
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Biodistribution and fate of core-labeled 125I polymeric nanocarriers prepared by Flash NanoPrecipitation (FNP)†
Christina
Tang
ab,
Jasmine
Edelstein
ab,
John L.
Mikitsh
c,
Edward
Xiao
ac,
Aaron H.
Hemphill
II
d,
Robert
Pagels
a,
Ann-Marie
Chacko‡
cd and
Robert
Prud’homme
*a
aDepartment of Chemical and Biological Engineering, Princeton University, Princeton, NJ, USA. E-mail: prudhomm@princeton.edu
bDepartment of Chemical and Life Science Engineering, Virginia Commonwealth University, Richmond, VA, USA
cDepartment of Radiology, Division of Nuclear Medicine and Clinical Molecular Imaging, University of Pennsylvania, Perelman School of Medicine, Philadelphia, PA, USA
dInstitute for Translational Medicine and Therapeutics, University of Pennsylvania, Perelman School of Medicine, Philadelphia, PA, USA
First published on 3rd March 2016
Abstract
Non-invasive medical imaging techniques based on radionuclide imaging are powerful platforms to track the fate of radiolabeled materials for diagnostic or drug delivery applications. Polymer-based nanocarriers tagged with non-standard radionuclides with relatively long half-lives (e.g.64Cu: t1/2 = 12.7 h, 76Br: t1/2 = 16.2 h, 89Zr: t1/2 = 3.3 d, 124I: t1/2 = 4.2 d) may greatly expand applications of nanomedicines in molecular imaging and therapy. However, radiolabeling strategies that ensure stable in vivo association of the radiolabel with the nanocarrier remain a significant challenge. In this study, we covalently attach radioiodine to the core of pre-fabricated nanocarriers. First, we encapsulated polyvinyl phenol within a poly(ethylene glycol) coating using Flash NanoPrecipitation (FNP) to produce stable 75 nm and 120 nm nanocarriers. Following FNP, we radiolabeled the encapsulated polyvinyl phenol with 125I via electrophilic aromatic substitution in high radiochemical yields (>90%). Biodistribution studies reveal low radioactivity in the thyroid, indicating minimal leaching of the radiolabel in vivo. Further, PEGylated [125I]PVPh nanocarriers exhibited relatively long circulation half-lives (t1/2α = 2.9 h, t1/2β = 34.9 h) and gradual reticuloendothelial clearance, with 31% of injected dose in blood retained at 24 h post-injection.
1. Introduction
Molecular imaging with radiolabeled compounds using positron emission tomography (PET) enables non-invasive visualization and quantification of ligand biodistribution and tissue-specific pharmacokinetics with high sensitivity (<M−10).1,2 Conventional radionuclides have short half-lives (e.g.15O: t1/2 = 2 min, 13N: t1/2 = 10 min, 11C: t1/2 = 20.4 min, 18F: t1/2 = 109.8 min), limiting the time frame for radiosynthesis and in vivo imaging.3–5 Nanocarrier applications for delivery by EPR (enhanced permeation and retention) or targeting require longer circulation times; and therefore require non-standard radionuclides with longer half-lives (e.g.64Cu: t1/2 = 12.7 h, 76Br: t1/2 = 16.2 h, 89Zr: t1/2 = 3.2 d 124I: t1/2 = 4.2 d).6,7There have generally been two approaches for PET imaging of nanocarriers. Most commonly, chelators have been introduced on the surfaces of the nanocarriers which are subsequently complexed with the PET cation.8,9 However, attachment of chelates and radionuclides to the surface of nanoprobes can add surface charge, triggering adsorption of plasma proteins and a RES response.3,10–13 Further, early detachment of these radionuclides from their nanoprobe anchors in vivo confounds accuracy of probe distribution results.14–26 Radionuclide detachment can occur due to kinetically unstable radiometal chelate systems or metabolic degradation of the nanocarrier-radiolabel linkage.8,27,28 The second approach is to synthesize the nanocarrier core as a metal oxide or sulfide of the PET cation. For example, Zhou et al. generated chelator-free PEGylated [64Cu]CuS nanocarriers, but the size was limited to 30 nm.29 The time required for the synthesis of these radioactive cores sacrifices radioactivity; the complexity of synthesis, and the difficulty in subsequent passivation of the metal particle with a biocompatible coating, make this approach problematic. Biocompatible coatings, such as by poly(ethylene glycol) (PEG), are required to delay reticuloendothelial (RES) clearance and improve circulation times.13,14,30,31
The particles and process we describe here are the first to address all of the limitations currently encountered in radionuclide-active nanocarriers. Ideally, nanocarriers and processes to produce them should have the following characteristics: (1) they can be produced over a range of sizes, (2) the radionuclides are covalently bound to the carrier by a straightforward and rapid process, (3) the core, rather than just the surface, is labeled to avoid addition of surface charge and increase radionuclide loading (surface attachment suffers from the surface to volume limitation), and finally, (4) the carrier requires a dense PEG coating to enable long circulation, and possibly provide a platform for targeting. Thus, our goal was to develop a strategy to radiolabel the core of pre-fabricated PEGylated nanocarriers. We focused on radiolabeling nanocarriers with the low-energy gamma emitter 125I (t1/2 = 60 days) to facilitate optimization of radiolabeling and to assess in vitro and in vivo properties.7 The same chemistry can be used to radiolabel nanocarriers with higher energy radioiodines, including 124I or 123I for PET or SPECT imaging, respectively. Since radioiodination of phenols has been well studied, we encapsulated poly-4-vinyl phenol (PVPh) within PEGylated nanocarriers to provide a stable nanocarrier core and site for radioiodine conjugation.
In Flash NanoPrecipitation (FNP) (Fig. 1), hydrophobic core material (e.g. PVPh) and amphiphilic block copolymer (e.g. PEG-b-polystyrene (PS)) are dissolved in an organic phase and rapidly mixed with a miscible aqueous anti-solvent. Rapid mixing causes nucleation and growth of the precipitating core material. The hydrophobic segment of the block copolymer adsorbs to the hydrophobic precipitate, arresting nanocarrier growth as the hydrophilic PEG block sterically stabilizes the colloidal nanocarriers.32,33 As a process of kinetic assembly, FNP provides control over nanocarrier size from 40 nm to 400 nm with narrow polydispersity.34,35 Following FNP, we performed 127I- or 125I-driven iodination reactions on the nanocarrier PVPh core to examine the accessibility of the encapsulated polyvinyl phenol for electrophilic aromatic substitution. The in vivo fate and biodistribution of the nanocarriers radiolabeled with 125I were studied with nanocarriers.
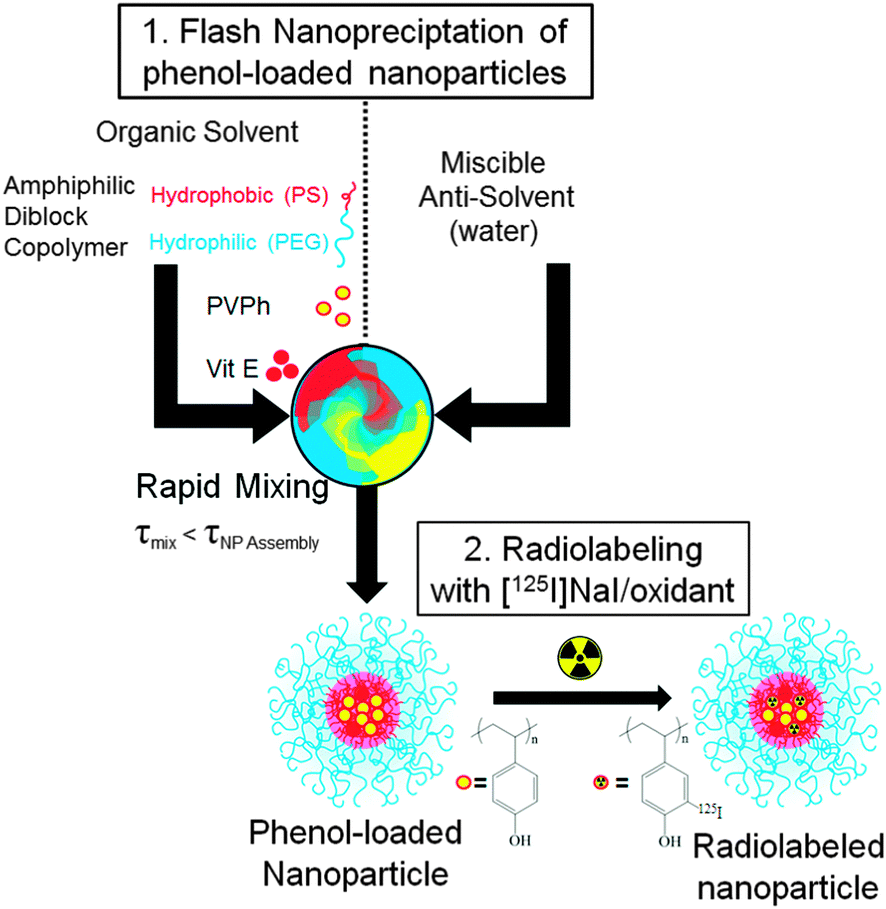 | ||
| Fig. 1 Schematic of nanocarrier synthesis by FNP and subsequent radiolabeling of the PVPh core with 125I. | ||
2. Experimental
Materials
ACS grade sodium iodide, D-α-tocopherol, and chloramine-T trihydrate were purchased from Sigma Aldrich. HPLC grade tertrahydrofuran (THF) was obtained from Fisher Scientific. Phosphate buffered saline without Ca2+ and Mg2+ was obtained from Lonza. Block copolymer, PEG5000-b-PS1600, where the subscript denotes the molecular weight of the block in kilo Daltons, was from Polymer Source (Dorval, QC, CAN) and PVPh1500–7000 was from Polysciences, Inc. (Warrington, PA). All materials were used as received. Water (MQ) was purified by 0.2 μm filtration and four-stage deionization to a resistivity of 17 MΩ or greater (NANOpure Diamond, Barnstead International, Dubuque, IA).Nanocarrier synthesis and characterization
FNP was performed using a hand-operated confined impinging jet mixer with dilution as previously described.36 The amphiphilic stabilizer PEG-b-PS and core material(s) PVPh and D-α-tocopherol were dissolved in THF (40 mg mL−1). The ratio of PVPh and D-α-tocopherol was varied to tune the size of the nanoparticles. The organic mixture (typically 1 mL) was rapidly mixed with an equal volume of NANOpure water in a manually operated confined impinging jet mixer (mixing Reynolds number of ∼1300)37 and collected into a stirred water reservoir. The final volume was typically 10 mL containing 10 vol% organic solvent. After FNP, organic solvent was removed from the suspensions via dialysis using 6–8 kDa MWCO dialysis tubing against 1 L of NANOpure water at room temperature, which was refreshed four times over 24 h. Nanocarriers were concentrated to 10 mg mL−1 using Amicon® Ultra 3 kDa MWCO centrifugal filters in a centrifuge.The hydrodynamic diameters and size distributions (PDI) of nanocarriers were measured with a ZetaSizer Nano Dynamic Light Scattering (DLS) device (Malvern Instruments, Worcestershire, UK). Measurements were done in triplicate at a wavelength of 633 nm and a scattering angle of 173°. Nanocarriers were diluted in NANOpure water at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 ratio to avoid multiple scattering. Samples for TEM were prepared by placing 5 μL of the nanocarrier dispersion on an Ultrathin Carbon Film on a Holey Carbon Support film on 400 mesh copper grid (Ted Pella, Inc., Redding, CA) and drying under ambient conditions. The samples were imaged using a Philips CM100 TEM (Eindhoven, The Netherlands) operated at an accelerating voltage of 100 kV.
:10 ratio to avoid multiple scattering. Samples for TEM were prepared by placing 5 μL of the nanocarrier dispersion on an Ultrathin Carbon Film on a Holey Carbon Support film on 400 mesh copper grid (Ted Pella, Inc., Redding, CA) and drying under ambient conditions. The samples were imaged using a Philips CM100 TEM (Eindhoven, The Netherlands) operated at an accelerating voltage of 100 kV.
Nanocarrier iodination
PEGylated PVPh nanocarriers (4 mg mL−1 in deionized water, 100% PVPh core) were iodinated via aromatic electrophilic substitution using a previously reported method.38 Briefly, sodium iodide (1 eq.) was added to nanocarriers and cooled on ice. Sodium hypochlorite (1 eq.) was added dropwise, every 10–15 s over 10 min. After 2 h at 0 °C, the reaction was quenched by the addition of excess sodium metabisulfite and neutralized using HCl. Following iodination, nanocarrier size was measured by DLS. The reaction mixture was dialyzed against water for 65 h to remove excess sodium iodide, sodium hypochlorite, and PBS salts, and then freeze-dried. Nanocarrier shell components were then redissolved in deuterated DMSO and analyzed using 13C-NMR on a Bruker Avance-III 500 MHz device (Bruker Biosciences, Inc. Billerica, MA).Nanocarrier radioiodination and radiolabel stability
PEGylated PVPh nanocarriers were radiolabeled with [125I]NaI (Perkin Elmer, Boston, MA) using an alternative method. The nanocarriers (200 μL, 10 mg mL−1 in deionized water) were combined with chloramine-T oxidant solution (2 μL, 17.6 mM in PBS) and [125I]NaI (approximately 250 μCi in 2 μL PBS) for 30 min with stirring. Unreacted free radioiodine was removed by using a 10 kDa MWCO Amicon ultracentrifuge filter device (Millipore Inc. Bellerica, MA, USA) at 16400 g for 15 min. The sample was washed with 300 μL PBS twice, and the purified radiolabeled nanocarriers were recovered by spin inversion. The radiochemical yield and radiochemical purity was assessed by digital autoradiography (DAR) (FLA-7000, FujiFilm Phosphorimager, Tokyo, Japan) of radioTLC SiO2 silica gel plates. TLC plates were run in 1:1 10% ammonium acetate/methanol and exposed for approximately 1.5 h to the phosophor plate (BAS 2000, Fujifilm) before imaging. Under these conditions, the nanocarrier remains at the origin whereas free radioiodine travels near the solvent front (ESI†).
To examine nanocarrier-associated radiolabel stability, triplicate samples of nanocarriers (5 μL, 215 μCi) were added to 100 μL PBS or 100 μL human serum, and incubated with mixing at 37 °C for 0 min, 5 min, 15 min, 30 min, 1 h, 2 h, 4 h, 6 h, 12 h, 24 h, and 48 h. A 1 μL sample was withdrawn at the indicated time point and then spotted for radio-TLC under the TLC conditions listed previously.
In vivo fate and biodistribution
All animal studies were carried out in accordance with the Guide for the Care and Use of Laboratory Animals as adopted by the US National Institutes of Health and approved by the University of Pennsylvania IACUC. Naive C57BL/6 mice (18–22 g) were anesthetized and nanocarriers (150 μL, 5 mg kg−1, 0.1 μCi μg−1) injected intravenously via the jugular vein (n = 3 per time point).Blood samples (approximately 50 μL each) were collected via retro-orbital bleeding from anesthetized mice at 2 min, 10 min, 30 min, 1 h, 6 h, and 48 h post-injection. Each sample was weighed, and then radioactivity was measured using a gamma counter (Wizard 2470 Perkin Elmer). Sample-associated radioactivity was measured and radioactivity levels were normalized to the amount of nano-particle bound radioactivity in the injected dose to obtain a fraction of the injected dose in the tissue of interest. Results are reported as either percent of injected dose per gram of tissue (% ID per g) or percent of injected dose per whole organ (% ID) where blood accounts for 7% of mouse body weight.14,39 Nanoparticle concentration in blood as a function of time post-injection was fit to a bi-exponential decay curve for a standard two compartment pharmacokinetic model. The α-, and β-half-lives were extrapolated to describing the blood distribution and clearance phase, respectively, using Prism 5.0 (GraphPad) software.
Based on the blood clearance, mice were euthanized at 5 min, 4 h, 10 h, 24 h, and 96 h post-injection and selected organs were harvested and weighed. The liver, spleen, kidney and thyroid were monitored to provide insight into in vivo particle stability over 96 hours. Sample-associated radioactivity was measured and radioactivity levels were normalized by the method described above. Results are reported as either percent of injected dose per gram of tissue (% ID per g) or percent of injected dose per whole organ (% ID), where bone accounts for 14% of mouse body weight, respectively.14,39 All data are expressed as mean ± standard deviation.
3. Results and discussion
Nanocarrier synthesis and characterization
PEGylated PVPh nanocarriers were produced using Flash NanoPrecipitation (FNP). PVPh was dissolved with the amphilphilic block copolymer PEG-b-PS in THF, and then rapidly mixed with deionized water (an antisolvent for PVPh). The PEG-b-PS self-assembled via hydrophobic interactions between the PS block and PVPh precipitates, while the PEG block sterically stabilizes the nanocarriers. As a process of kinetic assembly, FNP provides control over nanocarrier size – a property that influences in vivo performance. According to the literature, nanocarriers larger than 100 nm are easily sequestered in the sinusoids of the spleen and fenestra of the liver, whereas nanocarriers smaller than 20 nm are rapidly filtered by the kidney.3,40–44 To create nanocarriers that avoid rapid clearance by these mechanisms, our goal was to produce nanocarriers between 20 and 100 nm.A representative TEM micrograph of PEGylated PVPh nanocarriers is shown in Fig. 2a. The PVPh core of the spherical nanocarriers is evident in the TEM image. Nanocarriers appear smaller than the hydrodynamic radius measured with DLS because the PEG layer is not sufficiently electron dense to be visible with TEM. The PEG layer adds approximately 20 nm to the hydrodynamic diameter.32 The resulting PVPh-loaded nanocarriers were 120 ± 8 nm with a PDI of 0.228 ± 0.008.
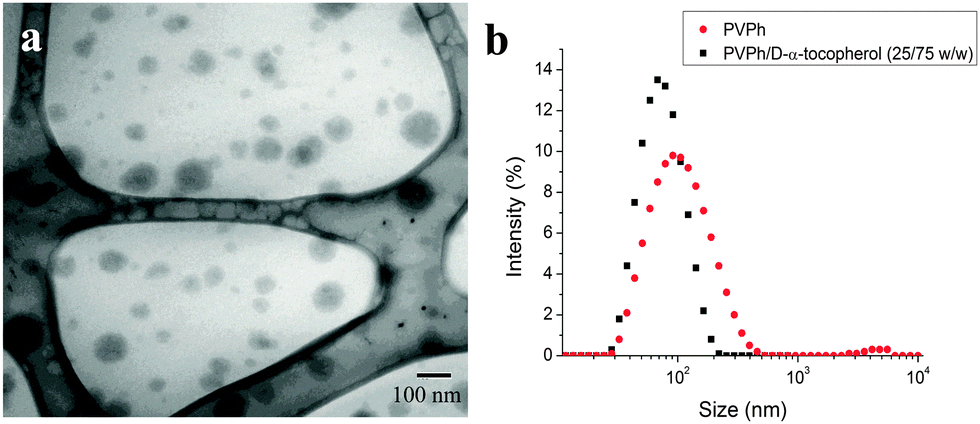 | ||
| Fig. 2 Characterization of PEGylated PVPh nanocarriers synthesized using FNP. (a) Representative TEM micrograph of nanocarriers. Nanocarriers appear smaller than the hydrodynamic radius measured with DLS because the PEG layer is not sufficiently electron dense to be visible with TEM. (b) Size distribution of nanocarriers with (79 ± 3 nm, PDI 0.151 ± 0.009) and without (120 ± 8 nm, PDI 0.228 ± 0.008) D-α-tocopherol. Coprecipitation of PVPh and D-α-tocopherol reduced resulting nanocarrier size. | ||
To reduce the size of the resulting nanocarriers, we co-precipitated the PVPh with D-α-tocopherol (vitamin E). D-α-Tocopherol acts as nucleating agent to seed particle growth via heterogeneous nucleation. Specifically, it lowers the activation energy for particle growth, induces nucleation, and controls the number of nuclei.45 Nanocarriers containing a 75:25 wt:wt D-α-tocopherol:PVPh core were 79 nm with a PDI of 0.151 ± 0.009 (Fig. 2b).
Nanocarrier iodination
Proteins and peptides containing tyrosine can be radiolabeled via electrophilic aromatic substitution of the phenol moiety. Initially, we use well-established iodination chemistry, using sodium iodide with sodium hypochlorite as the oxidant, to radioiodinate the PVPh core of the PEGylated nanocarriers.Our preliminary goal was to establish that the encapsulated PVPh could be iodinated via electrophilic aromatic substitution. Since the iodination procedure has been reported at a range of pH values,46,47 we verified that the phenol-containing nanocarriers were stable under potential reaction conditions. The nanocarrier dispersions were stable for at least 4 weeks in deionized water. Variable pH, from 3 to 10, did not significantly affect nanocarrier size. At pH 12, the ionization of phenol destabilizes the nanocarriers (ESI†). Nanocarrier size was not significantly affected by the iodination reaction under the specified conditions (ESI†).
Following the iodination reaction and purification, we verified that the PVPh core was radiolabeled in the ortho position. 13C NMR spectroscopy reveals that iodination causes an upfield shift of the carbon signal ortho to the phenol from 115 to 84 ppm.48 Di-iodination of polyvinyl phenol core is evident from the downfield shift of the ortho13C–I signal to 87 ppm (Fig. 3). Using one equivalent of non-radioactive 127I, 55% of the phenols were iodinated and 54% of the iodinated phenols were di-iodinated. This demonstrated that the iodination reaction reacts in the core of the nanocarrier, and not just on the surface. Furthermore, in higher magnification TEM images the contrast in the iodinated particles appeared uniform (i.e. uniformly dark from the higher contrast iodine) rather than the iodine being just in a shell on the surface. In separate experiments, no iodine-addition to PEG-b-PS under the same non-radioactive iodination conditions were observed by 13C NMR.
 | ||
| Fig. 3 Aromatic section of 13C-NMR spectrum of nanocarrier components redissolved in deuterated DMSO following iodination reaction and purification. Iodination causes an upfield shift of the carbon ortho to phenol from 115 to 84 ppm. Di-iodination of polyvinyl phenol core is evident from the downfield shift of the iodinated peak to 87 ppm. | ||
Nanocarrier radioiodination and radiolabel stability
Using a shorter, alternative procedure, we radiolabeled the PEGylated PVPh nanocarriers with gamma emitting 125I, as [125I]NaI, being the limiting reagent. The nanocarriers were combined with an oxidant, chloramine-T in PBS and [125I]NaI for 30 min with stirring. Following the reaction the nanocarriers were separated from unreacted free radioiodine by ultracentrifugation for 15 minutes (10 kDa MWCO Amicon ultracentrifuge filter device). The sample was washed with 300 μL PBS twice, and the purified radiolabeled nanocarriers were recovered by spin inversion. The total procedure time was 80–90 minutes comparable to previously reported radiolabeling experiments.14,17,49Given the results from non-radioactive nanocarrier labeling, our data suggest that radioiodine would be introduced ortho to the hydroxyl group of phenol.14,50 Under the same conditions as the radioiodination, no iodination of the PEG-b-PS was observed as confirmed by 13C NMR. Post-radioiodination, particles were ∼110 nm by DLS compared to 79 ± 3 nm (PDI 0.151 ± 0.009). The increase in particle size during radioiodination which was not observed in the initial iodination experiments, likely due to the difference in reaction conditions. In the initial iodination experiments, the sodium hypochlorite oxidant and iodine was added in excess to the particles and then the nanocarriers were isolated by protracted dialysis. In contrast, in the radioiodination the 125I is the limiting reagent with chloramine-T oxidant added in excess. This likely affects particle size.
Radioiodination occurred at high radiochemical yields (>90%) with high radiochemical purity (>99%). We achieved specific activities of 0.1 μCi μg−1 nanocarrier, which is sufficient for biodistribution studies. Only a small fraction (0.01–0.05%) of phenols needed to be radioiodinated because of the large concentration of phenols encapsulated in the nanocarrier core. However, nanocarriers with significantly higher specific activity can be easily synthesized via addition of more radioiodine, which was the stoichiometrically limiting reagent in this synthesis. This offers an advantage compared to chelation which often only enables attachment of tens of radionuclides per particle.51 High loading enables acquisition of high-quality images because nanoprobes will have a high signal to noise ratio at lower mass dosages.52
Radiochemical stability of radiolabeled nanocarriers in vitro was evaluated up to 48 h in PBS and human serum. No free radioiodine was detected with TLC, indicating minimal radiolabel leaching over 48 h (ESI†). This initial characterization indicates these nanocarriers are sufficiently stable for in vivo fate studies. Separate analysis of particle stability in vivo is discussed below.
In vivo fate and biodistribution
To determine the appropriate time points to evaluate biodistribution, we first examined the amount of radioactivity in the blood following injection. Blood levels of nanocarriers declined in a bi-phasic manner, with a distribution half-life (t1/2α) of 2.9 h and an elimination half-life (t1/2β) of 34.9 h (Fig. 4). Nearly a third of nanocarriers remained in circulation 24 h post-injection (31 ± 2% ID, 18 ± 2% ID per g). These circulation times are consistent with circulation times for FNP nanocarriers measured by alternate techniques.53,54 In contrast, previous studies of PEGylated radiolabeled nanocarriers produced by alternate techniques exhibit lower blood retention at 24 h (8–25% ID, 1–6% ID per g)29,55–61 and shorter circulation half-lives (t1/2α: 14 min–2.4 h, t1/2β: 6–22.5 h)29,56,58,62 (ESI,† Table S1). Our unique nanocarrier construct accounts for the longer circulation times. First, FNP imparts a dense PEG layer (2.25 nm2 per chain for a 5k PEG),37 effectively shielding the nanomaterial from opsonization. Second, FNP avoids the addition of surface charge to the nanocarriers, which inherently occurs for nanocarriers prepared by chelation, since for FNP the radiolabel is neutral and is in the core. Together, this likely delays a RES response that prematurely clears nanoprobes from the body.3,10–13 | ||
| Fig. 4 Blood clearance profile of radiolabeled nanocarriers following intravenous injection in healthy mice (n = 3 per time point). | ||
Based on the blood clearance half-lives, mice were euthanized at 5 min, 4 h, 10 h, 24 h, and 96 h post-injection and selected organs were harvested and weighed. The liver, spleen, kidney and thyroid were monitored to provide insight into in vivo particle stability over 96 hours. Decreasing radioactivity in the blood, heart, and lungs, and increasing radioactivity in the liver and spleen over time are consistent with gradual RES clearance for this size particle (Fig. 5).9,63,64 Although hepatic uptake dominates at 24 h post-injection (16 ± 2% ID per g), previous studies of PEGylated 64Cu-labeled nanocarriers ranging from 20–30 nm demonstrated higher liver uptake at 24 h, ranging from 22 to 31% ID per g.31,54
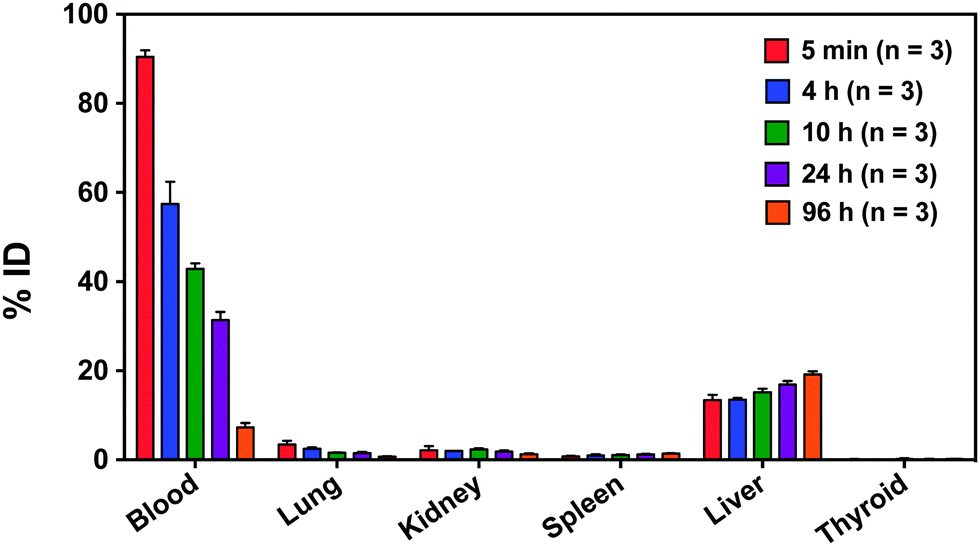 | ||
| Fig. 5 Biodistribution of radiolabeled nanocarriers in the blood, lung, kidney, spleen, liver, thyroid, and brain over time reported as % ID (n = 3). Decreasing radioactivity in the blood and increasing radioactivity in the liver and spleen over time is consistent with gradual RES clearance of nanocarriers and prolonged systemic circulation. Decreasing radioactivity in the kidney suggests minimal renal clearance. Low lung radioactivity suggests that there is no specific binding despite the high blood perfusion level. Low thyroid radioactivity suggests that 125I did not leach from nanocarrier core. | ||
Low kidney radioactivity (Fig. 5) confirms that the nanocarriers tend to avoid renal clearance, partially accounting for the prolonged circulation time. Low lung and heart radioactivity (Fig. 5) suggest that there is no specific binding of the nanocarriers despite the high blood perfusion level in these organ compartments.
At 24 h, the radioactivity in the thyroid is relatively low at 0.11 ± 0.09% ID, and at 96 h it only slightly increases to 0.16 ± 0.05% ID. Since free iodine naturally accumulates in the thyroid, low thyroid radioactivity over time indicates that minimal 125I leached from the radiolabeled nanocarrier core in vivo.65 Thus, PEGylated [125I]PVPh nanocarriers are circulating intact. For comparison, Novakova et al. injected the free polymer [125I] poly(D,L-lactide)-block-poly(ethylene oxide) into rats and found increasing uptake of radioactivity in the thyroid over 24 h.57 In their study, radioiodine was covalently attached to poly(D,L-lactide) via an added p-methoxyphenol end group.57 The high radioactivity in the thyroid 24 h post-injection (4 ± 2% ID) was attributed to release of radioiodine from hydrolysis of the biodegradable poly(D,L-lactide) block.57 Given that our particle components are non-biodegradable, free radioiodine would presumably result from dissociation and metabolism of the radiolabel covalently bound to the polyvinyl phenol in the core and would accumulate in the thyroid. However, thyroid radioactivity remains low over 96 hours particle stability in vivo over the evaluated time course.
The liver, spleen, kidney and thyroid were monitored to provide insight into in vivo particle stability over 96 hours. Examining the overall biodistribution: (1) the nanocarrier concentration in the liver and spleen increase as a function of time, (2) the radioactivity in the kidneys remains low, and (3) the radioactivity of the thyroid remains extremely low. Taken together, (1) and (2) indicate the particles are gradually cleared by the RES as expected for this size particle and the nanocarrier size appears to remain intact over 96 hours. Combined with the low thyroid radioactivity over 96 hours (3), the radiolabeled particles remain stable in circulation during evaluated time course.
The delayed RES clearance and relatively long circulation half-life in vivo, PEGylated [125I]PVPh nanocarriers, are a promising platform for molecular imaging. Introducing a phenol chemical handle via encapsulation of PVPh allows us to easily tag these materials with radioiodine to track their fate in vivo ultimately by non-invasive diagnostic means using PET or SPECT imaging.
4. Conclusions
We produced PEGylated PVPh nanocarriers via Flash NanoPrecipitation and successfully radiolabeled their phenol-containing core with 125I at high radiochemical yields (>90%). Radioiodination enabled facile characterization of in vivo nanocarrier fate and biodistribution. The nanocarriers demonstrated extended circulation half-lives (t1/2β = 2.9 h, t1/2β = 34.9 h) with 31% ID retained in the blood pool 24 h post-injection and gradual RES clearance (liver 17% ID (24 h), 19% ID (96 h); spleen 1.2% ID (24 h), 1.4% ID (96 h)) indicating the nanocarrier size appears to remain intact over 96 hours. Further, the very low thyroid radioactivity throughout the in vivo study indicate radiolabeled nanocarriers circulate intact over 96 hours. Given their delayed RES clearance and long circulation half-life in vivo, PEGylated [125I]PVPh nanocarriers are a promising platform for preclinical and translational imaging. This study provides a starting point for evaluation of the circulation and fate of nanoparticles produced by FNP based on size, charge, surface coating, and active targeting moieties. It leads directly to studies of targeting and biodistribution, which are extremely important as the field places more emphasis on targeted nanocarriers. The FNP process enables the preparation of targeted nanocarriers in a facile, and scalable manner.66,67 Introducing a phenol chemical handle via encapsulation of PVPh allows us to easily tag these nanomaterials with radioiodine to track their fate in vivo, ultimately by non-invasive means using PET or SPECT imaging.Acknowledgements
This work was supported by Princeton University's IP Accelerator Fund to RKP and Princeton University's Lidow Senior Thesis Fund to JNE. This work was also supported the National Institutes of Health (Award No. 1RO1CA155061-1 and Award No. R37AI-051214). Additional support was provided by the Summer Undergraduate Program for Educating Radiation Scientists at the University of Pennsylvania (SUPERS@PENN) to AHH and NIH NCATS through grant KL2TR00139 to AMC. We would like to thank Dr Istvan Pelczer for discussions regarding 13C NMR.References
- D. Psimadas, P. Georgoulias, V. Valotassiou and G. Loudos, J. Pharm. Sci., 2012, 7, 2271–2280 CrossRef PubMed.
- E. G. Solon, Cell Tissue Res., 2015, 360, 87–107 CrossRef PubMed.
- A. L. Branco de Barros, A. Tsourkas, B. Saboury, V. Cardoso and A. Alavi, EJNMMI Res., 2012, 2, 39 CrossRef PubMed.
- G. Ting, C. H. Chang and H. E. Wang, Anticancer Res., 2009, 29, 4107–4118 CAS.
- X. Sun, X. Huang, X. Yan, Y. Wang, J. Guo, O. Jacobson and D. Liu, ACS Nano, 2014, 8, 8438–8446 CrossRef CAS PubMed.
- L. Koehler, K. Gagnon, S. McQuarrie and F. Wuest, Molecules, 2010, 15, 2686–2718 CrossRef CAS PubMed.
- A.-M. Chacko and C. R. Divgi, Med. Chem., 2011, 7, 395–412 CrossRef CAS.
- M. Brechbiel, Q J Nucl Med Mol Imaging, 2008, 52, 166–173 CAS.
- K. Stockhofe, J. M. Postema, H. Schieferstein and T. L. Ross, Pharmaceuticals, 2014, 7, 392–418 CrossRef CAS PubMed.
- R. R. Arvizo, O. R. Miranda, D. F. Moyano, C. a. Walden, K. Giri, R. Bhattacharya, J. D. Robertson, V. M. Rotello, J. M. Reid and P. Mukherjee, PLoS One, 2011, 6, 3–8 Search PubMed.
- L. Thiele, B. Rothen-Rutishauser, S. Jilek, H. Wunderli-Allenspach, H. P. Merkle and E. Walter, J. Controlled Release, 2001, 76, 59–71 CrossRef CAS PubMed.
- C. He, Y. Hu, L. Yin, C. Tang and C. Yin, Biomaterials, 2010, 31, 3657–3666 CrossRef CAS PubMed.
- M. D. Howard, M. Jay, T. D. Dziubla and X. Lu, J. Biomed. Nanotechnol., 2008, 4, 133–148 CrossRef CAS.
- E. Simone, B. J. Zern, A. Chacko, J. L. Mikitsh, S. Muro, R. V Stan and V. R. Muzykantov, Biomaterials, 2012, 33, 5406–5413 CrossRef CAS PubMed.
- W. Cai, K. Chen, Z.-B. Li, S. S. Gambhir and X. Chen, J. Nucl. Med., 2007, 48, 1862–1870 CrossRef CAS PubMed.
- J. Choi, J. C. Park, H. Nah, S. Woo, J. Oh, K. M. Kim, G. J. Cheon, Y. Chang, J. Yoo and J. Cheon, Angew. Chem., Int. Ed., 2008, 47, 6259–6262 CrossRef CAS PubMed.
- S. E. A. Gratton, P. D. Pohlhaus, J. Lee, J. Guo, M. J. Cho and J. M. DeSimone, J. Controlled Release, 2007, 121, 10–18 CrossRef CAS PubMed.
- Y. Sun, M. Yu, S. Liang, Y. Zhang, C. Li, T. Mou, W. Yang, X. Zhang, B. Li, C. Huang and F. Li, Biomaterials, 2011, 32, 2999–3007 CrossRef CAS PubMed.
- A. Chrastina and J. E. Schnitzer, Int. J. Nanomed., 2010, 5, 653–659 Search PubMed.
- C. C. Lee, M. Yoshida, J. M. J. Fréchet, E. E. Dy and F. C. Szoka, Bioconjugate Chem., 2005, 16, 535–541 CrossRef CAS PubMed.
- X. Shao, H. Zhang, J. R. Rajian, D. L. Chamberland, P. S. Sherman, C. A. Quesada, A. E. Koch, N. A. Kotov and X. Wang, ACS Nano, 2011, 5, 8967–8973 CrossRef CAS PubMed.
- J. C. Park, M. K. Yu, G. Il An, S. Il Park, J. Oh, H. J. Kim, J. H. Kim, E. K. Wang, I. H. Hong, Y. S. Ha, T. H. Choi, K. S. Jeong, Y. Chang, M. J. Welch, S. Jon and J. Yoo, Small, 2010, 6, 2863–2868 CrossRef CAS PubMed.
- S. Ballot, N. Noiret, F. Hindré, B. Denizot, E. Garin, H. Rajerison and J. P. Benoit, Eur. J. Nucl. Med. Mol. Imaging, 2006, 33, 602–607 CrossRef CAS PubMed.
- S. J. Kennel, J. D. Woodward, A. J. Rondinone, J. Wall, Y. Huang and S. Mirzadeh, Nucl. Med. Biol., 2008, 35, 501–514 CrossRef CAS PubMed.
- M. R. McDevitt, D. Chattopadhyay, J. S. Jaggi, R. D. Finn, P. B. Zanzonico, C. Villa, D. Rey, J. Mendenhall, C. A. Batt, J. T. Njardarson and D. A. Scheinberg, PLoS One, 2007, 2, 907 Search PubMed.
- R. Rossin, S. Muro, M. J. Welch, V. R. Muzykantov and D. P. Schuster, J. Nucl. Med., 2008, 49, 103–111 CrossRef PubMed.
- H. Akizawa, T. Uehara and Y. Arano, Adv. Drug Delivery Rev., 2008, 60, 1319–1328 CrossRef CAS PubMed.
- R. Haubner and H.-J. Wester, Curr. Pharm. Des., 2004, 10, 1439–1455 CrossRef CAS PubMed.
- M. Zhou, R. Zhang, M. Huang, W. Lu, S. Song, M. P. Melancon, M. Tian, D. Liang and C. Li, J. Am. Chem. Soc., 2010, 132, 15351–15358 CrossRef CAS PubMed.
- A. Vonarbourg, C. Passirani, P. Saulnier and J. P. Benoit, Biomaterials, 2006, 27, 4356–4373 CrossRef CAS PubMed.
- R. Gref, A. Domb, P. Quellec, T. Blunk, R. H. Müller, J. M. Verbavatz and R. Langer, Adv. Drug Delivery Rev., 1995, 16, 215–233 CrossRef CAS PubMed.
- S. J. Budijono, B. Russ, W. Saad, D. H. Adamson and R. K. Prud'homme, Colloids Surf., A, 2010, 360, 105–110 CrossRef CAS.
- H. Shen, S. Hong, R. K. Prud'Homme and Y. Liu, J. Nanopart. Res., 2011, 13, 4109–4120 CrossRef CAS.
- S. M. D'Addio and R. K. Prud'homme, Adv. Drug Delivery Rev., 2011, 63, 417–426 CrossRef PubMed.
- R. A. Petros and J. M. DeSimone, Nat. Rev. Drug Discovery, 2010, 9, 615–627 CrossRef CAS PubMed.
- C. W. M. Jing Han, Z. Zhu, H. Qian, A. R. Wohl, C. J. Beaman and T. R. Hoye, J. Pharm. Sci., 2012, 101, 4018–4023 CrossRef PubMed.
- S. M. D'Addio, W. Saad, S. M. Ansell, J. J. Squiers, D. Adamson, M. Herrero-Alonso, A. R. Wohl, T. R. Hoye, C. W. Macosko, L. D. Meyer, C. Vauthier and R. K. Prud'homme, J. Controlled Release, 2012, 162, 208–217 CrossRef PubMed.
- K. J. Edgar and S. N. Falling, J. Org. Chem., 1990, 55, 5287–5291 CrossRef CAS.
- B. J. Zern, A. M. Chacko, J. Liu, C. F. Greineder, E. R. Blankemeyer, R. Radhakrishnan and V. Muzykantov, ACS Nano, 2013, 7, 2461–2469 CrossRef CAS PubMed.
- N. Nishiyama and K. Kataoka, Pharmacol. Ther., 2006, 112, 630–648 CrossRef CAS PubMed.
- O. M. Koo, I. Rubinstein and H. Onyuksel, Nanomedicine, 2005, 1, 193–212 CAS.
- M. Yokoyama, J. Exp. Clin. Med., 2011, 3, 151–158 CrossRef CAS.
- X. Duan and Y. Li, Small, 2013, 9, 1521–1532 CrossRef CAS PubMed.
- P. Decuzzi, B. Godin, T. Tanaka, S. Y. Lee, C. Chiappini, X. Liu and M. Ferrari, J. Controlled Release, 2010, 141, 320–327 CrossRef CAS PubMed.
- C. E. Figueroa, P. Reider, P. Burckel, A. A. Pinkerton and R. K. Prud'homme, Ther. Delivery, 2012, 3, 1269–1279 CrossRef CAS.
- N. M. Alexander, J. Biol. Chem., 1974, 249, 1946–1952 CAS.
- T. J. Tadashi Kometani and D. S. Watt, Tetrahedron Lett., 1985, 26, 2043–2046 CrossRef.
- W. B. Smith, T. W. Proulx and F. Worth, Org. Magn. Reson., 1976, 8, 205–207 CrossRef CAS.
- A. M. Chacko, W. Qu and H. F. Kung, J. Med. Chem., 2008, 51, 5690–5701 CrossRef CAS PubMed.
- A. E. Bolton and W. M. Hunter, Biochem. J., 1973, 133, 529–539 CrossRef CAS PubMed.
- G. Sun, J. Xu, A. Hagooly, R. Rossin, Z. Li, D. A. Moore, C. J. Hawker, M. J. Welch and K. L. Wooley, Adv. Mater., 2007, 19, 3157–3162 CrossRef CAS.
- E. M. Jagoda, J. J. Vaquero, J. Seidel, M. V. Green and W. C. Eckelman, Nucl. Med. Biol., 2004, 31, 771–779 CrossRef CAS PubMed.
- S. M. Ansell, S. A. Johnstone, P. G. Tardi, L. Lo, S. Xie, Y. Shu, T. O. Harasym, N. L. Harasym, L. Williams, D. Bermudes, B. D. Liboiron, W. Saad, R. K. Prud'homme and L. D. Mayer, J. Med. Chem., 2008, 51, 3288–3296 CrossRef CAS PubMed.
- N. M. Pinkerton, M. E. Gindy, V. L. Calero-DdelC, T. Wolfson, R. F. Pagels, D. Adler, D. Gao, S. Li, R. Wang, M. Zevon, N. Yao, C. Pacheco, M. J. Therien, C. Rinaldi, P. J. Sinko and R. K. Prud'homme, Adv. Healthcare Mater., 2015, 4, 1376–1385 CrossRef CAS PubMed.
- R. Rossin, D. Pan, K. Qi, J. L. Turner, X. Sun, K. L. Wooley and M. J. Welch, J. Nucl. Med., 2005, 46, 1210–1219 Search PubMed.
- C. Glaus, R. Rossin, M. J. Welch and G. Bao, Bioconjugate Chem., 2010, 21, 715–722 CrossRef CAS PubMed.
- K. Novakova, M. Laznicek, F. Rypacek and L. Machova, J. Bioact. Compat. Polym., 2002, 17, 285–296 CrossRef CAS.
- Y. Yamamoto, Y. Nagasaki, Y. Kato and Y. Sugiyama, J. Controlled Release, 2001, 77, 27–38 CrossRef CAS PubMed.
- Y. Zhao, D. Sultan, L. Detering, S. Cho, G. Sun, R. Pierce, K. L. Wooley and Y. Liu, Angew. Chem., Int. Ed., 2014, 53, 156–159 CrossRef CAS PubMed.
- M. P. Melancon, W. Lu, Z. Yang, R. Zhang, Z. Cheng, A. M. Elliot, J. Stafford, T. Olson, J. Z. Zhang and C. Li, Mol. Cancer Ther., 2008, 7, 1730–1739 CrossRef CAS PubMed.
- Y. Wang, Y. Liu, H. Luehmann, X. Xia, P. Brown, C. Jarreau, M. Welch and Y. Xia, ACS Nano, 2012, 6, 5880–5888 CrossRef CAS PubMed.
- G. Zhang, Z. Yang, W. Lu, R. Zhang, Q. Huang, M. Tian, L. Li, D. Liang and C. Li, Biomaterials, 2009, 30, 1928–1936 CrossRef CAS PubMed.
- M. T. Peracchia, E. Fattal, D. Desmaële, M. Besnard, J. P. Noël, J. M. Gomis, M. Appel, J. D'Angelo and P. Couvreur, J. Controlled Release, 1999, 60, 121–128 CrossRef CAS PubMed.
- D. Bazile, C. Prud'Homme, M. T. Bassoullet, M. Marlard, G. Spenlehauer and M. Veillard, J. Pharm. Sci., 1995, 84, 493–498 CrossRef CAS PubMed.
- M. J. Adam and D. S. Wilbur, Chem. Soc. Rev., 2005, 34, 153–163 RSC.
- M. Akbulut, P. Ginart, M. E. Gindy, C. Theriault, K. H. Chin, W. Soboyejo and R. K. Prud'homme, Adv. Funct. Mater., 2009, 19, 718–725 CrossRef CAS.
- S. M. D'Addio, S. Baldassano, L. Shi, L. Cheung, D. H. Adamson, M. Bruzek, J. E. Anthony, D. L. Laskin, P. J. Sinko and R. K. Prud'homme, J. Controlled Release, 2013, 168, 41–49 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5tb02172c |
| ‡ Current address: Duke-NUS Medical School, 169857, Singapore. |
| This journal is © The Royal Society of Chemistry 2016 |