. As a consequence of the increased cross-linking density, the degradation stability of C12-modified hydrogels increased by a factor of 4. Moreover, hydrophobic modification improved the hydrolytic resistance of maleimides; this also contributes to gel stability. The in vitro release of bevacizumab, which served as a model antibody, could be delayed for almost 60 days using modification with C12. Similar trends were observed for C6-modified 8armPEG40k hydrogels; however, the effects were considerably weaker. In summary, utilizing hydrophobic association and chemical cross-linking in tandem is a promising approach to create biodegradable hydrogels for delayed antibody release.
000 Da (8armPEG40k-furan and 8armPEG40k-maleimide) using the Diels–Alder (DA) reaction as a cross-linking mechanism. Hydrophobic 6-aminohexanoic acid (C6) and 12-aminododecanoic acid (C12) spacers were introduced between the polymer backbone and the functional end-groups; the influence on the gel properties was studied. Modification with C6 and C12 spacers induced hydrophobic interactions between the macromonomers leading to association and increased viscosity of the polymer solutions; both effects were influenced by the spacer length. In combination with DA cross-linking, hydrophobic derivatives of 8armPEG40k-furan and 8armPEG40k-maleimide led to hydrogels with improved properties. Upon introduction of C12 spacers, gelation of 8armPEG40k hydrogels occurred twice as fast. Interestingly, no effect was observed when only one of the two components had been modified. Our experiments suggest that the association of macromonomers by hydrophobic interactions facilitates chemical cross-linking via DA chemistry. This hypothesis is supported by calculations of the network mesh size and the Young's modulus of compression, which showed an increased cross-linking density of hydrophobically modified hydrogels. As a consequence of the increased cross-linking density, the degradation stability of C12-modified hydrogels increased by a factor of 4. Moreover, hydrophobic modification improved the hydrolytic resistance of maleimides; this also contributes to gel stability. The in vitro release of bevacizumab, which served as a model antibody, could be delayed for almost 60 days using modification with C12. Similar trends were observed for C6-modified 8armPEG40k hydrogels; however, the effects were considerably weaker. In summary, utilizing hydrophobic association and chemical cross-linking in tandem is a promising approach to create biodegradable hydrogels for delayed antibody release.
Introduction
Since the introduction of recombinant insulin1 more than three decades ago, the steady progress in pharmaceutical biotechnology has resulted in the launch of numerous biologics. In 2014 alone, 12 new biologics, such as recombinant proteins and monoclonal antibodies, were approved by the US Food and Drug Administration.2 Among these, monoclonal antibodies are exceptionally promising therapeutics as they provide great specificity and affinity toward their clinical targets.3–5 Indeed, therapeutic antibodies have significantly advanced the treatment of severe diseases, such as cancer,6 rheumatoid arthritis,7 or age-related macular degeneration.8 However, despite their therapeutic value, antibodies have two major shortcomings that limit potential applications.9 Firstly, antibodies are fragile molecules and, therefore, prone to aggregation10 and degradation.11 Secondly, most antibodies have to be administered by injection or infusion, as their oral bioavailability is generally low.12 To improve the compliance and reduce the administration frequency, innovative drug delivery systems that provide sustained, delayed or intermittent antibody release are needed.
Among these drug delivery systems, hydrogels are particularly suitable for controlled antibody release.13 Firstly, hydrogels can act as a drug depot that protects the incorporated protein from degradation. Secondly, the release rate can be tailored to the specific therapeutic needs; for example, hydrogels can prolong the dosing intervals by providing sustained or delayed antibody release. Hydrogels can be prepared from a wide range of natural and synthetic polymers via physical or chemical cross-linking.14,15 Physical cross-linking (e.g., via ionic or hydrophobic interactions) enables the preparation of in situ forming hydrogels that respond to external stimuli, such as ionic strength or temperature.16–19 These systems can be administered in liquid form and rapidly turn into a solid gel after injection. However, the resulting hydrogels are often considered not stable enough for sustained drug release.20–22 Chemically cross-linked hydrogels, on the other hand, provide superior mechanical properties and stability against dissolution. However, only few chemical reactions are suitable for in situ cross-linking;23 furthermore, controlling the cross-linking rate can be challenging. Gelation must be fast enough to completely encapsulate the drug after injection; on the other hand, rapidly gelling formulations can clog the needle and prevent the application of the gel. As a consequence, the combination of physical interactions and chemical cross-linking may open up new possibilities to design rapidly forming hydrogels with improved stability.
Recently, the Diels–Alder (DA) reaction has been introduced as a promising cross-linking mechanism for the preparation of degradable, poly(ethylene glycol) (PEG) based hydrogels.24–26 Since the DA reaction proceeds at body temperature in the absence of potentially toxic catalysts, it is in principle suitable for in situ cross-linking. However, the slow cross-linking rate can prove problematic and prevent the formation of a drug depot after injection.27 In conventional DA hydrogels, star-shaped PEG is end-functionalized with furyl and maleimide groups; gelation occurs by step-growth polymerization of complementary macromonomers. In this study, hydrophobic alkyl spacers were introduced between the PEG chains of eight-armed PEG and the reactive end-groups. In this way, amphiphilic macromonomers were created. The hydrophilic PEG core ensures water solubility, while the hydrophobic end-groups are expected to stick together and facilitate cross-linking via DA reaction. In the first part of this study, the association of amphiphilic macromonomers in aqueous solution was investigated by means of fluorescence spectroscopy and viscosity measurements. In the second part, hydrogels were prepared and the impact of physical interactions on gel formation, average network mesh size, and mechanical properties was studied. In the last part, the hydrogels were characterized regarding their swelling and degradation behavior. Finally, the gels were loaded with bevacizumab and the in vitro release was determined.
Materials and methods
Materials
Anhydrous 1,4-dioxane, dichloromethane, sodium bicarbonate (NaHCO3), sodium sulfate (Na2SO4), and triphenylphosphine were purchased from Acros Organics (Geel, Belgium). Boc-6-aminohexanoic acid was purchased from Bachem (Weil am Rhein, Germany). Diethyl ether was obtained from CSC Jäcklechemie (Nuremberg, Germany). Eight-armed poly(ethylene glycol) with a molecular mass of 40000 Da (8armPEG40k-OH) was purchased from JenKem Technology (Allen, TX, USA). Ceteareth-20 was obtained from Kolb Distribution Ltd (Hedingen, Switzerland). Acetyl chloride, 12-aminododecanoic acid, anhydrous dimethylformamide, anhydrous tetrahydrofuran, deuterated chloroform (CDCl3), N,N′-dicyclohexylcarbodiimide (DCC), diisopropyl azodicarboxylate, N,N-diisopropylethylamine, di-tert-butyl dicarbonate, fluorescamine, 3-(2-furyl)propanoic acid, N-hydroxysuccinimide (NHS), N-methoxycarbonylmaleimide, piperidine, and pyrene were received from Sigma-Aldrich (Taufkirchen, Germany). All other chemicals were obtained from Merck KGaA (Darmstadt, Germany). Bevacizumab (Avastin®, 25 mg mL−1, Roche Ltd, Basel, Switzerland) was kindly provided by the hospital pharmacy of the University of Regensburg (Germany). Purified water was freshly prepared using a Milli-Q water purification system (Millipore, Schwalbach, Germany).
1H-NMR spectroscopy
1H-NMR spectra were recorded in CDCl3 at 295 K using a Bruker Avance 300 spectrometer (Bruker BioSpin GmbH, Rheinstetten, Germany).
Synthesis of Boc-12-aminododecanoid acid
Boc-12-aminododecanoic acid was synthesized as described by Dorn et al.28 with the following modifications. 12-Aminododecanoic acid (9.3 mmol, 2.0 g) was suspended in 30 mL of absolute methanol containing triethylamine (11.2 mmol, 1.5 mL). Di-tert-butyl dicarbonate (9.3 mmol, 2.0 g) was dissolved in 10 mL of absolute methanol and added to the suspension. The mixture was heated to 60 °C and refluxed under stirring for 12 hours, leading to a clear solution. The solvent was removed under reduced pressure and the residue was dissolved in ethyl acetate. The solution was extracted four times with 0.25 M HCl. The organic phase was dried over anhydrous Na2SO4 and filtered. The solvent was evaporated to give a white solid (94% yield). The complete Boc-protection of the primary amino groups was confirmed using a fluorescamine assay.29
Synthesis of unmodified macromonomers (Scheme 1)
Eight-armed PEG-amine with a molecular mass of 40000 Da (8armPEG40k-NH2) was synthesized as previously described.30 The resulting PEG-amine was then functionalized with furyl (8armPEG40k-furan) or maleimide groups (8armPEG40k-maleimide) as described for 8armPEG20k-NH2.24 However, higher reagent concentrations were required due to the lower concentrations of amino groups. For the synthesis of 8armPEG40k-furan, 16 equivalents of 3-(2-furyl)propanoic acid, NHS, DCC, and NaHCO3 were used; for the preparation of 8armPEG40k-maleimide, 40 equivalents of N-methoxycarbonylmaleimide were used. The degree of end-group functionalization was 75–80% for 8armPEG40k-furan and 65–70% for 8armPEG40k-maleimide, as determined by 1H-NMR. The degree of functionalization was reproducible and sufficient to enable cross-linking.
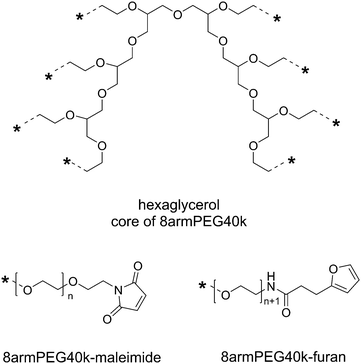 |
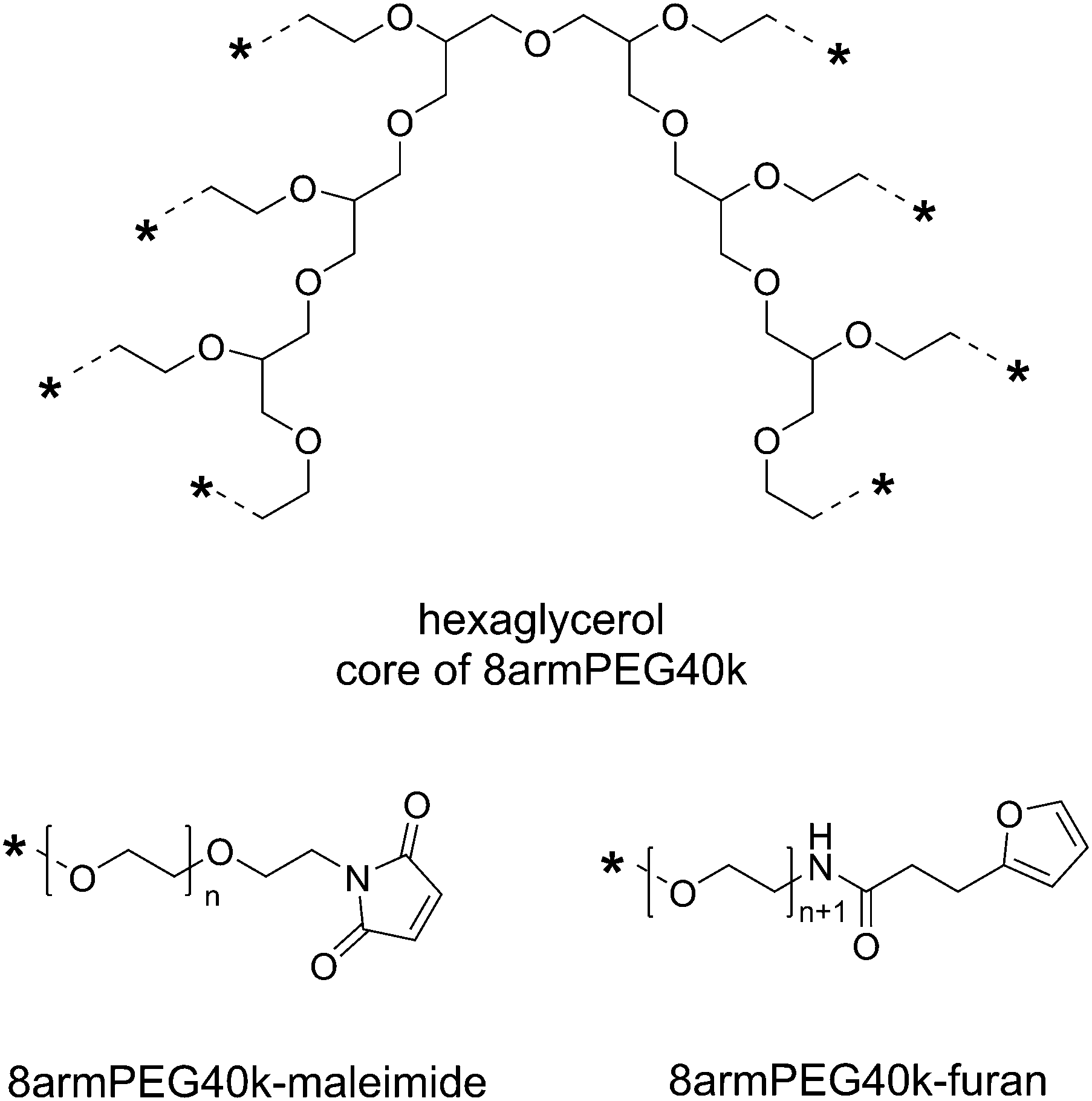
| | Scheme 1 Functional end-groups of 8armPEG40k-maleimide and 8armPEG40k-furan. Both macromonomers were synthesized from 8armPEG40k-OH (molecular mass 40000 Da, hexaglycerol core) as previously described. | |
Synthesis of hydrophobically modified macromonomers (Scheme 2)
8armPEG40k-NH2 was first reacted with Boc-6-aminohexanoic acid as previously described for 8armPEG20k-NH2.26 The complete functionalization with Boc-6-aminohexanoic acid was confirmed by a fluorescamine assay. Following this procedure, the obtained 8armPEG40k-C6-NH-Boc was deprotected (8armPEG40k-C6-NH2) and subsequently functionalized with furyl (8armPEG40k-C6-furan) or maleimide groups (8armPEG40k-C6-maleimide) as described above. 8armPEG40k-C12-NH-Boc, 8armPEG40k-C12-furan and 8armPEG40k-C12-maleimide were synthesized in the same way using Boc-12-aminododecanoic acid instead of Boc-6-aminohexanoic acid. The degree of end-group functionalization was 65–70% for 8armPEG40k-C6-furan and 8armPEG40k-C12-furan, and 60–65% for 8armPEG40k-C6-maleimide and 8armPEG40k-C12-maleimide, as determined by 1H-NMR. The degree of functionalization was reproducible and sufficient to enable cross-linking.
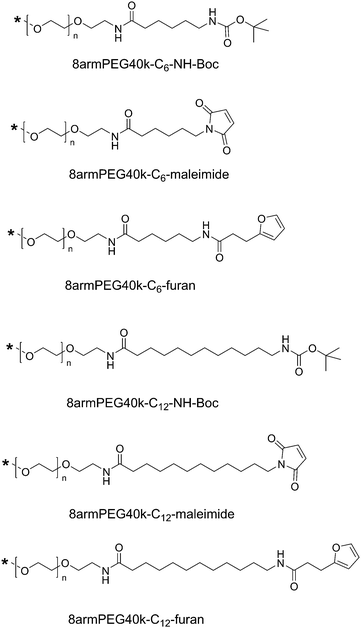 |
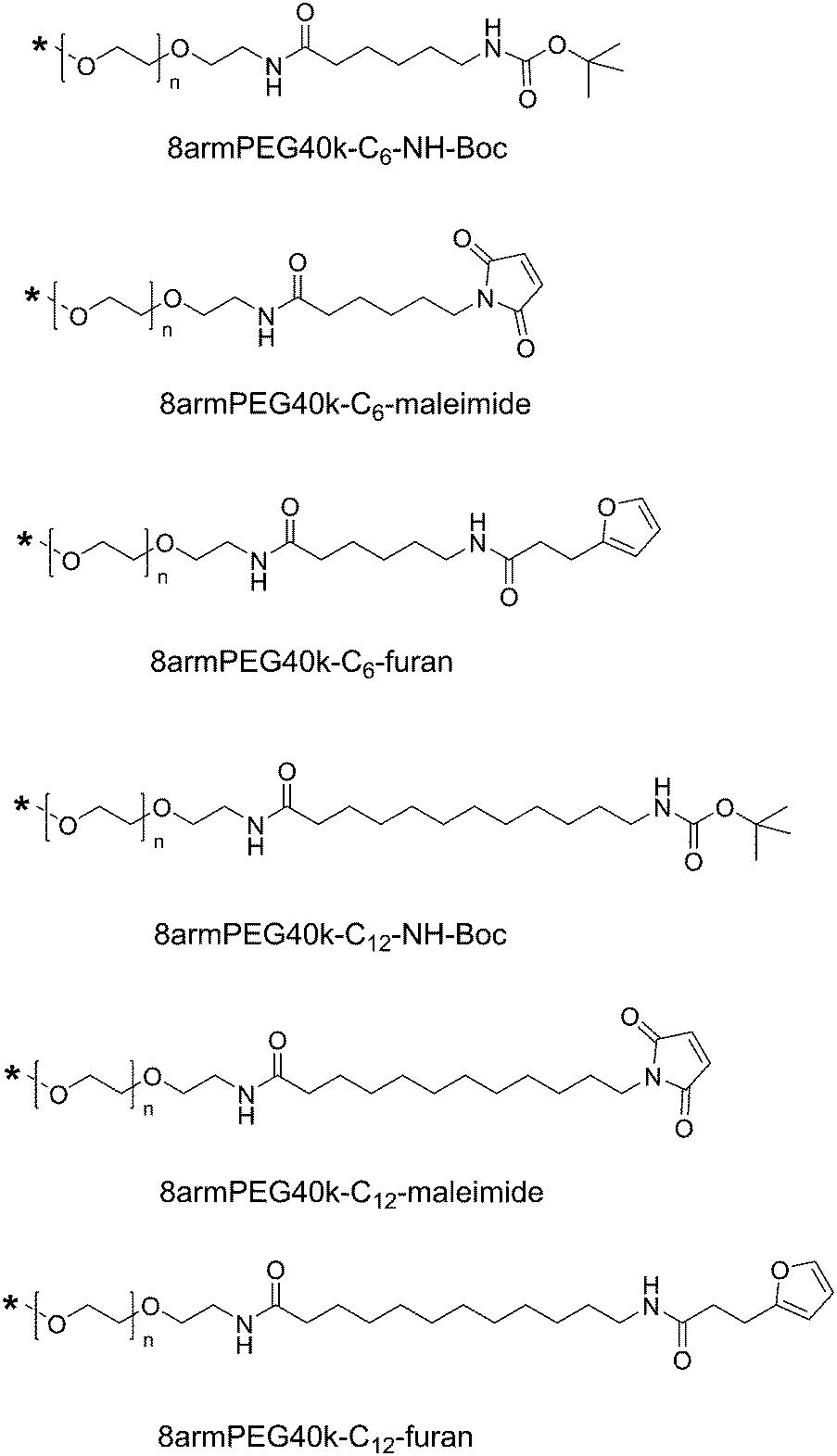
| | Scheme 2 End-groups of hydrophobically modified macromonomers. Hydrophobic 6-aminohexanoic acid (C6) or 12-aminododecanoic acid (C12) spacers were introduced between the PEG backbone and the functional end-groups. | |
Determination of the critical micelle concentration
To check for the presence of micellar structures, emission spectra of pyrene in pure water, a micellar solution of ceteareth-20, and a 20% (w/V) solution of 8armPEG40k-C12-NH-Boc were recorded at an excitation wavelength of 339 nm, using a Perkin Elmer LS-55 fluorescence plate reader (Perkin Elmer, Wiesbaden, Germany). The emission spectra were normalized to the peak at 383 nm (I3); the results of five independent measurements were averaged. To determine the critical micelle concentration (CMC), aqueous solutions of 8armPEG40k-OH, 8armPEG40k-C6-NH-Boc, and 8armPEG40k-C12-NH-Boc were prepared; concentrations of 0.1%, 0.25%, 0.5%, 1.0%, 2.5%, 5.0%, 7.5%, 10%, 15%, and 20% were used. First, 30 mg of pyrene was dissolved in 750 μL of acetone. Equivalents of this solution corresponding to 1.0 mg of pyrene were transferred to amber glass vials and the acetone was allowed to evaporate. Then, 200 μL of the polymer solutions were added and the samples were incubated on an orbital shaker for 24 hours. Following this, 100 μL of each sample were transferred into a 96-well plate (Greiner Bio-One, Frickenhausen, Germany) and the fluorescence intensity of pyrene was determined at an excitation wavelength of 339 nm and an emission wavelength of 390 nm. A linear regression was performed for both the non-ascending and the ascending part of the graph; the CMC was determined from the intersection of the two lines.
Viscosity measurements
Aqueous solutions of 8armPEG40k-OH, 8armPEG40k-C6-NH-Boc, and 8armPEG40k-C12-NH-Boc were prepared; the polymer concentrations were 2% (w/V) and 20% (w/V). Viscosity measurements were performed on a TA Instruments Discovery HR-2 rheometer (TA Instruments, Eschborn, Germany) at a constant temperature of 25 °C. The instrument was equipped with a 20 mm parallel steel plate; the sample volume was 350 μL. The shear rate was increased from 0.1 to 10.0 s−1. The TA Instruments Trios software was used to calculate the viscosity from the relation between shear rate and stress assuming Newtonian flow behavior.
Hydrogel preparation and characterization
For hydrogel preparation, equal molar amounts of 8armPEG40k-furan and 8armPEG40k-maleimide, 8armPEG40k-C6-furan and 8armPEG40k-C6-maleimide, or 8armPEG40k-C12-furan and 8armPEG40k-C12-maleimide were separately dissolved in water. Gel formation was initiated by mixing complementary macromonomer solutions; the overall polymer concentrations were 5% (w/V), 10% (w/V) and 15% (w/V). Rheological characterization was performed using a TA Instruments AR 2000 rheometer (TA Instruments, Eschborn, Germany) as previously described.24 Storage (G′) and loss modulus (G′′) were recorded over time; the crossover of G′ and G′′ was regarded as the gel point. The average mesh size (ξ) was determined as previously described.26
Young's modulus of compression
To characterize the mechanical properties of hydrogels, the Young's modulus of compression (E) was determined. For this purpose, gel cylinders with a volume of 250 μL were prepared as described above. The gel samples were cross-linked for 6 hours at 37 °C. The polymer concentrations were 5% (w/V), 10% (w/V) and 15% (w/V). Diameter and height of the gel cylinders were determined using a caliper. The samples were then subjected to a compression test on an Instron 5542 materials testing machine (Norwood, MA, USA). For this purpose, the gel cylinders were placed upright on the lower steel plate of the instrument. The upper steel plate was set to compress the samples at a velocity of 1 mm min−1. The compressive force (N) and the compressive strain (%) were recorded over time. The Young's modulus of compression was determined from the linear part of the curve between 10% and 50% compression using the Instron Bluehill Software (Norwood, MA, USA). Data is presented as mean ± standard deviation, based on the results of three independent samples.
Swelling, degradation and antibody release
Swelling and degradation of 8armPEG40k, 8armPEG40k-C6 and 8armPEG40k-C12 hydrogels was studied in phosphate buffer, pH 7.4 at 37 °C as previously described.26 The polymer concentrations were 5% (w/V), 10% (w/V) and 15% (w/V). For the release experiments, 8armPEG40k, 8armPEG40k-C6 and 8armPEG40k-C12 hydrogels were loaded with bevacizumab (Avastin®); cross-linking was carried out in 5 mM acetate buffer, pH 5.0. The polymer concentration was 10% (w/V); the bevacizumab loading was 5 mg mL−1. The in vitro release of bevacizumab was determined in phosphate buffer, pH 7.4 at 37 °C as previously described.26
Hydrolytic stability of maleimides
In order to investigate the hydrolytic stability of maleimides, 20.0 mg of 8armPEG40k-maleimide, 8armPEG40k-C6-maleimide or 8armPEG40k-C12-maleimide were dissolved in 2.0 mL of 50 mM phosphate buffer, pH 7.4. The polymer solutions were transferred into 10 mm quartz cuvettes. The absorbance at 299 nm was recorded for 900 min using a Kotron UVIKON® 941 spectrophotometer (Kotron Instruments S.p.A., Milan, Italy). The cuvette holder was equipped with a heating jacket; the temperature was set to 37 °C.
Statistical analysis
All results are presented as mean ± standard deviation, based on the data obtained from at least n = 3 samples. Statistical significance was determined by means of one-way ANOVA, followed by Tukey's post hoc test using GraphPad Prism 6.0 (GraphPad Software Inc., La Jolla, CA, USA). Differences were considered statistically significant at p < 0.05.
Results and discussion
Association of macromonomers in aqueous solution
The major goal of this study was to prepare in situ forming hydrogels for drug delivery by combining physical interactions with DA chemistry. In order to induce hydrophobic interactions between the macromonomers, hydrophilic 8armPEG40k-NH2 was first reacted with Boc-6-aminohexanoic acid (C6) or Boc-12-aminododecanoic acid (C12). After deprotection, the hydrophobically modified PEG was functionalized with furyl and maleimide groups, respectively. While it has already been shown in our previous publication that introducing hydrophobic C6 spacers gives hydrogels with increased stability to degradation,26 the present study aimed at providing a deeper mechanistic understanding of the molecular processes responsible for the improved gel properties. Furthermore, we wanted to investigate whether the gel properties, such as gel time and stability to degradation, depend on the spacer length. In the first experiment, the hydrophobic interactions between the synthesized macromonomers were investigated. To this end, the association of 8armPEG40k-C6-NH-Boc or 8armPEG40k-C12-NH-Boc in water was studied using pyrene as a probe molecule. The Boc-protected polymers were chosen since they were uncharged. Furthermore, their association is solely due to hydrophobic interactions; covalent cross-linking via DA reaction cannot take place.
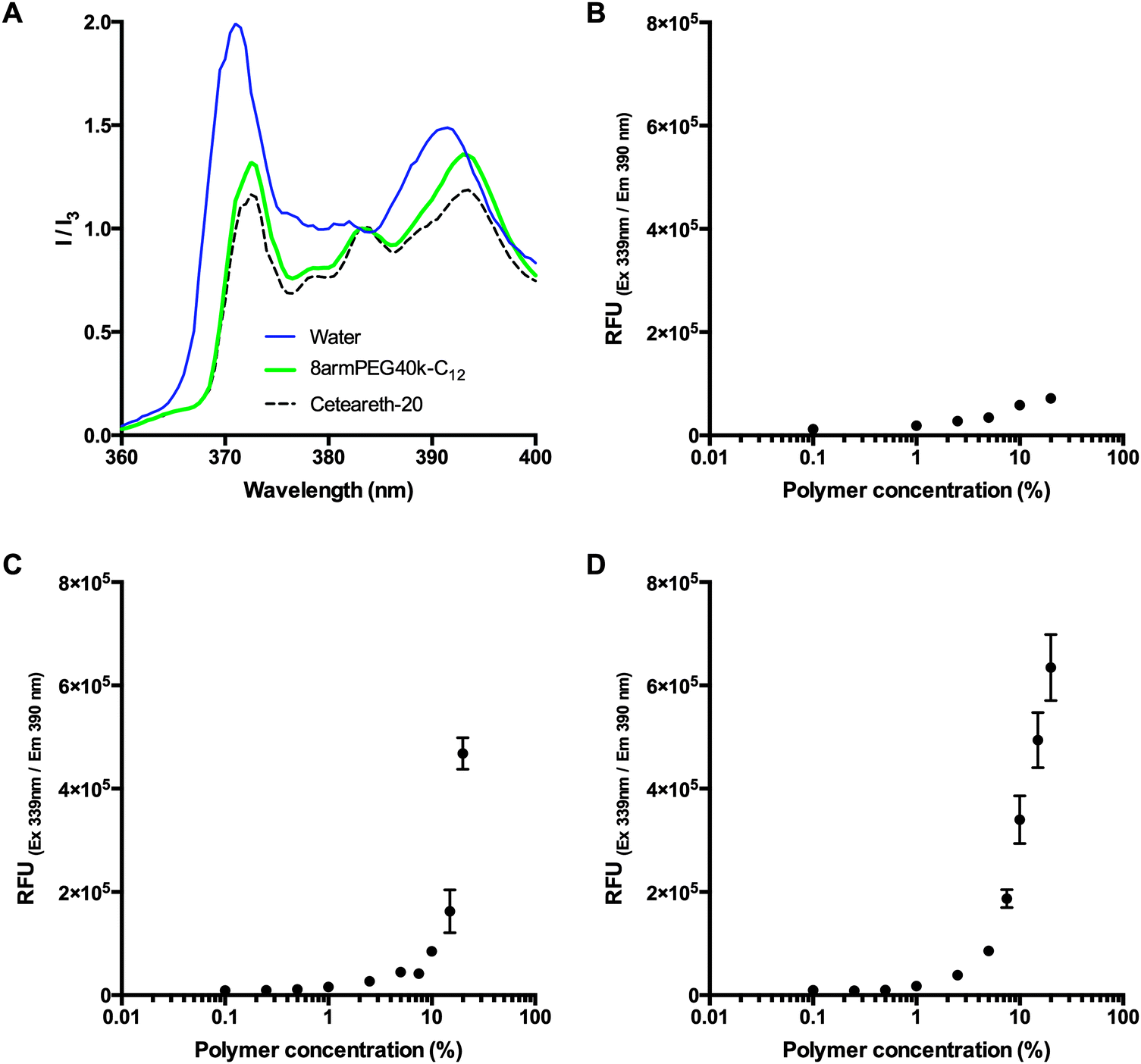
To check for the existence of polymeric micelles, the emission spectra of pyrene in pure water (negative control), a micellar solution of the detergent ceteareth-20 (positive control), and a 20% (w/V) solution of 8armPEG40k-C12-NH-Boc were compared. Pyrene is commonly used to probe the formation of micelles as its fluorescence intensity is greatly influenced by the hydrophobicity of the environment. The emission spectra were normalized to the peak at 383 nm (I3). Fig. 1A shows that, in comparison to water, the relative intensity at 372 nm (I1) strongly decreased for both ceteareth-20 and 8armPEG40k-C12-NH-Boc. A decrease in I1 has been reported as an indicator for micelle formation.31–33 In addition, the ratio between I1 and I3 was determined. In hydrophobic environments, the ratio is relatively low (e.g., 0.53 in hexane), whereas in hydrophilic environments, the ratio is significantly higher (e.g., 1.88 in water). In ceteareth-20 and 8armPEG40k-C12-NH-Boc, values of 1.12 and 1.26 have been determined. I1/I3 values of about 1.2 have been reported to be typical for associative polymers and surfactant micelles in aqueous solutions.34 This suggests that pyrene is located within hydrophobic domains of ceteareth-20 and 8armPEG40k-C12-NH-Boc, respectively. To follow the formation of micelles, pyrene fluorescence was measured in aqueous solutions of 8armPEG40k-OH, 8armPEG40k-C6-NH-Boc and 8armPEG40k-C12-NH-Boc at concentrations ranging from 0.1% (w/V) to 20% (w/V). As expected, in solutions of 8armPEG40k-OH, no micelle formation was detected (Fig. 1B). However, in solutions of 8armPEG40k-C6-NH-Boc and 8armPEG40k-C12-NH-Boc, a sharp increase in the fluorescence intensity was detected above a certain concentration (Fig. 1C and D). This indicates the beginning of polymer association and the formation of micelles. The critical micelle concentrations (CMCs) were determined as 10.4% (2.3 μM) and 3.3% (0.73 μM) for 8armPEG40k-C6-NH-Boc and 8armPEG40k-C12-NH-Boc, respectively. In comparison, for poloxamer 407, an amphiphilic poly(ethylene oxide)-b-poly(propylene oxide)-b-poly(ethylene oxide) copolymer, CMC values of 0.26–0.8% (w/V) have been determined;35 for linear PEGs end-capped with n-hexadecyl groups, CMC values of 2–5 μM have been reported.36 These values compare with the results reported in the present study.
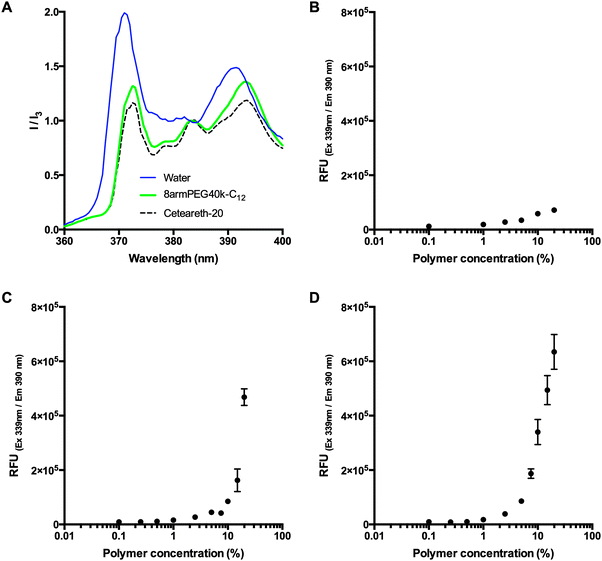 |
| | Fig. 1 Emission spectra of pyrene in pure water (negative control), a micellar solution of ceteareth-20 (positive control), and a 20% (w/V) solution of 8armPEG40k-C12-NH-Boc (A). The spectra are normalized to the fluorescence intensity at 383 nm (I3); a decrease of the peak at 372 nm (I1) is an indicator for micelle formation. Pyrene fluorescence in aqueous solutions of 8armPEG40k-OH (B), 8armPEG40k-C6-NH-Boc (C) and 8armPEG40k-C12-NH-Boc (D) at concentrations ranging from 0.1% (w/V) to 20% (w/V). A sharp increase in the fluorescence intensity (C and D) indicates the beginning of polymer association. | |
Fig. 1 clearly shows that modification of 8armPEG40k-NH2 with hydrophobic residues leads to hydrophobic interactions between the macromonomers. Moreover, the interactions become stronger as the length of the hydrophobic alkyl chain increases, as indicated by the lower CMC. It, therefore, seems reasonable to employ even longer alkyl spacers, such as 18-aminooctadecanoic acid, to amplify the effect. However, it should be kept in mind that increasing the spacer length can affect the water solubility of the polymer. In fact, another hydrophobically modified PEG was synthesized by reacting 8armPEG40k-C12-NH2 with further molecules of 12-aminododecanoic acid. However, the obtained macromonomer (8armPEG40k-C12-C12-NH2) was almost insoluble in water (data not shown). As water solubility of the macromonomers is a prerequisite for the formation of hydrogels, this derivative was not included in the present study.
Viscosity of macromonomer solutions
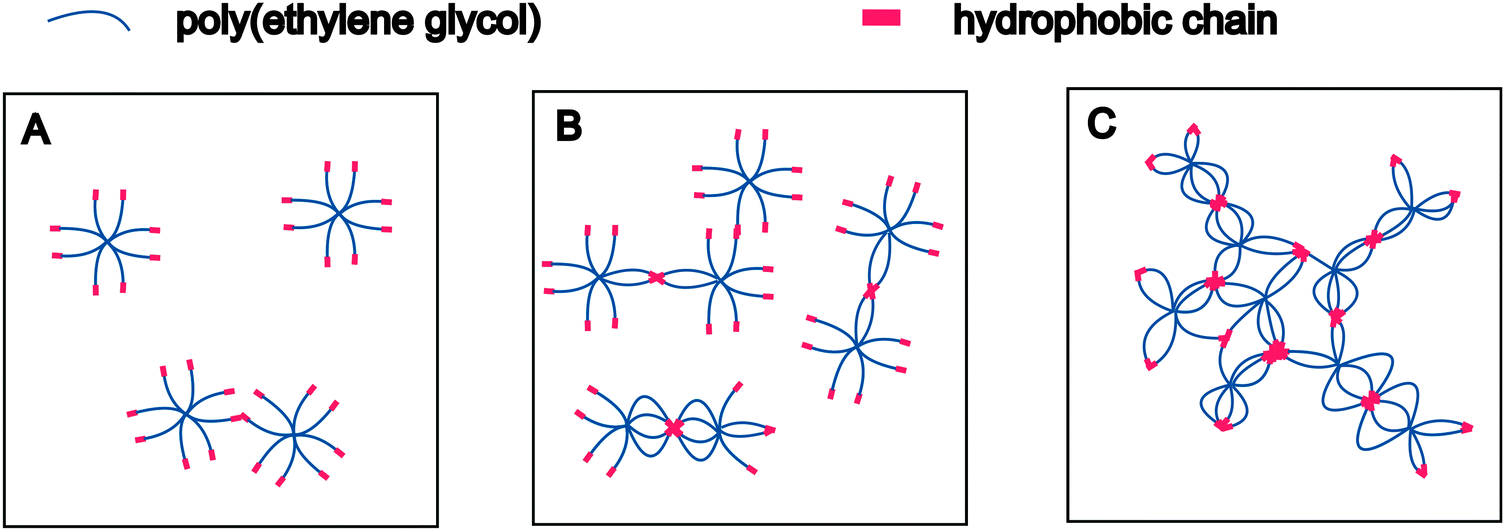
The presence of polymeric micelles above a certain macromonomer concentration is an interesting finding. To understand possible effects on hydrogel formation, the mechanism of polymer association should be discussed in more detail. It is well established that water-soluble polymers with hydrophobic end-modification form flower- or star-like micelles,37,38i.e., associates with a dense hydrophobic core surrounded by a corona of hydrophilic polymer chains.39 In addition, amphiphilic polymers are known to increase the viscosity of aqueous solutions; they are, therefore, referred to as associative thickeners.40,41 This behavior can be explained by the transient network theory.42 Water-soluble polymers with “sticky” hydrophobic end-groups associate in water to form physically stabilized networks in which the cross-links are weak enough to constantly break and re-combine. Winnik and Yekta reviewed the available literature and discussed how the formation of micelles and transient networks can explain the behavior of associative polymers in aqueous solution.43 Based on this combined theory, the association of 8armPEG40k-C6 and 8armPEG40k-C12 macromonomers solution can be described as follows. At very low concentrations, isolated macromonomers exist that only engage in transient random interactions (Fig. 2A). Therefore, solutions of 8armPEG40k-C6-NH-Boc or 8armPEG40k-C12-NH-Boc should exhibit nearly the same viscosity as solutions of polymers without hydrophobic end-modification. However, with increasing concentration, interactions between individual macromonomers become more likely and the hydrophobic end-groups start to associate (Fig. 2B). With further increasing concentration, more and more micelles are formed. These micelles are ultimately bridged by polymer chains resulting in the formation of transient networks that consequently increase the viscosity of the solution (Fig. 2C). Solutions of 8armPEG40k-C6-NH-Boc or 8armPEG40k-C12-NH-Boc should, therefore, exhibit much higher viscosities than equally concentrated solutions of polymers without hydrophobic end-modification.
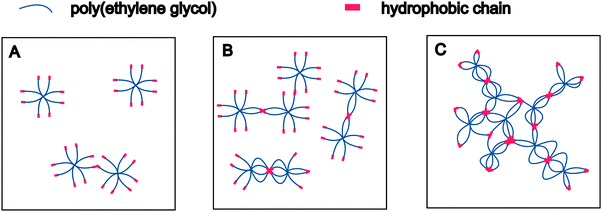 |
| | Fig. 2 Association of hydrophobically modified eight-armed PEG in aqueous solution at increasing concentrations. Isolated macromonomers engage in random interactions at low macromonomer concentrations (A). With increasing concentration, interactions become more likely and the hydrophobic end-groups start to associate (B). Formation of bridged micelles and transient networks at high macromonomer concentrations (C). | |
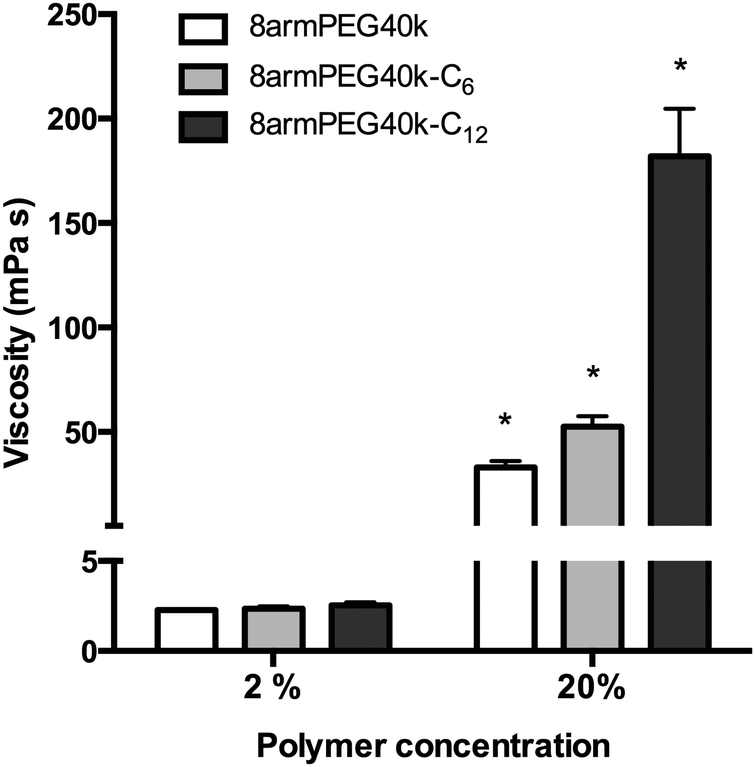
To test this hypothesis, aqueous solutions of 8armPEG40k-OH, 8armPEG40k-C6-NH-Boc and 8armPEG40k-C12-NH-Boc were prepared; their viscosities were measured at room temperature (Fig. 3). At polymer concentrations of 2% (w/V), viscosities of about 2 mPa s were determined. There were no significant differences between the polymer types, suggesting no additional physical interactions between macromonomers with hydrophobic end-groups. After increasing the polymer concentrations to 20% (w/V), i.e., above the CMC of 8armPEG40k-C6-NH-Boc and 8armPEG40k-C12-NH-Boc, the viscosities of all polymer solutions had increased accordingly. However, compared with 8armPEG40k-OH, solutions of 8armPEG40k-C6-NH-Boc and 8armPEG40k-C12-NH-Boc showed significantly higher viscosities. For example, the viscosity of a 20% (w/V) solution of 8armPEG40k-C6-NH-Boc was 1.6-times higher than the viscosity of an equally concentrated solution of 8armPEG40k-OH. This finding clearly demonstrates the existence of additional physical interactions between 8armPEG40k-C6 macromonomers. The effect was even more pronounced when 8armPEG40k-C12-NH-Boc was used; the viscosity was 5.5-times higher than for 8armPEG40k-OH. The differences between 8armPEG40k-C6-NH-Boc and 8armPEG40k-C12-NH-Boc were expected based on the determined CMC values (10.4% vs. 3.3%). Macromonomers end-capped with long alkyl chains (e.g., 12-aminododecanoic acid) show a higher tendency for hydrophobic interaction than those end-capped with short chains (e.g., 6-aminohexanoic acid). In summary, it can be concluded that star-shaped PEG molecules end-capped with hydrophobic alkyl chains are capable of physical interactions; the association of macromonomers with hydrophobic end-groups is likely to influence hydrogel formation.
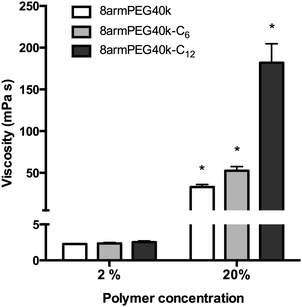 |
| | Fig. 3 Viscosity of aqueous solutions of 8armPEG40k-OH, 8armPEG40k-C6-NH-Boc and 8armPEG40k-C12-NH-Boc at 25 °C. * indicates statistically significant differences to all other groups of the same polymer concentration. | |
Acceleration of gel formation by hydrophobic association
For the following experiments, hydrogels were prepared from macromonomers functionalized with furyl and maleimide end-groups; different alkyl spacer lengths were used. In the first experiment, the effect of macromonomer association on the gel time of DA hydrogels was studied. The DA reaction allows the preparation of covalently cross-linked, degradable hydrogels. In contrast to other cross-linking reactions, such as radical polymerization or the copper(I)-catalyzed azide–alkyne cycloaddition, the use of potentially toxic initiators or catalysts can be avoided. This is important for applications such as controlled protein release or cell encapsulation. However, a major disadvantage of the DA reaction is the comparably low cross-linking rate at ambient temperature. Consequently, it would be beneficial if gel formation were accelerated by hydrophobic interactions between macromonomers.
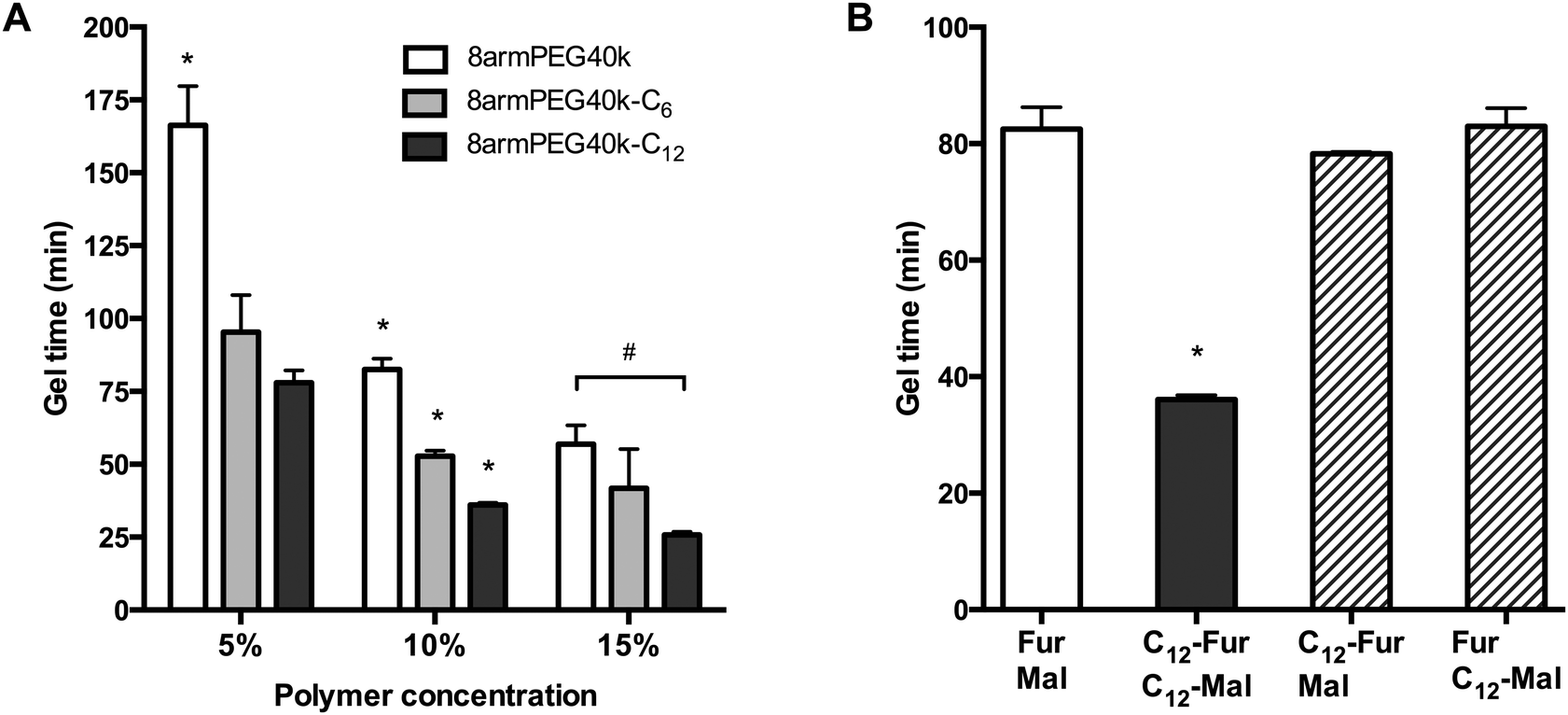
As shown in Fig. 4A, the gel times of plain 8armPEG40k-hydrogels were 166 min (5% polymer), 83 min (10% polymer) and 57 min (15% polymer). Upon modification with 12-aminododecanoic acid, the gel times decreased on average by 55%, with gel points at 78 min (5% polymer), 36 min (10% polymer) and 26 min (15% polymer) being determined. Apparently, the association of macromonomers by hydrophobic interactions promotes the cross-linking process independent from the polymer concentration. In comparison, modification with 6-aminohexanoic acid also influenced the gel formation rate, but the effect was much weaker. This is in line with the previous results, which showed that both macromonomer association and viscosity enhancement depend on the spacer length. To elucidate the contribution of macromonomer association on gel formation, a further experiment was done (Fig. 4B). When both 8armPEG40k-C12-furan and 8armPEG40k-C12-maleimide were used, gelation proceeded twice as fast as in hydrogels made from unmodified macromonomers, i.e., 8armPEG40k-furan and 8armPEG40k-maleimide. However, when 8armPEG40k-C12-furan was combined with unmodified 8armPEG40k-maleimide, or vice versa, no significant acceleration of gelation was observed. Although association of 8armPEG40k-C12-furan (or 8armPEG40k-C12-maleimide) occurs at the tested concentration, this does apparently not affect cross-linking via DA reaction. We, therefore, believe that hydrophobic modification of both macromonomers (i.e., 8armPEG40k-furan and 8armPEG40k-maleimide) is required to accelerate gel formation. It is conceivable that the association of 8armPEG40k-C12-furan and 8armPEG40-C12-maleimide into mixed micelles brings the reactive furyl and maleimide groups into close proximity. This makes the cross-linking process more efficient and accelerates gel formation.
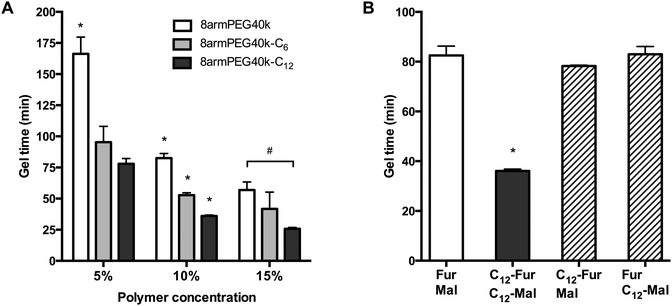 |
| | Fig. 4 Gel time of 8armPEG40k, 8armPEG40k-C6 and 8armPEG40k-C12 hydrogels at 37 °C (A). Influence of hydrophobic interactions on gel formation (B). Gel formation only proceeded twice as fast when both components (i.e., 8armPEG40k-furan or 8armPEG40k-maleimide) were modified with hydrophobic C12 spacers (C12-Fur/C12-Mal). The polymer concentration was 10% (w/V). * indicates statistically significant differences to all other groups of the same polymer concentration; # indicates statistically significant differences between the specified groups. | |
Network mesh size and mechanical properties
To study the influence of hydrophobic modification on the cross-linking density of hydrogels, the average network mesh size (ξ) of 8armPEG40k, 8armPEG40k-C6 and 8armPEG40k-C12 hydrogels was determined as previously described.24 In brief, the gel volume after cross-linking, Vgc, and after swelling in water, Vgs, was determined by weighing cylindrical gel samples in air and hexane. Afterwards, the gel samples were freeze-dried and the volume of the dry polymer, Vp, was determined. After calculating the polymer fraction of the hydrogel after cross-linking (v2c = Vp/Vgc) and in the swollen state (v2s = Vp/Vgs), the number of moles of elastically active chains within the network, ve, was calculated using a modified version of the Flory–Rehner eqn (1).44–46| | 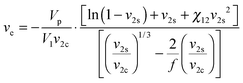 | (1) |
The calculation requires several parameters, namely the molar volume of water (V1 = 18 mL mol−1), the branching factor of the macromonomers (f = 8), and the polymer–water interaction parameter (χ12). The parameter χ12 describes the average extent of interaction between a macromonomer and water; it depends on the hydrophilic lipophilic balance (HLB) of the macromonomer. For amphiphilic macromonomers that are composed of both ethylene oxide (EO) and hydrocarbon chains (HC), χ12 can be calculated according to:47| | 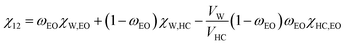 | (2) |
In this equation, ωEO represents the weight fraction of the ethylene oxide chain; VW and VHC are the molar volumes of water and the hydrocarbon part of the macromonomer. The water–ethylene oxide interaction parameter (χW,EO) was set to 0.4; the water–hydrocarbon interaction parameter (χW,HC) was set to 2.0 and the term χHC,EO (VW/VHC) was taken as 0.4. Based on these values, the interaction parameters for 8armPEG40k-C6 (χ12 = 0.42) and 8armPEG40k-C12 in water (χ12 = 0.45) were calculated; for unmodified 8armPEG40k hydrogels, the PEG–water interaction parameter (χ12 = 0.40) was used. Together with the average bond length along the PEG backbone (l = 0.146 nm), the molecular mass of the PEG repeating unit (Mr = 44 g mol−1), the total mass of PEG in the hydrogel (mp), and the Flory characteristic ratio (Cn = 4), ve allows the calculation of ξ according to eqn (3):48
| | 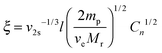 | (3) |
The data listed in Table 1 shows that ξ is decreasing with increasing polymer concentration. The effect was more pronounced in 8armPEG40k and 8armPEG40k-C6 hydrogels than in 8armPEG40k-C12 hydrogels. The modification of 8armPEG40k with 6-aminohexanoic acid spacers only had a minor influence on ξ. In contrast to that, modification with more hydrophobic 12-aminododecanoic spacers significantly decreased ξ. Apparently, the association of 8armPEG40k-C12-furan and 8armPEG40k-C12-maleimide results in more densely cross-linked hydrogels with less network defects.
Table 1 Average network mesh size (ξ) and Young's modulus of compression (E) of 8armPEG40k, 8armPEG40k-C6 and 8armPEG40k-C12 hydrogels at concentrations of 5, 10 and 15% (w/V)
| Polymer type |
Concentration (%) |
ξ (nm) |
E (kPa) |
| 8armPEG40k |
5 |
25.9 ± 0.2 |
8.1 ± 4.1 |
| 10 |
19.6 ± 0.1 |
33.2 ± 4.0 |
| 15 |
17.5 ± 0.1 |
87.2 ± 19.6 |
|
|
| 8armPEG40k-C6 |
5 |
24.3 ± 1.0 |
25.0 ± 4.0 |
| 10 |
20.3 ± 0.6 |
50.3 ± 5.4 |
| 15 |
17.4 ± 1.7 |
117.5 ± 13.1 |
|
|
| 8armPEG40k-C12 |
5 |
18.2 ± 0.6 |
38.9 ± 4.6 |
| 10 |
15.4 ± 0.1 |
79.6 ± 12.1 |
| 15 |
14.3 ± 0.2 |
172.8 ± 28.4 |
Since cross-linking density and material stiffness are closely related, the association of 8armPEG40k-C12-furan and 8armPEG40k-C12-maleimide should also be reflected in the mechanical properties of the hydrogels. To confirm this relation, the Young's modulus of compression (E) was determined. It is defined as the ratio of compressive stress to compressive strain and frequently used to describe the mechanical properties of materials.49,50 As expected, the modification of macromonomers with hydrophobic residues had a strong effect on the mechanical properties of hydrogels. For example, the Young's modulus of 8armPEG40k-C12 hydrogels (10% and 15% polymer concentration) was twice as high as that of unmodified 8armPEG40k hydrogels. The effect was even more pronounced at low polymer concentrations; the Young's modulus of 5% (w/V) 8armPEG40k-C12 hydrogels was approximately five times higher than that of unmodified 8armPEG40k hydrogels (Table 1). As already observed in the other experiments, modification with 6-aminohexanoic acid residues resulted in much weaker effects. In summary, using hydrophobically modified macromonomers for hydrogel preparation resulted in higher cross-linking densities, as indicated by the values of ξ and E. The association of hydrophobically modified macromonomers to micelles and transient networks is thought to make the cross-linking process more efficient. As a consequence, more densely cross-linked networks are formed and the number of imperfections, such as elastically inactive chains, is reduced.
Swelling, degradation and antibody release
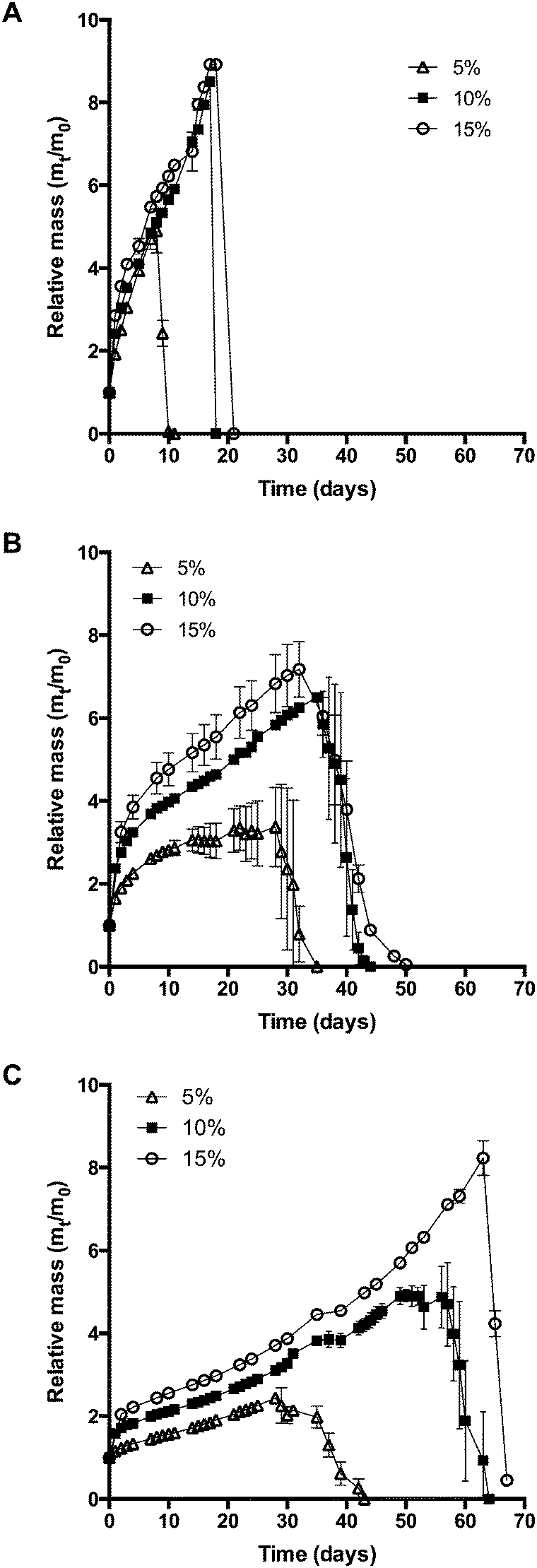
As discussed in the previous section, using hydrophobically modified macromonomers increases the cross-linking density of hydrogels. Since the cross-linking density has a strong influence on hydrogel degradation, the swelling behavior of gel cylinders in phosphate buffer pH 7.4 was monitored over time at 37 °C (Fig. 5). As expected, the swelling and degradation behavior of hydrogels was dependent on the polymer concentration. Furthermore, we found that hydrophobically modified macromonomers yielded hydrogels with increased stability to degradation. For example, at a concentration of 10% (w/V), 8armPEG40k hydrogels showed a 9-fold mass increase during incubation, whereas the mass of 8armPEG40k-C6 and 8armPEG40k-C12 hydrogels increased only 6-fold and 4-fold, respectively. The gel samples were completely degraded after 20 (8armPEG40k hydrogels), 40 (8armPEG40k-C6 hydrogels) and 60 days (8armPEG40k-C12 hydrogels). In summary, upon introduction of hydrophobic residues, the swelling of hydrogels proceeds more slowly and the mass increase is less pronounced. In addition, hydrogels made of hydrophobically modified macromonomers showed considerably higher resistance to degradation than unmodified hydrogels.
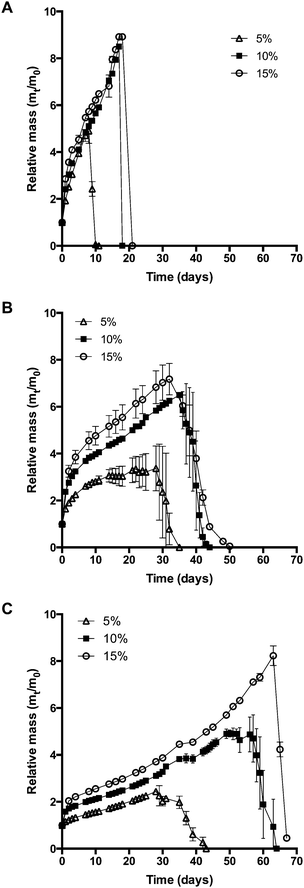 |
| | Fig. 5 Swelling and degradation of 8armPEG40k (A), 8armPEG40k-C6 (B), 8armPEG40k-C12 hydrogels (C) in phosphate buffer, pH 7.4 at 37 °C. | |
This may be due to several reasons. On the one hand, the extent of interaction between water and polymer chains has an impact on hydrogel swelling. The interaction between water and purely hydrophilic polymers is preferred over interactions with polymers that comprise both hydrophilic and hydrophobic parts. The presence of hydrophobic domains within the hydrogel network may, therefore, limit the overall degree of swelling.51–53 In addition, hydrogels made of hydrophobically modified macromonomers have a higher crosslinking density than 8armPEG40k hydrogels. This directly translates into reduced swelling and improved stability to degradation. Finally, the degradation rate of DA hydrogels is governed by the hydrolytic stability of the involved maleimide groups.25 It has been shown that degradation occurs by retro-Diels–Alder (rDA) reaction and subsequent ring-opening hydrolysis of the newly generated maleimide groups. The rate of the rDA reaction is low at 37 °C; however, ring-opening hydrolysis of maleimide to unreactive maleamic acid shifts the DA/rDA equilibrium and significantly accelerates the degradation rate. To check the influence of hydrophobic modification, the hydrolytic stability of 8armPEG40k-maleimide, 8armPEG40k-C6-maleimide and 8armPEG40k-C12-maleimide was determined in phosphate buffer, pH 7.4 at 37 °C (Fig. 6). Assuming a pseudo-first order reaction, rate constants (kobs) and half-lives (t1/2) were calculated by fitting the integrated form of eqn (4) to the experimental data.
| | 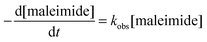 | (4) |
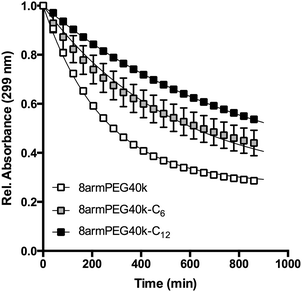 |
| | Fig. 6 Absorbance of 8armPEG40k-maleimide, 8armPEG40k-C6-maleimide and 8armPEG40k-C12-maleimide in phosphate buffer, pH 7.4 at 37 °C. The absorbance decreased due to ring-opening hydrolysis of maleimide; hydrophobic modification resulted in improved hydrolytic resistance. Experimental data are indicated by symbols; solid lines represent the best fits of eqn (4). | |
The results listed in Table 2 show that hydrophobic modification increases the stability of maleimide groups. The half-lives of 8armPEG40k-C6-maleimide and 8armPEG40k-C12-maleimide were 2.2 and 3.4 times longer than the half-life of unmodified 8armPEG40k-maleimide. These results are in good agreement with literature data, which also showed improved hydrolytic stability of N-alkyl substituted maleimides.54–57 Electron-donating N-substituents, such as alkyl groups, stabilize the C–N–C bond of maleimide; moreover, the enolization of the carbonyl group is accelerated by N-alkyl substituents. Both effects retard the nucleophilic attack by hydroxide ions.54
Table 2 Calculated rate constants (kobs) and half-lives (t1/2) for the ring-opening reaction of 8armPEG40k-maleimide, 8armPEG40k-C6-maleimide and 8armPEG40k-C12-maleimide in phosphate buffer, pH 7.4 at 37 °C
| Polymer type |
k
obs (min−1) |
t
1/2 (min) |
R
2
|
| 8armPEG40k-maleimide |
4.06 × 10−3 |
170 |
0.9986 |
| 8armPEG40k-C6-maleimide |
1.89 × 10−3 |
367 |
0.9853 |
| 8armPEG40k-C12-maleimide |
1.19 × 10−3 |
585 |
0.9998 |
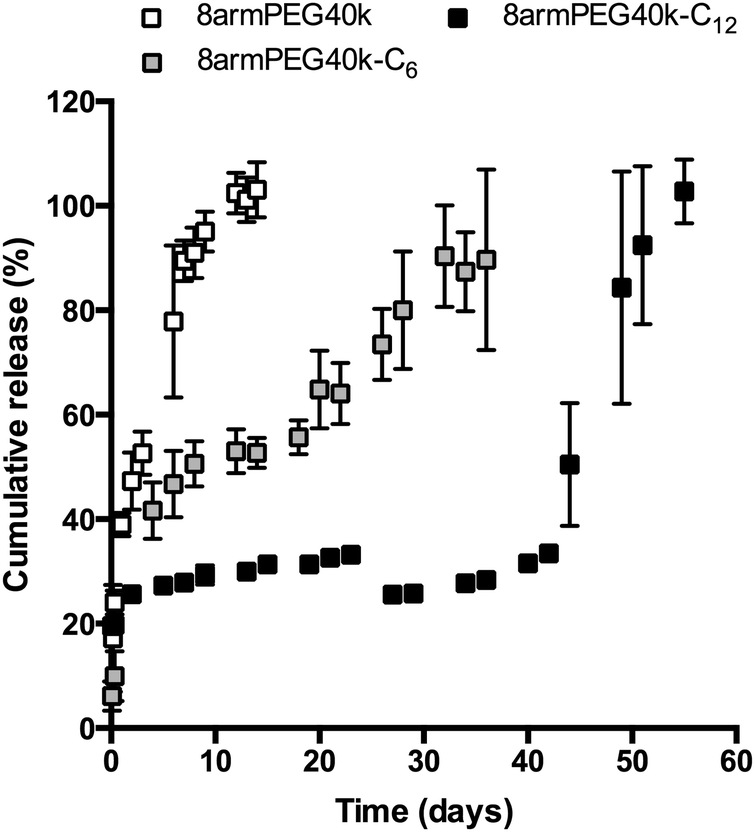
In the final experiment, hydrogels were loaded with bevacizumab (Avastin®), which served as a model antibody. The release of bevacizumab from 8armPEG40k, 8armPEG40k-C6 and 8armPEG40k-C12 hydrogels is shown in Fig. 7; the polymer concentration was 10% (w/V) in all groups. As it was expected from the previous studies, the release rate was strongly influenced by the length of the hydrophobic spacer. Release from unmodified 8armPEG40k hydrogels was comparatively fast; the incorporated bevacizumab was completely released after 10 days. In contrast to 8armPEG40k, release from 8armPEG40k-C6 and 8armPEG40k-C12 hydrogels was significantly delayed. Both release profiles had a hyperbolic shape; however, their time scales were different. The first phase was characterized by fast, diffusion-controlled release. This was followed by a plateau during which little or no protein release was observed. Apparently, the incorporated protein is physically trapped within the polymer network. In addition, a small fraction of the protein is immobilized during cross-linking by Michael-type addition to 8armPEG40k-C6-maleimide and 8armPEG40k-C12-maleimide. The third phase of release starts with the onset of hydrogel degradation. Once a critical mesh size had been exceeded after 20 (8armPEG40k-C6) and 45 days (8armPEG40k-C12), the entrapped bevacizumab was suddenly released; the release was completed after 30 (8armPEG40k-C6) and 55 days (8armPEG40k-C12). Although it is obvious that bevacizumab release is mainly controlled by degradation, some discrepancies appear when the swelling and release data are compared. For example, 8armPEG40k-C12 hydrogels started to degrade after approximately 55 days, while the onset of antibody release was after approximately 45 days. A possible explanation for the observed time difference between the beginning of degradation and the onset of antibody release may be the fact that the incorporation of proteins into hydrogels can lead to network imperfections, which consequently affect the degradation rate. This explanation seems likely, as a correlation between the extent of network defects and the incorporation of cells58 or nanoparticles59 into hydrogels has already been described. In summary, the obtained in vitro data suggests that 8armPEG40k-C6 and 8armPEG40k-C12 hydrogels may be suitable for controlled protein release in general, and for delayed antibody release in particular.
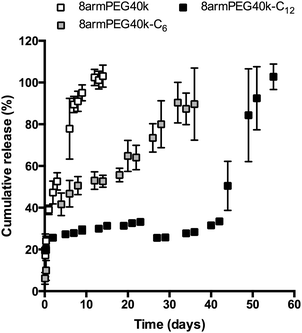 |
| | Fig. 7 Release of bevacizumab from 8armPEG40k, 8armPEG40k-C6 and 8armPEG40k-C12 hydrogels at a concentration in phosphate buffer, pH 7.4 at 37 °C. The polymer concentration was 10% (w/V) in all groups. | |
Conclusion
Cross-linking via DA chemistry is a promising method to prepare hydrogels for controlled release of therapeutic proteins. However, before their application in pre-clinical studies, the hydrogels still need to be optimized regarding their gel points, stability to degradation and time frames of drug release. In this study, it has been demonstrated that the combination of hydrophobic interactions and DA chemistry can contribute to improved hydrogel properties, such as faster gelation and increased stability to degradation. Modification of hydrophilic PEG with alkyl chains induces hydrophobic interactions between the macromonomers, which result in the formation of micellar structures and transient networks. The resulting inner structure of the polymer solution has been shown to facilitate cross-linking via DA chemistry. Using hydrophobically modified macromonomers, therefore, results in hydrogels with improved properties, in particular if long alkyl chains (e.g., C12) are employed. For example, hydrogels prepared from 8armPEG40k-C12-furan and 8armPEG40k-C12-maleimide were characterized by significantly faster gelation, smaller mesh sizes, higher cross-linking densities, increased resistance to mechanical stress and improved stability to degradation. As a consequence, the in vitro release of incorporated proteins is considerably delayed; for example, bevacizumab was released from 8armPEG40k-C12 hydrogels after almost 60 days. Interestingly, the observed effects have been demonstrated to correlate with the length of the hydrophobic moieties. Short alkyl chains (e.g., C6) result in much weaker effects regarding gelation time, stability to degradation and time frame of drug release. Thus, hydrogel characteristics can be tailored by employing shorter or longer alkyl chains for hydrophobic modification. In summary, the combination of hydrophobic association and chemical cross-linking is a promising approach to improve the properties of hydrogels and bring them closer to clinical application.
Acknowledgements
Financial support from the German Research Foundation (DFG), grant number BR 5132/1-2 is gratefully acknowledged.
References
- D. V. Goeddel, D. G. Kleid, F. Bolivar, H. L. Heyneker, D. G. Yansura, R. Crea, T. Hirose, A. Kraszewski, K. Itakura and A. D. Riggs, Proc. Natl. Acad. Sci. U. S. A., 1979, 76, 106–110 CrossRef CAS.
- C. Morrison, Nat. Biotechnol., 2015, 33, 125–128 CrossRef CAS PubMed.
- A. L. Nelson, E. Dhimolea and J. M. Reichert, Nat. Rev. Drug Discovery, 2010, 9, 767–774 CrossRef CAS PubMed.
- P. J. Hudson and C. Souriau, Nat. Med., 2003, 9, 129–134 CrossRef CAS PubMed.
- T. J. Vaughan, A. J. Williams, K. Pritchard, J. K. Osbourn, A. R. Pope, J. C. Earnshaw, J. McCafferty, R. A. Hodits, J. Wilton and K. S. Johnson, Nat. Biotechnol., 1996, 14, 309–314 CrossRef CAS PubMed.
- M. V. Mehren, G. P. Adams and L. M. Weiner, Annu. Rev. Med., 2003, 54, 343–369 CrossRef PubMed.
- R. Maini, E. W. St Clair, F. Breedveld, D. Furst, J. Kalden, M. Weisman, J. Smolen, P. Emery, G. Harriman, M. Feldmann and P. Lipsky, Lancet, 1999, 354, 1932–1939 CrossRef CAS.
- The CATT Research Group, N. Engl. J. Med., 2011, 364, 1897–1908 CrossRef PubMed.
- P. Chames, M. Van Regenmortel, E. Weiss and D. Baty, Br. J. Pharmacol., 2009, 157, 220–233 CrossRef CAS PubMed.
- S. J. Shire, Z. Shahrokh and J. Liu, J. Pharm. Sci., 2004, 93, 1390–1402 CrossRef CAS PubMed.
- J. Vlasak and R. Ionescu, mAbs, 2014, 3, 253–263 CrossRef.
- R. J. Keizer, A. D. R. Huitema, J. H. M. Schellens and J. H. Beijnen, Clin. Pharmacokinet., 2010, 49, 493–507 CrossRef CAS PubMed.
- T. Vermonden, R. Censi and W. E. Hennink, Chem. Rev., 2012, 112, 2853–2888 CrossRef CAS PubMed.
- W. B. Liechty, D. R. Kryscio, B. V. Slaughter and N. A. Peppas, Annu. Rev. Chem. Biomol. Eng., 2010, 1, 149–173 CrossRef CAS PubMed.
- T. R. Hoare and D. S. Kohane, Polymer, 2008, 49, 1993–2007 CrossRef CAS.
- Y. Qiu and K. Park, Adv. Drug Delivery Rev., 2012, 64, 49–60 CrossRef.
- L. Klouda and A. G. Mikos, Eur. J. Pharm. Biopharm., 2008, 68, 34–45 CrossRef CAS PubMed.
- S. R. Van Tomme, G. Storm and W. E. Hennink, Int. J. Pharm., 2008, 355, 1–18 CrossRef CAS PubMed.
- C. He, S. W. Kim and D. S. Lee, J. Controlled Release, 2008, 127, 189–207 CrossRef CAS PubMed.
- D. Cohn, A. Sosnik and A. Levy, Biomaterials, 2003, 24, 3707–3714 CrossRef CAS PubMed.
- A. Sosnik and D. Cohn, Biomaterials, 2004, 25, 2851–2858 CrossRef CAS PubMed.
- M. Katakam, W. R. Ravis, D. L. Golden and A. K. Banga, Int. J. Pharm., 1997, 152, 53–58 CrossRef CAS.
- C.-C. Lin and K. S. Anseth, Pharm. Res., 2008, 26, 631–643 CrossRef PubMed.
- S. Kirchhof, F. P. Brandl, N. Hammer and A. M. Goepferich, J. Mater. Chem. B, 2013, 1, 4855–4864 RSC.
- S. Kirchhof, A. Strasser, H.-J. Wittmann, V. Messmann, N. Hammer, A. M. Goepferich and F. P. Brandl, J. Mater. Chem. B, 2015, 3, 449–457 RSC.
- S. Kirchhof, M. Gregoritza, V. Messmann, N. Hammer, A. M. Goepferich and F. P. Brandl, Eur. J. Pharm. Biopharm., 2015, 96, 217–225 CrossRef CAS PubMed.
- M. Gregoritza and F. P. Brandl, Eur. J. Pharm. Biopharm., 2015, 97, 438–453 CrossRef CAS PubMed.
- I. T. Dorn, K. R. Neumaier and R. Tampé, J. Am. Chem. Soc., 1998, 120, 2753–2763 CrossRef CAS.
- S. Udenfriend, S. Stein, P. Bohlen, W. Dairman, W. Leimgruber and M. Weigele, Science, 1972, 178, 871–872 CAS.
- F. Brandl, M. Henke, S. Rothschenk, R. Gschwind, M. Breunig, T. Blunk, J. Tessmar and A. Göpferich, Adv. Eng. Mater., 2007, 9, 1141–1149 CrossRef.
- G. Basu Ray, I. Chakraborty and S. P. Moulik, J. Colloid Interface Sci., 2006, 294, 248–254 CrossRef CAS PubMed.
- R. Hennig, A. Veser, S. Kirchhof and A. Goepferich, Mol. Pharmaceutics, 2015, 12, 3292–3302 CrossRef CAS PubMed.
- K. Kalyanasundaram and J. K. Thomas, J. Am. Chem. Soc., 1977, 99, 2039–2044 CrossRef CAS.
- E. Kumacheva, Y. Rharbi, M. A. Winnik, L. Guo, K. C. Tam and R. D. Jenkins, Langmuir, 1997, 13, 182–186 CrossRef CAS.
- Q. Gao, Q. Liang, F. Yu, J. Xu, Q. Zhao and B. Sun, Colloids Surf., B, 2011, 88, 741–748 CrossRef CAS PubMed.
- Y. Wang and M. A. Winnik, Langmuir, 1990, 6, 1437–1439 CrossRef CAS.
- A. Yekta, B. Xu, J. Duhamel, H. Adiwidjaja and M. A. Winnik, Macromolecules, 1995, 28, 956–966 CrossRef CAS.
- E. Alami, M. Almgren, W. Brown and J. Franois, Macromolecules, 1996, 29, 2229–2243 CrossRef CAS.
- A. Halperin, Macromolecules, 1987, 20, 2943–2946 CrossRef CAS.
- R. D. Hester and D. R. Squire, J. Coat. Technol., 1997, 69, 109–114 CrossRef CAS.
- T. Annable, J. Rheol., 1993, 37, 695–726 CrossRef CAS.
- F. Tanaka and S. F. Edwards, Macromolecules, 1992, 25, 1516–1523 CrossRef CAS.
- M. A. Winnik and A. Yekta, Curr. Opin. Colloid Interface Sci., 1997, 2, 424–436 CrossRef CAS.
-
P. J. Flory, Principles of polymer chemistry, Cornell University Pr., Ithaca, N.Y., 10 edn, 1953 Search PubMed.
- J. C. Bray and E. W. Merrill, J. Appl. Polym. Sci., 1973, 17, 3779–3794 CrossRef.
- D. L. Elbert, A. B. Pratt, M. P. Lutolf, S. Halstenberg and J. A. Hubbell, J. Controlled Release, 2001, 76, 11–25 CrossRef CAS PubMed.
- B. Kronberg and P. Stenius, J. Colloid Interface Sci., 2010, 102, 410–417 CrossRef.
- T. Canal and N. A. Peppas, J. Biomed. Mater. Res., 1989, 23, 1183–1193 CrossRef CAS PubMed.
- K. S. Anseth, C. N. Bowman and L. Brannon-Peppas, Biomaterials, 2003, 17, 1647–1657 CrossRef.
- S. C. Owen, S. A. Fisher, R. Y. Tam, C. M. Nimmo and M. S. Shoichet, Langmuir, 2013, 29, 7393–7400 CrossRef CAS PubMed.
- T. Inoue, G. Chen, K. Nakamae and A. S. Hoffman, J. Controlled Release, 1997, 49, 167–176 CrossRef CAS.
- Y. Zhang, C.-Y. Won and C.-C. Chu, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 4554–4569 CrossRef CAS.
- Y. Yin, Y. Yang and H. Xu, J. Appl. Polym. Sci., 2002, 83, 2835–2842 CrossRef CAS.
- S. Matsui and H. Aida, J. Chem. Soc., Perkin Trans. 2, 1978, 1277–1280 RSC.
- C. P. Ryan, M. E. B. Smith, F. F. Schumacher, D. Grohmann, D. Papaioannou, G. Waksman, F. Werner, J. R. Baker and S. Caddick, Chem. Commun., 2011, 47, 5452–5454 RSC.
- R. I. Nathani, V. Chudasama, C. P. Ryan, P. R. Moody, R. E. Morgan, R. J. Fitzmaurice, M. E. B. Smith, J. R. Baker and S. Caddick, Org. Biomol. Chem., 2013, 11, 2408–2411 CAS.
- R. P. Lyon, J. R. Setter, T. D. Bovee, S. O. Doronina, J. H. Hunter, M. E. Anderson, C. L. Balasubramanian, S. M. Duniho, C. I. Leiske, F. Li and P. D. Senter, Nat. Biotechnol., 2014, 32, 1059–1062 CrossRef CAS PubMed.
- N.-J. Cho, M. Elazar, A. Xiong, W. Lee, E. Chiao, J. Baker, C. W. Frank and J. S. Glenn, Biomed. Mater., 2009, 4, 011001 CrossRef PubMed.
- W. Lee, N. J. Cho, A. Xiong, J. S. Glenn and C. W. Frank, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 20709–20714 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6tb00223d |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a