
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Construction of a tissue-specific transcription factor-tethered extracellular matrix protein via coiled-coil helix formation
SokeLee
Siew
,
Mami
Kaneko
,
Masayasu
Mie
* and
Eiry
Kobatake
Department of Environmental Chemistry and Engineering, Interdisciplinary Graduate School of Science and Engineering, Tokyo Institute of Technology, 4259 Nagatsuta, Midori-ku, Yokohama 226-8502, Japan. E-mail: mie.m.aa@m.titech.ac.jp; Fax: +81-45-924-5779; Tel: +81-45-924-5414
First published on 11th March 2016
Abstract
Tissue-specific transcription factors are key regulators of cellular differentiation. Previously, we succeeded in introducing basic helix–loop–helix tissue-specific transcription factor proteins into cells to induce cellular differentiation. Based on these results, we decided to focus on the use of tissue-specific transcription factor proteins in the construction of biomaterials. In this proof-of-concept study, we demonstrate the construction of a tissue-specific transcription factor-tethered extracellular matrix protein. Here, the tissue-specific transcription factor Olig2 was tethered to a designed artificial extracellular matrix protein via coiled-coil helix formation. Tethered Olig2 was introduced into mouse embryonic carcinoma P19 cells attached to our designed extracellular matrix protein, and was shown to exhibit the ability to induce neural differentiation.
Introduction
The design of novel biomaterials that supply both cell adhesion molecules and signaling molecules for proliferation and differentiation is important for advancing the field of tissue engineering. For the construction of such biomaterials, synthetic organic polymers and proteins have been introduced into artificial extracellular matrices (ECMs).1One of the strategies for the design of artificial ECMs is based on the combination of a stable structural unit and active functional units that control cellular functions such as cell adhesion, proliferation and differentiation.1–3 The widely utilized active functional unit is cell-adhesive peptides such as RGD.4,5 These peptides were introduced to various structural units by chemical strategies or genetic engineering. Fittkau et al. demonstrated that the polyethylene glycol (PEG) hydrogels chemically modified with RGD and YIGSR peptides enhanced the migration of microvascular endothelial cells compared to RGD alone.6 A combination of functional units has showed synergistic effects.7,8 To improve the functional capacities of these artificial ECMs, growth factors were attached to artificial ECMs, both covalently and non-covalently.9–12 For the construction of biomaterials, combinations of ECMs and growth factors have been well studied owing to the fact that some growth factors are bound to ECMs under physiological conditions.13–15 Generally, strategies employed for the construction of biomaterials have mimicked these physiological conditions. Thus, while a combination of growth factors and ECMs is a reasonable strategy for the construction of artificial biomaterials, such a strategy is often restricted to certain varieties of artificial ECMs. Besides the growth factors, bioactive proteins which regulate cellular functions should be focused for the combination of ECMs to expand the varieties of artificial ECMs.
Tissue-specific transcription factors are key regulators of cellular differentiation. Forced expression of tissue-specific transcription factors by gene transfection is able to induce differentiation of specific lineages.16 Recently, the differentiation of various cell types was induced via the transfection of a combination of several transcription factor genes.17–20 In addition to gene transfection, transcription factor proteins can also be introduced into cells.21–27 Generally, the introduction of proteins into cells has been difficult. However, short peptides called protein transduction domains (PTDs) or cell penetrating peptides (CPPs) facilitate protein penetration into cells, so that a protein of interest can be delivered into cells upon fusion with a PTD.28 Moreover, some kinds of transcription factor proteins can be introduced into cells without addition of a PTD because they have PTDs within their primary sequences.21,23–25 In our previous studies, the basic helix–loop–helix (bHLH) tissue-specific transcription factor proteins NeuroD2, MyoD and Olig2 were introduced into cells without external PTDs, and were shown to induce cellular differentiation.23–25 The fact that transcription factor proteins can be introduced into cells leads us to the idea that transcription factor proteins could potentially be used for the construction of biomaterials as similar to growth factors. Immobilization of transcription factor proteins represents a novel strategy for the construction of biomaterials, and has the possibility to expand the varieties of biomaterials.
In this study, a novel strategy for the construction of biomaterials is introduced by tethering a tissue-specific transcription factor protein to an artificial ECM protein. Here, oligodendrocyte and motor neuron specific transcription factor Olig2 were tethered to a previously constructed artificial ECM protein via coiled-coil helix formation (Scheme 1). Coiled-coil interactions are utilized for the noncovalent connection of two macromolecules in the design of biomaterials.29 Coiled-coil interactions were used not only for design of self-assembled biomaterials, but also for tethering of growth factors.30–35 In this experiment, helix peptides, named ACID-p1 (helix A) and BASE-p1 (helix B), were fused to Olig2 and artificial ECM proteins, respectively. These peptides designed by O'shea et al. can form stable coiled-coil interactions.36 Using these peptides, we have designed the growth factor-tethered artificial ECMs.33–35 The artificial ECM named EAEC, previously constructed in our laboratory, is comprised of 12 repeats of the APGVGV peptide motif derived from elastin as a structural unit (E12), as well as the AG73 peptide sequence derived from laminin and the C3 peptide sequence which binds to neural cell adhesion molecules (NCAMs) derived from the synthetic peptide library, for cell adhesion and enhanced neurite outgrowth.35 As a proof of concept, tethered Olig2 was internalized into mouse embryonic carcinoma P19 cells and the ability to induce neural differentiation was investigated.
 | ||
| Scheme 1 Schematic depiction of this study. Tissue-specific transcription factor protein is tethered to an artificial ECM protein via coiled-coil helix formation. | ||
Materials and methods
Construction of plasmids
The plasmid pET-His6-helixA-Olig2 was constructed as follows. The helixA sequence with a histidine tag was amplified from pET-His6-helixA-scVEGF121, which was previously constructed in our laboratory using primer sets GCGCTATCATGCCATACCGC and GCTAGCTTGAGCGAGCTCTTTCTCCA.34 The amplified fragment was cloned into pUC18 and its sequence was confirmed. The resulting plasmid, pUC18-PfM I-His6-helixA-Nhe I, was digested with PfM I and Nhe I. The digested fragment was inserted into pET-His6-Olig2, also constructed in our previous study, and digested with the same restriction enzymes.25 The resulting plasmid for protein expression in E. coli was named pET-His6-helixA-Olig2.Construction of the plasmids pET-His-EAEC, for expression of His-EAEC protein (EAEC), and pET-His EAEC-HB, for expression of the helix B peptide fused-EAEC protein (EAEC-HB), has been described in our previous publication.35
Expression and purification of constructed proteins
E. coli KRX competent cells were transformed with pET-His6-helixA-Olig2 for expression of the His6-helixA-Olig2 protein (HA-Olig2). Transformed cells were cultured in Luria–Bertani (LB) media with 50 μg ml−1 of ampicillin at 37 °C until an OD660 of approximately 0.6 was reached. Protein expression was induced via addition of rhamnose (0.1%) and isopropyl-β-D(−)-thiogalactopyranoside (IPTG, 0.5 mM).After induction of protein expression, cells were harvested by centrifugation and the cell pellets were resuspended in phosphate buffer (16 mM Na2HPO4, 4 mM NaH2PO4, pH 7.6) with 150 mM NaCl. Then, resuspended cells were lysed by sonication (Biorupter). The cell lysates were separated into soluble and insoluble fractions by centrifugation and the supernatants were added to His-select Nickel affinity Resin (SIGMA). After rotation for 45 min at 4 °C, samples were washed with wash buffer (0.5 M NaCl, 20 mM phosphate buffer, pH 7.6) containing 10 mM imidazole. The proteins were subsequently eluted with wash buffer containing 500 mM imidazole, and the resulting solutions were dialyzed against phosphate buffered saline (PBS: 137 mM NaCl, 8.1 mM Na2HPO4, 2.7 mM KCl, 1.47 mM KH2PO4, pH7.4) using a Slide-A-lyzer dialysis cassette (Pierce: MW 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, 0.5–3 ml).
000, 0.5–3 ml).
For expression of EAEC and EAEC-HB, pET-His-EAEC and pET-His-EAEC-HB plasmids were transfected into E. coli BL21(DE3) competent cells. Transformed E. coli cells were cultured in LB medium with ampicillin. Cells were grown to the mid-log phase at 37 °C, at which time IPTG was added to a final concentration of 1 mM. Cultures were grown at 16 °C for an additional 4 h. Then, cells were harvested by centrifugation and resuspended in PBS. The resuspended cells were subsequently lysed by sonication. The cell lysates were separated into soluble and insoluble fractions by centrifugation. Proteins were purified from the insoluble fraction after solubilization in PBS with 8 M urea. Solubilized inclusion bodies were applied to His-select Nickel affinity Resins and incubated for 45 min. After washing with wash buffer containing 10 mM imidazole and 4 M urea, proteins were eluted by phosphate buffer with 500 mM imidazole and 4 M urea. The resulting protein solutions were dialyzed using a Slide-A-Lyzer dialysis cassette (MW 10000, 0.5–3 ml) against PBS.
The purified proteins were analyzed by SDS-PAGE (15% acrylamide gels) and concentrations were determined using a BCA assay kit (Pierece).
Cell culture
The mouse embryonic carcinoma cell line P19 (Riken Bioresource Center) was cultured in αMEM including 10% (v/v) fetal bovine serum (FBS), penicillin (100 units per ml) and streptomycin (0.1 mg ml−1). During induction of differentiation, cells were cultured in DMEM/Ham's F-12 with 2% (v/v) B-27™ Supplement Minus Vitamin A (Gibco), and subsequently incubated in a 37 °C humidified chamber (95% (v/v) air and 5% (v/v) CO2).Evaluation of the cellular membrane transduction ability of soluble HA-Olig2
For evaluation of the transduction ability of HA-Olig2, purified protein was modified with Oregon Green 488 carboxylic acid and succinimidyl ester (molecular probes) according to the manufacturer's instructions. P19 cells, seeded at 7 × 104 cells per 35 mm tissue culture dish (BD), were incubated for 12 h with a fluorescently labeled protein at a final concentration of 0.2 μM. Cells were subsequently trypsinized and inoculated on 35 mm glass base dishes (Iwaki). After 2–3 hours, the cells were observed using laser scanning confocal microscopy (Olympus: FV300 system).Induction of neural differentiation by soluble HA-Olig2
P19 cells were plated onto a 6-well tissue culture plate (BD) at 6 × 104 cells per well. After overnight incubation, the medium was changed to DMEM/Ham's F-12 containing 2% (v/v) B-27™ Supplement minus Vitamin A, and HA-Olig2 or synthetic helix A peptide (HA peptide : AQLEKELQALEKENAQLEWELQALEKELAQ) purchased from MBL (Japan) was added to the cells. The final protein or peptide concentration was 0.1 μM. The same procedure was performed every 24 hours.After 5 days of culture, cells were immunostained with anti-β-III tubulin antibody (R&D systems) and anti-mouse Alexa 488 conjugated antibody (molecular probes), as described in our previous study.25 Nuclei were stained with Hoechst 33258.
Cell-adhesion activity of EAEC-HB
Cell-adhesion assays were performed in a 3.5 cm suspension culture dish (SUMILON MS-1135R). Purified proteins (EAEC, EAEC-HB, E12), at a concentration of 1 nM, or laminin, (SIGMA) at a concentration of 1 μg ml−1, were added to the surface of the dishes. After incubation for 1 h at 37 °C, unbound proteins were removed by washing. Then P19 cells were seeded at a density of 5 × 104 cells per dish followed by incubation for 24 h at 37 °C. Finally, non-attached cells were removed by washing, while attached cells, in 4 randomly chosen fields of a dish, were counted using a microscope. Each assay was repeated 3 times.HA-Olig2 tethering via a coiled-coil helix structure
To coat the surface of the wells, 100 nM of EAEC or EAEC-HB was added to a 96-well plate (costar 3361) for non-tissue culture and incubated overnight at 4 °C. After removal of unbound protein, the plate was washed with PBS containing 0.05% of Tween 20 (PBS-T) followed by blocking with 3% BSA for 1 hour. Next, various concentrations (10 nM, 100 nM, 1 μM) of HA-Olig2 were added, and the plates were incubated for 2 h at 37 °C. A solution of 100 μl of 1/2000 diluted rabbit anti-Olig2 antibody (Abcam) was allowed to react for 40 min at room temperature. After washing with PBS-T, peroxidase-conjugated anti-rabbit IgG antibody was added and incubated for 40 min at room temperature. Finally, the plates were washed with PBS-T and the HRP substrate was added (Pierece). The reaction was stopped after 3 min by the addition of 1 M HCl, at which point the absorbance at 450 nm was measured using a microplate reader.For inhibition of HA-Olig2 binding to EAEC-HB, the EAEC-HB coated plate was treated with the HA peptide (1 μM) before the addition of HA-Olig2 (100 nM), and the plate was incubated for 2 h at 37% followed by washing with PBS-T.
Induction of neural differentiation by tethered HA-Olig2
HA-Olig2 (1 μM) was tethered to an EAEC-HB-coated 6-well suspension culture plate (100 nM), and P19 cells in αMEM containing 10% FBS were inoculated at a cell density of 5 × 104 cells per well. After overnight incubation, the medium was changed to DMEM/Ham's F-12 containing 2% (v/v) B-27™ Supplement minus Vitamin A. Cells were immunostained for 5 days of culture with anti-β-III tubulin antibody, as previously mentioned.Results and discussion
Expression and purification of helix-fused proteins
In our previous study, basic fibroblast growth factor (bFGF), single chain vascular endothelial growth factor (scVEGF) and epidermal growth factor (EGF) were tethered to artificial ECMs via coiled-coil helix formation.33–35 Here, instead of growth factors, a tissue-specific transcription factor protein was tethered to an artificial ECM protein. For tethering, Olig2 was fused to the helix A peptide (Fig. 1A). EAEC-HB, which exhibits both cell adhesive and neurite outgrowth enhancement activities, was employed as the artificial ECM.35 The helix-fused proteins were expressed in E. coli. HA-Olig2 (37.3 kDa) was expressed in the soluble fraction and purified HA-Olig2 was analyzed by SDS-PAGE (Fig. 1B). The purity of HA-Olig2 was slightly low. However, the main band in the lane was regarded as HA-Olig2. EAEC-HB (23.1 kDa) was purified from the insoluble fraction in a manner similar to that used in our previous study (Fig. 1C).35 These proteins appeared larger. These discrepancies in molecular weights were shown in our previous experiments in the case of proteins without helix peptides.25,35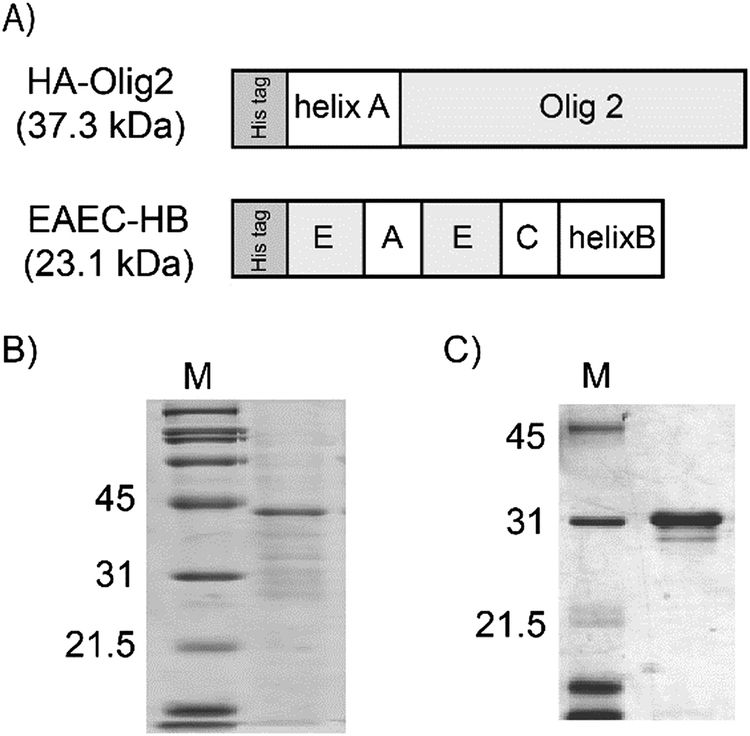 | ||
| Fig. 1 Design and expression of constructed fusion proteins. EAEC is designed with 12 repeats of the elastin-derived APGVGV sequence denoted as E, laminin derived peptide, RKRLQVQLSIRT, denoted as A, and NCAM binding synthetic peptide, ASKKPKRNIKA, denoted as C (A). Results of SDS-PAGE analysis. Purified HA-Olig2 (B) and EAEC-HB (C). The lanes M indicate the molecular weight marker. | ||
Evaluation of the cellular membrane transduction ability of soluble HA-Olig2
Olig2 exhibits the ability to be transduced into cells by its own PTD.25 Here, it was evaluated whether HA-Olig2 also retained this transduction ability. HA-Olig2 was modified with Oregon Green 488 and added to P19 cells. Similar to Olig2 without helix A, HA-Olig2 showed fluorescence signals (Fig. 2A). Before observation, cells were treated with trypsin and re-inoculated on a glass base dish to remove adsorbed HA-Olig2 on the cell membrane. From the cross-sectional view, it was confirmed that the fluorescence signal was derived from internalized HA-Olig2 (Fig. 2B). These results demonstrated that fusion with the helix A peptide preserves the transduction ability of HA-Olig2. | ||
| Fig. 2 (A) Cellular membrane transduction ability of soluble HA-Olig2. Proteins were modified with Oregon Green 488. Control cells were incubated with Oregon Green 488. The scale bar represents 50 μm. (B) Cross-sectional view of the cell with HA-Olig2. The scale bar represents 10 μm. | ||
After confirming the transduction ability of HA-Olig2, the effects of helix A on the induction of neural differentiation were evaluated (Fig. 3). In our previous study, we have shown that Olig2 protein has ability to induce neural differentiation after internalization.25 Prior to protein addition, cells seeded onto tissue culture dishes were attached to each other. However, cells were dispersed within 24 hours by the addition of HA-Olig2. This phenomenon was not observed in cells cultured without HA-Olig2. In culture with the HA peptide alone, some cells were dispersed, but most of the cells were still attached to each other similar to cells cultured without additives. After 5 days of culture, P19 cells cultured without additives and with the HA peptide alone did not show the elongation of neuritis, and remained attached to each other. From these results, it is found that the HA peptide itself does not have an ability to induce neural differentiation. In contrast, cells cultured with HA-Olig2 were found to exhibit elongated neurites. This was also confirmed via immunostaining of a neural marker with anti-β-III tubulin antibody. The neurite-elongated cells were shown to yield a fluorescence signal. These results suggest that HA-Olig2 was able to retain its transduction ability and induce neural differentiation, even upon fusion with helix A.
 | ||
| Fig. 3 Neural differentiation induction ability of soluble HA-Olig2. Cells were cultured without additives (control), with the HA peptide alone (HA peptide) and with HA-Olig2 (HA-Olig2). After 5 days of culture, cells were stained with anti-β-III tubulin antibody (green). Nuclei were stained with Hoechst 33258 (blue). The scale bar represents 50 μm. | ||
Evaluation of P19 cell binding to EAEC-HB
In our previous study, EAEC was found to exhibit cell adhesive activity in rat pheochromocytoma cell (PC12) culture.35 Herein, the binding of P19 cells to EAEC-HB was evaluated (Fig. 4). Before seeding of P19 cells, dishes were coated with E12 protein consisting only of (APGVGV)12, laminin, EAEC and EAEC-HB. After coating with the proteins, cells were seeded and incubated for 24 h. Prior to cell-counting, un-attached cells were removed by washing with PBS. Then, the number of cells was counted using microscopy. Cells seeded onto the dish without protein, as well as those coated with the E12 protein, were not attached to the dishes. On the other hand, cells seeded onto dishes coated with EAEC, either with or without helix B, were well attached, similar to those of the laminin-coated dishes. These results suggest that P19 cells exhibited a similar ability to attach to EAEC-HB as to that of PC12 cells. | ||
| Fig. 4 Cell adhesion activity of EAEC-HB. (A) Cells were cultured on a protein-coated plate. The scale bar represents 100 μm. (B) Number of attached cells. Cells were counted in 4 randomly chosen fields using microscopy. Each assay was repeated 3 times. Error bars shown indicate the standard deviation. | ||
HA-Olig2 tethering to EAEC-HB via coiled-coil helix formation
To confirm tethering of HA-Olig2 to EAEC-HB via coiled-coil helix formation, various concentrations of HA-Olig2 were added to dishes coated with 100 nM of either EAEC-HB or EAEC. Tethered HA-Olig2 was detected via the anti-Olig2 antibody. When HA-Olig2 was added to a surface coated with EAEC without helix B, the signal was increased gradually, as a function of the protein concentration. This is most likely the result of non-specific binding between EAEC and the helix peptide of HA-Olig2 due to hydrophobic interactions. However, the signal derived from the combination of HA-Olig2 and EAEC-HB was increased remarkably. Fig. 5 shows the differences observed for tethered HA-Olig2 at 100 nM. The signal derived from the combination of HA-Olig2 and EAEC-HB showed the highest signal among these conditions. To confirm tethering of HA-Olig2 to EAEC-HB via coiled-coil helix formation, EAEC-HB on the dish surface was treated with the HA peptide before the addition of HA-Olig2. In our previous study, we have confirmed that binding between FGF fused with helix A (HA-FGF) and the artificial ECM fused with helix B (ERE-HB) was due to the formation of coiled-coil structures between helix A and helix B by the addition of the competitive helix A or helix B peptide.33 As expected, the signal derived from the combination of HA-Olig2 and EAEC-HB treated with the HA peptide was lower than that of HA-Olig2 and EAEC-HB. These results suggest that HA-Olig2 was indeed tethered to EAEC-HB via coiled-coil helix formation. | ||
| Fig. 5 Evaluation of tethered HA-Olig2. After coating with EAEC or EAEC-HB (100 nM), HA-Olig2 (100 nM) was added (EAEC, EAEC-HB). For inhibition of HA-Olig2 binding, EAEC-HB was treated with the HA peptide before the addition of HA-Olig2 (EAEC-HB treated with HA peptide). Tethered HA-Olig2 was detected using anti-Olig2 antibody. | ||
Induction of neural differentiation by tethered HA-Olig2
Finally, neural differentiation of P19 cells was induced by tethered HA-Olig2. The P19 cells were attached well on EAEC-HB with HA-Olig2 though the number of attached cells was lower than EAEC-HB without HA-Olig2 (data not shown). It was supposed that cell adhesive domains were covered with tethered HA-Olig2. After 5 days of culture, cells were stained with anti-β-III tubulin antibody. The P19 cells that were seeded onto the tissue culture dish did not show morphological changes, and stained with anti-β-III tubulin antibody (Fig. 6A). When P19 cells were seeded onto the EAEC coated dish, a few cells showed neurite outgrowth (Fig. 6B). It is suggested that spontaneous differentiation was enhanced by AG73 and C3 peptides in EAEC. On the other hand, when P19 cells were seeded onto EAEC-HB with HA-Olig2, many cells exhibited elongated neurites, as confirmed by immunostaining (Fig. 6C). These results suggest that tethered HA-Olig2 protein was able to introduce into cells and induce neural differentiation of P19 cells. The mechanism of neural differentiation induced by tethered HA-Olig2 is still under investigation. Cells cultured in the medium derived from the dish coated with EAEC-HB tethering HA-Olig2 did not show neural morphological changes, but showed the dispersion of cells similar to culture with soluble HA-Olig2 (data not shown). It was suggested that HA-Olig2 was released from EAEC-HB and internalized into cells. When cells were cultured on EAEC-HB tethering HA-Olig2, the local concentration of released HA-Olig2 would be enough for induction of differentiation. | ||
| Fig. 6 Induction of neural differentiation by tethered HA-Olig2. Cells were stained with anti-β-III tubulin antibody (green). Nuclei were stained with Hoechst 33258 (blue). (A) Control culture and cells cultured on a tissue culture plate. (B) Cells cultured on EAEC without HA-Olig2. (C) Cells cultured on HA-Olig2 tethered EAEC-HB. The scale bar represents 100 μm. | ||
To our knowledge, this is the first report of the construction of biomaterials using transcription factor proteins.
Conclusions
In this study, a novel strategy for the construction of biomaterials was developed. A tissue-specific transcription factor protein was tethered to an artificial extracellular matrix protein via formation of a coiled-coil helix structure. The tethered tissue-specific transcription factor protein was introduced into cells and was shown to exhibit the ability to induce cellular differentiation. This strategy may provide a new option for the construction of biomaterials.Acknowledgements
This work was supported by Grants-in-Aid for Scientific Research from Japan Society for the Promotion of Science (JSPS).References
- M. P. Lutolf and J. A. Hubbell, Nat. Biotechnol., 2005, 23, 47–55 CrossRef CAS PubMed.
- R. L. DiMarco and S. C. Heilshorn, Adv. Mater., 2012, 24, 3923–3940 CrossRef CAS PubMed.
- J. H. Collier, J. S. Rudra, J. Z. Gasiorowski and J. P. Jung, Chem. Soc. Rev., 2010, 39, 3413–3424 RSC.
- U. Hersel, C. Dahmen and H. Kessler, Biomaterials, 2003, 24, 4385–4415 CrossRef CAS PubMed.
- E. Kobatake, K. Onoda, Y. Yanagida and M. Aizawa, Biomacromolecules, 2000, 1, 382–386 CrossRef CAS PubMed.
- M. H. Fittkau, P. Zilla, D. Bezuidenhout, M. P. Lutolf, P. Human, J. A. Hubbell and N. Davies, Biomaterials, 2005, 26, 167–174 CrossRef CAS PubMed.
- M. Nakamura, M. Mie, H. Mihara, M. Nakamura and E. Kobatake, Biomaterials, 2008, 29, 2977–2986 CrossRef CAS PubMed.
- M. Nakamura, M. Mie, H. Mihara, M. Nakamura and E. Kobatake, J. Biomed. Mater. Res., 2009, 91B, 425–432 CrossRef CAS PubMed.
- Z. Ma, C. Gao, Y. Gong and J. Shen, Biomaterials, 2005, 26, 1253–1259 CrossRef CAS PubMed.
- I. Elloumi, R. Kobayashi, H. Funabashi, M. Mie and E. Kobatake, Biomaterials, 2006, 27, 3451–3458 CrossRef CAS PubMed.
- R. Mammadov, B. Mammadov, M. O. Guler and A. B. Tekinay, Biomacromolecules, 2012, 13, 3311–3319 CrossRef CAS PubMed.
- E. A. Phelps, D. M. Headen, W. R. Taylor, P. M. Thule and A. J. Garcia, Biomaterials, 2013, 34, 4602–4611 CrossRef CAS PubMed.
- R. R. Chen and D. J. Mooney, Pharm. Res., 2003, 20, 1103–1112 CrossRef CAS.
- R. O. Hynes, Science, 2009, 326, 1216–1219 CrossRef CAS PubMed.
- J. Taipale and J. Keski-Oja, FASEB J., 1997, 11, 51–59 CAS.
- A. B. Lassar, B. M. Paterson and H. Weintraub, Cell, 1986, 47, 649–656 CrossRef CAS PubMed.
- Q. Zhou, J. Brown, A. Kanarek, J. Rajagopal and D. A. Melton, Nature, 2008, 455, 627–632 CrossRef CAS PubMed.
- M. Ieda, J. D. Fu, P. Delgado-Olguin, V. Vedantham, Y. Hayashi, B. G. Bruneau and D. Srivastava, Cell, 2010, 142, 375–386 CrossRef CAS PubMed.
- T. Vierbuchen, A. Ostermeier, Z. P. Pang, Y. Kokubu, T. C. Südhof and M. Wernig, Nature, 2010, 463, 1035–1041 CrossRef CAS PubMed.
- S. Sekiya and A. Suzuki, Nature, 2011, 475, 390–393 CrossRef CAS PubMed.
- H. Noguchi, H. Kaneto, G. C. Weir and S. Bonner-Weir, Diabetes, 2003, 52, 1732–1737 CrossRef CAS PubMed.
- H. Noguchi, S. Bonner-Weir, F. Y. Wei, M. Matsushita and S. Matsumoto, Diabetes, 2005, 54, 2859–2866 CrossRef CAS PubMed.
- T. Noda, R. Kawamura, H. Funabashi, M. Mie and E. Kobatake, J. Biotechnol., 2006, 126, 230–236 CrossRef CAS PubMed.
- T. Noda, T. Fujino, M. Mie and E. Kobatake, Biochem. Biophys. Res. Commun., 2009, 382, 473–477 CrossRef CAS PubMed.
- M. Mie, M. Kaneko, F. Henmi and E. Kobatake, Biochem. Biophys. Res. Commun., 2012, 427, 531–536 CrossRef CAS PubMed.
- M. S. Sung, J. Y. Mun, O. Kwon, K. S. Kwon and D. B. Oh, Biochem. Biophys. Res. Commun., 2013, 437, 156–161 CrossRef CAS PubMed.
- K. Yomogida, S. Wu, B. Baravati, C. Avendano, T. Caldwell, B. Maniaci, Y. Zhu and C. Q. Chu, Biochem. Biophys. Res. Commun., 2013, 434, 263–267 CrossRef CAS PubMed.
- A. van den Berg and S. F. Dowdy, Curr. Opin. Biotechnol., 2011, 22, 888–893 CrossRef CAS PubMed.
- M. Pechar and R. Pola, Biotechnol. Adv., 2013, 31, 90–96 CrossRef CAS PubMed.
- N. Mehrban, B. Zhu, F. Tamagnini, F. I. Young, A. Wasmuth, K. L. Hudson, A. R. Thomson, M. A. Birchall, A. D. Randall, B. Song and D. N. Woolfson, ACS Biomater. Sci. Eng., 2015, 1, 431–439 CrossRef CAS PubMed.
- C. Boucher, J. C. Ruiz, M. Thibault, M. D. Buschmann, M. R. Wertheimer, M. Jolicoeur, Y. Durocher and G. De Crescenzo, Biomaterials, 2010, 31, 7021–7031 CrossRef CAS PubMed.
- M. Pechar, R. Pola, R. Laga, K. Ulbrich, L. Bednarova, P. Malon, I. Sieglova, V. Kral, M. Fabry and O. Vanek, Biomacromolecules, 2011, 12, 3645–3655 CrossRef CAS PubMed.
- E. Kobatake, R. Takahashi and M. Mie, Bioconjug. Chem., 2011, 22, 2038–2042 CrossRef CAS PubMed.
- Y. Assal, M. Mie and E. Kobatake, Biomaterials, 2013, 34, 3315–3323 CrossRef CAS PubMed.
- M. Mie, S. Sasaki and E. Kobatake, Biomed. Mater., 2014, 9, 015004 CrossRef PubMed.
- E. K. O'Shea, K. J. Lumb and P. S. Kim, Curr. Biol., 1993, 3, 658–667 CrossRef.
| This journal is © The Royal Society of Chemistry 2016 |